

Summary
Single particle cryo-electron microscopy (cryo-EM) is a technique aimed at structure determination of large macromolecular complexes in their unconstrained, physiological conditions. The power of the method has been demonstrated in selected studies where for highly symmetric molecules the resolution attained permitted backbone tracing. However, most molecular complexes appear to exhibit intrinsic conformational variability necessary to perform their functions. Therefore, it is now increasingly recognized that sample heterogeneity constitutes a major methodological challenge for cryo-EM. To overcome it dedicated experimental and particularly computational multi-particle approaches have been developed. Their applications point to the future of cryo-EM as an experimental method uniquely suited to visualize the conformational modes of large macromolecular complexes and machines.
Introduction
Cryo-electron microscopy (cryo-EM) in combination with digital image processing (single particle approach) is well suited to determine structures of large macromolecular complexes and machines at intermediate resolution (6-30 Å) [1]. In the course of single particle image processing, thousands to hundreds of thousands or even millions of particle projections are combined to yield an averaged, 3-D density map [2]. Structure determination using single particle technique is based on the premise that the specimen can be purified such that the macromolecular complexes exist in a soluble, isolated and structurally identical form. Only then the EM images can be treated as if they are projections of a single particle. Since modern microscopes can deliver resolution better than 1Å, the short exposure time imposed by the possible radiation damage to the specimen is one major obstacle and precludes one from routinely achieving sufficient total Signal-to-Noise Ratio (SNR) that would result in atomic resolution of determined structures. Indeed, structures that permit backbone tracing of the entire secondary structure are at hand, at least for highly symmetric particles such as (icosahedral) virus capsids [3-5] or GroEL [6].
However, it became increasingly clear in recent years that the main premise of the single particle approach of the macromolecular complex existing in multiple identical copies of the same structure is rarely if ever fulfilled. On one hand, there is compositional heterogeneity. Macromolecular assemblies are instable to some extent. As biochemical purification protocols are often a compromise between intactness of the complex and purity, the random loss of certain components during preparation can result in subpopulations of incomplete or damaged complexes. On the other hand, there is conformational heterogeneity. Even relatively stable macromolecular machines such as the ribosome undergo large conformational changes during their functional cycle and therefore can occur in a mixture of conformational states [7-11]. Therefore, for most of the macromolecular complexes studied, sample heterogeneity appears to be a major obstacle in structure determination in addition to difficulties caused by low SNR of the data. Depending on the extent of the heterogeneity and the degree to which the variability in the structure is localized, there are several ways in which the differences between the individual complexes can influence the resulting 3-D map because also steps of the image processing (alignment, classification, determination of Euler angle) can be adversely affected.
A variability of the complex that is not localized but is uniformly distributed over its structure should not affect the alignment in a major way. This kind of variability will mainly affect the high spatial frequencies in the reconstructed object, resulting in decreased resolution. Nevertheless, the 3-D map will be correct in its general features. Another possibility is that a small region of the complex is significantly more flexible than the bulk of the structure. In this case the alignment of projection data will be dominated by the major – and invariable - part of the complex, yielding basically the correct structure, except that the spatial resolution within the region of the flexible domain will be lower than the average resolution of the cryo-EM map. In addition, flexible components may have lower densities than the average density of the map (as a result of “conformational averaging”), thus becoming invisible at the standard density threshold selected to visualize the complex. Substoichiometric occupancy of components of the complex (for example ligands) will result in similar effects precluding their direct visualization. However, the most serious problem appears to be caused by a third type of heterogeneity caused by major conformational variability manifesting itself in coexistence of multiple substates of the complex in the sample. The average over such a mixture will not represent the structure of the complex faithfully. It is not even the case that the solved structure is a simple average of the individual complexes whose projections were recorded in their basic states, since the 3D projection alignment will most likely introduce additional errors in orientation parameters.
In the presence of strong compositional and/or conformational heterogeneity it can be prohibitively difficult to determine a structure using single particle EM technique. The adverse influence of even small variations in the structure of the macromolecular complexes on the 3D reconstruction is becoming increasingly dominant at high spatial frequencies and thus hampers the improvement of the resolution of a given specimen. Therefore, it is now recognized that heterogeneity in the cryo-EM data set is a major limiting factor in structure determination by single particle cryo-EM and a variety of both experimental and computational methods have been proposed to address the problem. However, it is also recognized that although admittedly posing a methodological challenge, the analysis and visualization of conformational modes of a macromolecular assembly in solution yields biological information of utmost importance. In what can be considered multi-particle cryo-EM, one expects new information about the structural dynamics of macromolecular complexes and machines to emerge in the years to come.
Structural heterogeneity of the specimen significantly complicates computational methodology required for structure determination. First of all, one has to establish without any doubt that such variability is indeed present in the data and that the difficulties in structure determination or refinement are not caused by low quality of images or simply by mistakes in computational protocols employed. This is not at all a trivial problem, because reliable tools available to validate a cryo-EM map in the absence of external information are still lacking. Nevertheless, considerable progress has been made to tackle the problem of sample heterogeneity and a variety of approaches have been developed. In the following, we will give an overview of computational multi particle approaches. Advances in sample purifications [12] and grid preparation [13] are also of great significance in this context, but have to remain beyond the scope of this review.
2D Analysis
Careful analysis of EM projection images in 2D is always recommended as 2D single particle methods are in many instances theoretically sound and much better developed than their 3D counterparts, not to mention much shorter time of calculations. This is particularly true for computational tools relevant for the analysis of conformational flexibility, such as calculation of variance fields of images, both in real and in Fourier spaces [14], eigenanalysis of variance/covariance matrix of the data, or cluster analysis of aligned images. The most straightforward approach is to compare similarly looking 2D averages and interpret the differences as the evidence for flexibility of the complex [15,16].
While the direct analysis of average and variance fields may be in practice cumbersome, the eigenanalysis proved to be an indispensable tool in cryo-EM [17]. Originally, the method was used to detect staining artifacts in the data [18] or slight misalignments due to “rocking” of the molecule on the support grid [17]. However, in the absence of obvious artifacts, it is possible to interpret the results of eigenanlysis in terms of conformational states of the molecule [19].
As powerful as 2D approach can be, it is not in wide use due to difficulties with unequivocal interpretation of the result. A subset of 2D images distinguished by a high degree of homogeneity has to be identified in a larger set of images with the help of clustering techniques. However, what is a homogeneous subset is a subjective decision and while clustering techniques can serve as guiding tools, it is impossible to support conclusions by any statistical analysis. The observed “flexibility” might be quite likely due to grouping of projection views at slightly different angular directions and in the absence of 3D structure this possibility cannot be disproved. Nevertheless, analysis of 2D data supported by PCA methodology can provide unique insight into conformational stability of the sample and in recent years undergoes resurgence, particularly in conjunction with higher dimensional methods.
From 2D to 3D: Using Tilted Data
A logical extension of analysis of 2D data is to expand the findings into 3D structure determination in this way confirming that the variability observed in 2D translates itself into presence of conformers of the protein in 3D. The most robust approaches are based on experimental determination of some of the necessary orientation parameters, such as in the Random Conical Tilt (RCT) technique [20]. By taking at the electron microscope two pictures of the same area of the grid, one tilted to 50° - 60°, the second untilted, the technique provides immediately robust estimate of two Eulerian angles of projection images. The third one is established by 2D alignment of the selected particle images. Moreover, it is possible (and advisable) to apply cluster analysis to aligned untilted 2D images in order to verify that they indeed are projections of the same 3D molecule. Presence of distinct 2D class averages opens up a possibility to compute 3D reconstructions corresponding to all of them. A direct consequence is that it becomes possible to find orientations and merge independent reconstructions reducing artifacts resulting from missing Fourier information, the so-called missing cone problem of RCT-derived structures [21]. More importantly, it also becomes possible to deduce presence of conformers by simply comparing different RCT reconstructions obtained from the same sample [22-25]. Similar results are obtained using the alternative “orthogonal tilt” data collection method [26].
From 2D to 3D: Common Lines Approaches, Focused classification and more
More often than not collection of high-tilt cryo-EM data is extremely challenging and even if problems with charging and other instabilities are overcome, data is of unsatisfactory quality. Thus, there is significant effort invested into development of computational methods that utilize only untilted images. The goal is to deliver properly sorted homogeneous subsets of 2D projection images while at the same time establishing 3D geometrical relations between them. Methods in this class rely on the central section theorem that states that Fourier transforms of 2D projections of a 3D object are central section of the 3D Fourier transform of this object. It is straightforward consequence that Fourier transforms of any two projections intersect along a line, henceforth called a common line. One can find such 3D orientations of 2D projections that pair-wise discrepancies between respective common lines are minimized, which yields an ab initio model of the structure [27,28]. Further, given the discrepancies it should be in principle possible to simultaneously sort out data into homogeneous subsets [29]. In a simpler application of common lines, it is assumed that orientations of projections are known, and discrepancies along common lines are only used for clustering of the 2D data [30]. The approach can be very effective, as in case of demonstration of the “breathing” core of the pyruvate dehydrogenase complex [9]. While promising, methods based on common lines methodology suffer from poor performance for low SNR data.
As the methods based on common lines methodology are not particularly robust, it is tempting to utilize larger amount of information included in the data, i.e., not only discrepancies along selected 1D lines in Fourier space, but between entire 2D images. This approach is based on “angular continuity” principle, which is a common-sense observation that 2D projections of a 3D object that have similar angular directions are similar, while those whose angular directions are far apart are not necessarily similar (for symmetric objects, this obviously holds for asymmetric unit only), and these projection direction dependent dissimilarities should be reflected by any discrepancy measure. This principle was used for ab initio structure determination and proved to be quite robust [31]. If the structure is expected to be in multiple states, one can perform clustering of 2D projection data assigned to the same (or almost the same) projection direction and hope that dissimilarities between class averages corresponding to different substates are larger than those between class averages corresponding to the same substate but having different (but close) angular directions. All what remains is to mutually reconcile the class averages such that the angular consistency is maintained. As long as the heterogeneity is caused by the presence or absence of components of the complex (ligands), one can expect the complexes to have different overall molecular masses, resulting in projections at the same angular direction that have different average. In this case, the mutual reconciliation is achieved by labeling clusters within each of the angular sets according to the average pixel density, which finally leads to calculation of multiple initial 3D structures of the complex. The method was proposed as “focused classification”, the name owing to the idea of restricting of regions of interest in 2D based on regions of high variability in 3D [33]. Similar reasoning was applied to the problem of sorting molecules by size, in which case the sorting of data was supplemented by the PCA analysis of 2D projection images [34]. The latter approach was later extended to the detection of two conformers (with and without a ligand) [32,35]. While the initial formulation of the principle of focused classification was general [33], all applications so far were restricted to detection of two classes of presence and absence of a ligand. It remains to be seen whether more general applications will be shown and to which extent such methods can be extended to studies of conformational flexibility of macromolecular complexes.
Sorting in 3D: Supervised Classification
Studies of conformational variability based on initial analysis of 2D cryo-EM projection data proceed in stages, each involving decisions that are somewhat subjective. In order to have a more straightforward and objective approach, it is preferable to attack the problem directly in 3D. When the imaged sample is a mixture of multiple conformers whose respective basic structures are known or can be guessed, “supervised classification” is a method relatively easy to use. One simply computationally project the known (or guessed) 3D templates into exhaustive sets of angular directions, compare all cryo-EM 2D data with all templates and assigns data to the thus established most similar template. This approach has been used to enhance the occupancy of a ribosomal ligand [36] or to distinguish between conformational states of the ribosome related by a ratchet-like rotation [7]. However, the question of the bias introduced by the choice of initial templates has to be addressed very carefully. Moreover, as substantial prior knowledge is required, it is difficult to use this method to explore new systems or to discover unexpected behavior of the specimen.
Sorting in 3D: Multi-Reference (or Competitive) Refinement
The bias of initial 3D templates can be mitigated, if not eliminated altogether, by employment of a 3D version of a multi-reference alignment, quite successfully used in 2D work [37]. In this method, each 2D EM projection data is aligned with all 2D reference projections of template volumes (whose number is in principle arbitrary). In addition to orientation parameters obtained as in 3D projection alignment, the values of similarity measures are analyzed and the data is assigned to the group of the most similar 3D template with appropriate orientation parameters and updated template volumes are computed [8,10,33,35,38-42]. The process is iterated until stable structures are obtained. The method constitutes a version of unsupervised classification in which class membership and orientation parameters are estimated simultaneously [28].
In order to make the method computational feasible, the initial 3D templates are not derived randomly from the data, as is standard in K-means clustering, but determined instead by one of the 2D to 3D approaches, for example by the focused classification [33]. In the case of the viral capsid Heymann and colleagues used a linear combination of previously determined cryo-EM maps of two processed and unprocessed capsids in order to obtain a gallery of initial 3D templates [39]. In the study of the fatty acid synthetase the initial 3D templates were obtained by normal mode analysis of the structure determined from the entire data set of projection images [8]. In addition, Brink and co-workers tested the bias introduced by the selection of initial models and in this case the results of the refinement proved to be robust. In the case of ribosomal complexes, a variation of the method was introduced in which the multi-particle refinement is initiated by addition of a “neutral” template (in this case a vacant ribosome) to the pool of determined complexes [43,44]. After convergence of the multi-reference alignment, the pool of templates is again increased until stable sub-populations are obtained.
As in 2D case, the multi-particle refinement method is a version of K-means clustering procedure that is invariant with respect to projection-to-volume registration (see Appendix in [28]). It therefore inherits advantages and disadvantages of respective methods and compounds them due to the fact that both algorithms are combined. Multi-reference alignment performs very well when the number of 3D templates sought is known and is not too high, good initial guesses of all 3D templates are available, the individual structures are not too dissimilar, and the 2D projection data are distributed evenly among groups. The main drawback is the substantial computational demand, particular when one has to explore a number of different partitions or templates are featureless.
Sorting in 3D: Maximum-Likelihood Approach
A version of multi-reference alignment inspired by the principle of Maximum-Likelihood (ML) was introduced with the goal of mitigating the initial reference bias and improving convergence properties [45]. It was based on the initial ML method for alignment of EM data by Sigworth, who considered a likelihood function of the model parameters that includes a conditional probability function of observing the outcome model (2D or 3D average of EM data) given a set of orientation parameter values [46]. Further, the method was extended to finite-mixture model that includes membership of each 2D projection image to one of the discrete classes, thus obtaining a ML version of the multi-reference alignment algorithm [45,47]. These algorithms assume a model in which orientation parameters are “fuzzied” by treating them as random variables and the Expectation-Maximization (E-M) recursive parameter estimation algorithm includes integrations of images over the continuous random distributions of orientation parameters sought. This has a distinct advantage of improving the convergence properties, i.e., the danger of producing artifactual structures with low SNR data is somewhat mitigated, but it also makes the method extremely computationally costly. In order to make the method computationally manageable, in an ad hoc improvement, a heuristic reduction of the search space in rotation angles was introduced. In the 3D case, further simplification includes the search for Eulerian angles carried out only over rather sparsely distributed set of projection directions (angular step set to 10°) [47]. As the simplifications are essentially heuristic, the convergence properties of the algorithm remain untested. A promising development is to use a locally adaptive algorithm, in which advantage is taken of smoothness of the correlation function in order to improve the computational efficiency of the method [48].
While the computational requirements restrict applicability of 3D ML approach to intermediate resolution analysis, the initial results are promising. The ML-based analysis of 70S ribosome complexed with elongation factor G revealed heterogeneity of the sample similar to the one obtained earlier using supervised classification, but without relying on prior knowledge of the content of the sample [45].
3D Variance analysis in real space
A comprehensive approach to 3D analysis of conformational heterogeneity requires methodology for establishing presence of variability in the 3D map (real-space variance in the reconstruction), deciding the plausible number of 3D structures in the sample, and constructing good initial guesses of these 3D templates. Such an approach is being advanced using statistical resampling technique of bootstrap in conjunction with PCA and clustering [14,49,50]. The first step, the calculation of the voxel-by-voxel variance for a structure computed as a 3-D reconstruction from the set of 2D projections is based on a very simple premise. By assuming that voxels in the reconstructed 3D map can be considered weighted sums of pixels in 2D projections, the variances and covariances can be calculated using a variant of the bootstrap technique in which a new set of projections is selected with replacements from a given set of projection data. This selection process is repeated large number times, for each resampled set of projections a corresponding 3D volume is calculated, and the voxel-by-voxel (bootstrap) variance of these volumes is calculated. The “correct” variance is obtained from the bootstrap variance using a simple algebraic relation [50]. The regions in 3D map that have high variance indicate possible flexibility of the complex [49,51] or substoichiometric binding of a ligand (Fig.1). This information can be used to initialize 3D K-means procedure, for example in the form of “focused classification” [33]. Better insight can be gained by taking advantage of the fact that the variances/covariances of bootstrap volumes are related to those of the original volume in the same way. Thus, it is possible to perform eigenanalysis of the bootstrap volumes and derive eigenvectors (here called eigenvolumes) that reflect variability of the studied complex [52]. Further, eigenvolumes can be computationally projected in the directions of 2D data and inner product between the pairs of eigenvolumes' projections and EM projection data yields factorial coordinates of the latter, which tie 2D projection data to the common 3D reference framework and to the determined variability of the structure. This step also results in a dramatic reduction of the complexity of the problem, as usually few dominating eigenvolumes (around ten) are sufficient to explain the dominant variability of the structure. The reduced dimensionality of the data set makes it possible to conduct exhaustive clustering studies of the factorial coordinates and determine the number and the structures of initial 3D templates. These are subsequently refined using the established methods of 3D K-means and multi-reference alignment (Fig.2). The described methodology is quite appealing in that the determination of initial templates and probably more importantly of their number is separated from computationally costly 3D multi-reference alignment and also in that eigenvolumes immediately yield insight into expected heterogeneity of the data set. However, high-resolution applications of the methodology have to be yet demonstrated.
Figure 1.
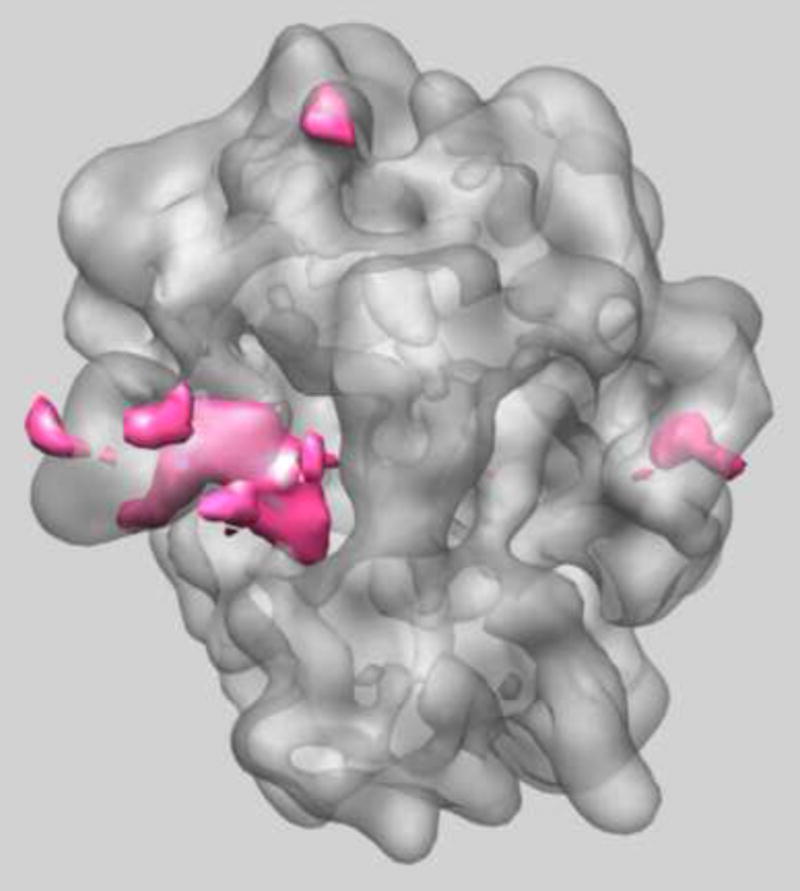
Real-space variance of Thermus thermophilus 70S ribosome in which the ternary complex of elongation factor Tu (EF-Tu), tRNA and guanine nucleotide has been trapped on the ribosome using the antibiotic kirromycin [44]. The structure was originally determined to 6.4 Å from a data set of 323,688 2D projection images that were pre-selected by multi-reference refinement from a total of 586,329 projection images. The 3D variance map was calculated from 170,000 bootstrap volumes. The regions of high variance include location of E-site tRNA and L1 protuberance (left) and also part of the stalk base (right) showing there is still localized heterogeneity in these regions in agreement with our previous analysis [44].
Figure 2.
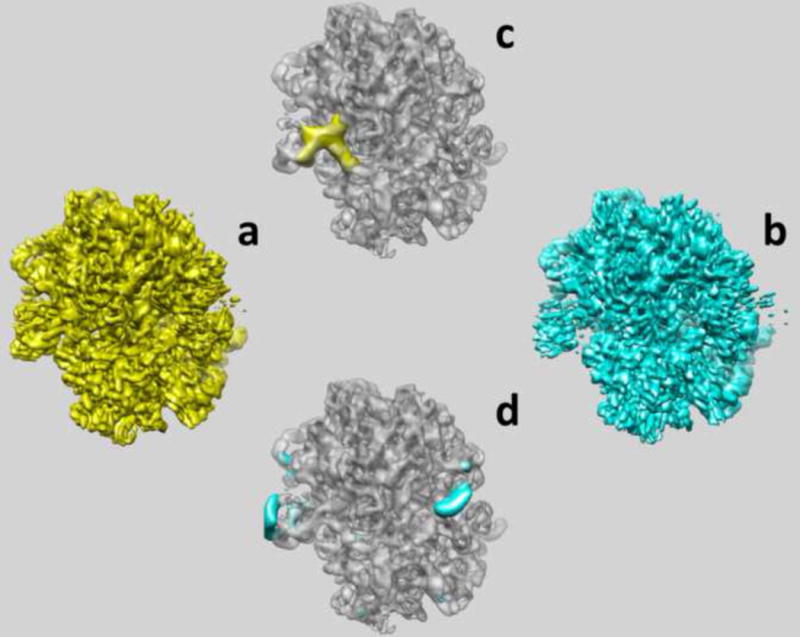
The results of multi-reference alignment of two seed volumes obtained from clustering of factorial coordinates of 2D projection data in space of eigenvolumes. (a) First sub-state determined from 211,641 projection images at 6.3Å resolution contains E-site t-RNA. (b) Second sub-state determined from 112,047 projection images at 6.4Å resolution contains only EF-Tu. (c) Difference map in which second state was subtracted from the first one clearly demonstrates the location and shape of E-site tRNA (yellow). (d) Difference map in which first state was subtracted from the second one in indicates swinging of L1 protein (on the left) and the stalk base (on the right) (blue).
From its inception, single particle cryo-EM rested on the hypothesis that the particles imaged would correspond to complexes of identical composition and conformation. It has become progressively clear that this fundamental assumption is rarely (if ever) realized. Although originally proposed as a means for calculating 3D variance maps that would serve to assess the quality and validity of a cryo-EM reconstruction, the resampling analysis we propose to develop will actually relax the requirement for imaging identical particles, and become a powerful tool for studying the conformational behavior of large macromolecular assemblies. It is important to note the fundamental difference between the resampling approach and the 3D multi-reference analysis proposed by others and us [7,8,11,31,39,47,50]. To be computationally feasible, previous methods required an assumption about the number of conformers, and reasonable guesses about the structures of those conformers. In contrast, the resampling approach requires no prior information, relying instead on the calculation and clustering of factorial coordinates of projection images based on eigenanalysis of alignment parameters for an entire data set. In summary, our approach provides a very efficient and elegant way to establishing the number of conformers in the data set, and information about the structure of conformers is derived directly and objectively from the EM data.
Conclusions
Delivery of the promise of single particle reconstruction, i.e., routine delivery of atomic resolution protein structures in close-to-native conditions, although at hand, requires exponentially growing effort. Moreover, without exceptions, complexes that are on the way to be determined by cryo-EM to atomic resolution (3-4Å) were already crystallized: Gro-EL, virus capsids, TMV, ribosome. While comparative studies are of great value and interest, it is possible that for cryo-EM to succeed at an atomic level the complex has to be sufficiently large and rigid that X-rays crystallographic effort is also likely to succeed. Combining this possibility with the realization that atomic-resolution cryo-EM work requires costly high-end instrumentation and very significant human and computational resources, it is quite possible that the future of single particle analysis lies elsewhere.
It is indeed the observation of conformational heterogeneity where cryo-EM reconstruction proved to be particularly efficient and uniquely useful. As we hopefully succeeded in demonstrating, many if not most of the recent EM structure determination projects contain the demonstration, discussion, or hints at plausibility of conformational variability of the complex. As we are currently learning also from other biophysical techniques, e.g. single-molecule fluorescence resonance energy transfer (smFRET) measurements, the dynamic behaviour of macromolecular machines such as the ribosome appears to be even more complex than anticipated. As shown by smFRET studies [53] and multi-particle cryo-EM [41,44,54,55] functional ribosomal complex can exhibit an intrinsic conformational heterogeneity, i.e. the ribosome can spontaneously adopt distinct native state conformations that are in equilibrium at room temperature. Thus, a metastable energy landscape view of the function of ribosomes and other macromolecular machines is emerging similar to models that have been developed for folding and dynamics of smaller proteins [53].
With such a proliferation of conformational variability findings by EM it is striking that there are virtually no mathematical methods to assess their validity, short of extending the resolution of the complex to the level at which secondary elements become identifiable. It is of serious concern to observe that claims of conformational variability are at the same time used to explain artifactual appearance of limited resolution EM maps or any observed differences with respect to X-ray crystallographic maps. The standard method of assessing quality of EM results is based on evaluating their self-consistency with the help of the Fourier Shell Correlation (FSC) methodology. It is generally known that entirely artifactual maps can boast impressive resolution, as evaluated by the FSC and that resolution estimated by the FSC for a plausibly looking map is not a proof that the map actually has valid information to such a frequency limit. Nevertheless, there is no shortage of widely exaggerated resolution claims that subsequently propagate to subsequent analysis of the map in terms of fitting of X-ray elements or claims of conformational variability. As the awareness of such dangers is growing many researchers embark on significant effort in order to support the findings.
Future work on methodology of conformational modes studies by multi particle analysis has to be accompanied by vigorous efforts to develop new validation methods for the field. Taking into account the central role that structural dynamics of macromolecules plays in understanding biological processes on molecular level, we have no doubt that the next frontier of structural biology is the development of reliable methods, both experimental and computational, to study these phenomena. Cryo-EM, in its ability to capture images of molecules in their unconstrained, close-to-native states, is perfectly suited to become a leading tool in studies of dynamical phenomena in macromolecules, particularly in application to large oligomeric complexes.
Acknowledgments
We thank Marek Kimmel for critical reading of the manuscript. This work was supported by grants from the NIH R01 GM 60635 (to PAP), the DFG (SFB 740 and SFB 449 to CMTS), by the European Union 3D-EM Network of Excellence and by the European Union and Senatsverwaltung für Wissenschaft, Forschung und Kultur Berlin (UltraStructureNetwork, Anwenderzentrum) (to CMTS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frank J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. New York: Oxford University Press; 2006. [Google Scholar]
- 2.Vainshtein BK, Penczek PA. Three-dimensional reconstruction. In: Shmueli U, editor. International Tables for Crystallography. 3. B. Springer; 2008. pp. 366–375. Reciprocal Space. [Google Scholar]
- 3.Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC, Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. Proc Natl Acad Sci U S A. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang W, Baker ML, Jakana J, Weigele PR, King J, Chiu W. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–1134. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- 5.Yu X, Jin L, Zhou ZH. 3.88 A structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature. 2008;453:415–419. doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ludtke SJ, Baker ML, Chen DH, Song JL, Chuang DT, Chiu W. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Gao HX, Valle M, Ehrenberg M, Frank J. Dynamics of EF-G interaction with the ribosome explored by classification of a heterogeneous cryo-EM dataset. Journal of Structural Biology. 2004;147:283–290. doi: 10.1016/j.jsb.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Brink J, Ludtke SJ, Kong YF, Wakil SJ, Ma JP, Chiu W. Experimental verification of conformational variation of human fatty acid synthase as predicted by normal mode analysis. Structure. 2004;12:185–191. doi: 10.1016/j.str.2004.01.015. [DOI] [PubMed] [Google Scholar]; (*) An interesting combination of theoretical prediction of conformational variability based on normal mode analysis together with experimental visualization of conformational modes by multi particle cryo-EM.
- 9.Zhou ZH, McCarthy DB, O'Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14802–14807. doi: 10.1073/pnas.011597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rye HS, Roseman AM, Chen S, Furtak K, Fenton WA, Saibil HR, Horwich AL. GroEL-GroES cycling: ATP and nonnative polypeptide direct alternation of folding-active rings. Cell. 1999;97:325–338. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- 11.Mellwig C, Bottcher B. Dealing with particles in different conformational states by electron microscopy and image processing. J Struct Biol. 2001;133:214–220. doi: 10.1006/jsbi.2001.4340. [DOI] [PubMed] [Google Scholar]
- 12.Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, Hartmuth K, Deckert J, Hauer F, Wolf E, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 13.Kelly DF, Abeyrathne PD, Dukovski D, Walz T. The Affinity Grid: a pre-fabricated EM grid for monolayer purification. J Mol Biol. 2008;382:423–433. doi: 10.1016/j.jmb.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penczek PA. Variance in three-dimensional reconstructions from projections. In: Unser M, Liang ZP, editors. Proceedings of the IEEE International Symposium on Biomedical Imaging; July 7-10; Washington, DC. 2002. pp. 749–752. [Google Scholar]
- 15.Wagenknecht T, Grassucci R, Berkowitz J, Forneris C. Configuration of interdomain linkers in pyruvate dehydrogenase complex of Escherichia coli as determined by cryoelectron microscopy. J Struct Biol. 1992;109:70–77. doi: 10.1016/1047-8477(92)90069-m. [DOI] [PubMed] [Google Scholar]
- 16.Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM. A major conformational change in p97 AAA ATPase upon ATP binding. Molecular Cell. 2000;6:1485–1490. doi: 10.1016/s1097-2765(00)00144-1. [DOI] [PubMed] [Google Scholar]
- 17.van Heel M, Frank J. Use of multivariate statistics in analysing the images of biological macromolecules. Ultramicroscopy. 1981;6:187–194. doi: 10.1016/0304-3991(81)90059-0. [DOI] [PubMed] [Google Scholar]
- 18.Boisset N, Radermacher M, Grassucci R, Taveau JC, Liu W, Lamy J, Frank J, Lamy JN. Three-dimensional immunoelectron microscopy of scorpion hemocyanin labeled with a monoclonal Fab fragment. Journal of Structural Biology. 1993;111:234–244. doi: 10.1006/jsbi.1993.1053. [DOI] [PubMed] [Google Scholar]
- 19.Frank J, Bretaudiere JP, Carazo JM, Verschoor A, Wagenknecht T. Classification of images of biomolecular assemblies: a study of ribosomes and ribosomal subunits of Escherichia coli. Journal of Microscopy. 1988;150:99–115. doi: 10.1111/j.1365-2818.1988.tb04602.x. [DOI] [PubMed] [Google Scholar]
- 20.Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. Journal of Microscopy. 1987;146:113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- 21.Penczek PA, Grassucci RA, Frank J. The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy. 1994;53:251–270. doi: 10.1016/0304-3991(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 22.Golas MM, Sander B, Will CL, Luhrmann R, Stark H. Major conformational change in the complex SF3b upon integration into the spliceosomal U11/U12 di-snRNP as revealed by electron cryomicroscopy. Molecular Cell. 2005;17:869–883. doi: 10.1016/j.molcel.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Radermacher M, Ruiz T, Clason T, Benjamin S, Brandt U, Zickermann V. The three-dimensional structure of complex I from Yarrowia lipolytica: a highly dynamic enzyme. J Struct Biol. 2006;154:269–279. doi: 10.1016/j.jsb.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radermacher M. Journal of Structural Biology. 2009 in press. [Google Scholar]
- 25.Brignole EJ, Smith S, Asturias FJ. Conformational flexibility of metazoan fatty acid synthase enables catalysis. Nat Struct Mol Biol. 2009;16:190–197. doi: 10.1038/nsmb.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leschziner AE, Saha A, Wittmeyer J, Zhang Y, Bustamante C, Cairns BR, Nogales E. Conformational flexibility in the chromatin remodeler RSC observed by electron microscopy and the orthogonal tilt reconstruction method. Proc Natl Acad Sci U S A. 2007;104:4913–4918. doi: 10.1073/pnas.0700706104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–124. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- 28.Penczek PA, Zhu J, Frank J. A common-lines based method for determining orientations for N > 3 particle projections simultaneously. Ultramicroscopy. 1996;63:205–218. doi: 10.1016/0304-3991(96)00037-x. [DOI] [PubMed] [Google Scholar]
- 29.Herman GT, Kalinowski M. Classification of heterogeneous electron microscopic projections into homogeneous subsets. Ultramicroscopy. 2008 doi: 10.1016/j.ultramic.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Hall RJ, Siridechadilok B, Nogales E. Cross-correlation of common lines: a novel approach for single-particle reconstruction of a structure containing a flexible domain. Journal of Structural Biology. 2007;159:474–482. doi: 10.1016/j.jsb.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pullan L, Mullapudi S, Huang Z, Baldwin PR, Chin C, Sun W, Tsujimoto S, Kolodziej S, Stoops JK, Lee JC, et al. The endosome-associated protein Hrs is hexameric and controls cargo sorting as a “master molecule”. Structure. 2006;14:661–671. doi: 10.1016/j.str.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Fu J, Gao H, Frank J. Unsupervised classification of single particles by cluster tracking in multi-dimensional space. Journal of Structural Biology. 2007;157:226–239. doi: 10.1016/j.jsb.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 33.Penczek PA, Frank J, Spahn CMT. A method of focused classification, based on the bootstrap 3-D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 2006;154:184–194. doi: 10.1016/j.jsb.2005.12.013. [DOI] [PubMed] [Google Scholar]; (*) 3D variance analysis, focused classification and multi particle refinement are combined to detect heterogeneity and sort the data into more homogeneous subsets.
- 34.White HE, Saibil HR, Ignatiou A, Orlova EV. Recognition and separation of single particles with size variation by statistical analysis of their images. Journal of Molecular Biology. 2004;336:453–460. doi: 10.1016/j.jmb.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Elad N, Clare DK, Saibil HR, Orlova EV. Detection and separation of heterogeneity in molecular complexes by statistical analysis of their two-dimensional projections. J Struct Biol. 2008;162:108–120. doi: 10.1016/j.jsb.2007.11.007. [DOI] [PubMed] [Google Scholar]; (*) A method to address the problem of heterogeneity has been developed based on analysis of EM projection images in 2D. The images are sorted by two consecutive stages of multivariate statistical analysis to dissect image variations caused by different projection directions from local structural changes.
- 36.Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, Frank J. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO Journal. 2002;21:3557–3567. doi: 10.1093/emboj/cdf326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) Here, supervised classification was used to enhance the density of the ternary complex, which due to sub-substochimetric binding to the ribosome appeared fragmented in the reconstruction of the full data set. In the template structure, an EF-G complex was used, which is similar but not identical to the ternary complex. In this way the authors could control reference bias.
- 37.van Heel M, Stoffler-Meilicke M. Characteristic views of E. coli and B. stearothermophilus 30S ribosomal subunits in the electron microscope. EMBO Journal. 1985;4:2389–2395. doi: 10.1002/j.1460-2075.1985.tb03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen DH, Song JL, Chuang DT, Chiu W, Ludtke SJ. An expanded conformation of single-ring GroEL-GroES complex encapsulates an 86 kDa substrate. Structure. 2006;14:1711–1722. doi: 10.1016/j.str.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Heymann JB, Cheng N, Newcomb WW, Trus BL, Brown JC, Steven AC. Dynamics of herpes simplex virus capsid maturation visualized by time-lapse cryo-electron microscopy. Nature Structural Biology. 2003;10:334–341. doi: 10.1038/nsb922. [DOI] [PubMed] [Google Scholar]; (**) In a pioneering study, multi-particle refinment enabled time-resolved cryo-EM to follow capsid maturation of a virus with icosahedral symmetry.
- 40.Schüler M, Connell SR, Lescoute A, Giesebrecht J, Dabrowski M, Schroeer B, Mielke T, Penczek PA, Westhof E, Spahn CMT. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nature Structural and Molecular Biology. 2006;13:1092–1096. doi: 10.1038/nsmb1177. [DOI] [PubMed] [Google Scholar]; (*) By taking into account sample heterogeneity, it was possible to improve the resolution for eukaryotic 80S ribosomes. Based on the cryo-EM map at 7.3Å resolution and additional biochemical knowledge, the complete chain of an IRES RNA (189 nucleotides) could be traced.
- 41.Klaholz BP, Myasnikov AG, Van Heel M. Visualization of release factor 3 on the ribosome during termination of protein synthesis. Nature. 2004;427:862–865. doi: 10.1038/nature02332. [DOI] [PubMed] [Google Scholar]
- 42.Simonetti A, Marzi S, Myasnikov AG, Fabbretti A, Yusupov M, Gualerzi CO, Klaholz BP. Structure of the 30S translation initiation complex. Nature. 2008;455:416–420. doi: 10.1038/nature07192. [DOI] [PubMed] [Google Scholar]
- 43.Connell SR, Topf M, Qin Y, Wilson DN, Mielke T, Fucini P, Nierhaus KH, Spahn CM. A new tRNA intermediate revealed on the ribosome during EF4-mediated back-translocation. Nat Struct Mol Biol. 2008;15:910–915. doi: 10.1038/nsmb.1469. [DOI] [PubMed] [Google Scholar]
- 44.Schuette JC, Murphy FVt, Kelley AC, Weir JR, Giesebrecht J, Connell SR, Loerke J, Mielke T, Zhang W, Penczek PA, et al. GTPase activation of elongation factor EF-Tu by the ribosome during decoding. EMBO J. 2009;28:755–765. doi: 10.1038/emboj.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) Despite successful affinity purification of a ribosomal complex, which resulted in nearly 100% occupancy with a ligand, sample heterogeneity was still present due to intrinsic conformational modes of the ribosome. After multi-particle refinement, the resolution of the major subpopulation could be pushed to 6.4 Å (0.5 cutoff, no interpolation of resolution), the highest resolution obtained so far using cryo-EM for ribosomes or other objects lacking symmetry.
- 45.Scheres SH, Gao H, Valle M, Herman GT, Eggermont PP, Frank J, Carazo JM. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nature Methods. 2007;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]; (*) The introduction of maximum likelihood optimization to 3D-EM.
- 46.Sigworth FJ. A maximum-likelihood approach to single-particle image refinement. Journal of Structural Biology. 1998;122:328–339. doi: 10.1006/jsbi.1998.4014. [DOI] [PubMed] [Google Scholar]
- 47.Scheres SHW, Nunez-Ramirez R, Gomez-Llorente Y, Martin CS, Eggermont PPB, Carazo JM. Modeling experimental image formation for likelihood-based classification of electron microscopy. Structure. 2007;15:1167–1177. doi: 10.1016/j.str.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tagare HD, Sigworth F, Barthel A. Fast, adaptive expectation-maximization alignment for Cryo-EM. Med Image Comput Comput Assist Interv Int Conf Med Image Comput Comput Assist Interv. 2008;11:855–862. doi: 10.1007/978-3-540-85990-1_103. [DOI] [PubMed] [Google Scholar]
- 49.Zhang W, Kimmel M, Spahn CM, Penczek PA. Heterogeneity of large macromolecular complexes revealed by 3D cryo-EM variance analysis. Structure. 2008;16:1770–1776. doi: 10.1016/j.str.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) Technical improvements (CTF correction, imporved normalization of the EM images) are described that allow the calcultion of the 3D variance in real space using the bootstrap method at higher resolution.
- 50.Penczek PA, Chao Y, Frank J, Spahn CMT. Estimation of variance in single particle reconstruction using the bootstrap technique. Journal of Structural Biology. 2006;154:168–183. doi: 10.1016/j.jsb.2006.01.003. [DOI] [PubMed] [Google Scholar]; (**) It is shown that statistical resampling methods and in particular the bootstrap technique can be used to estimate the voxel by voxel variance of an 3D EM reconstruction in realspace.
- 51.Grob P, Cruse MJ, Inouye C, Peris M, Penczek PA, Tjian R, Nogales E. Cryo-electron microscopy studies of human TFIID: Conformational breathing in the integration of gene regulatory cues. Structure. 2006;14:511–520. doi: 10.1016/j.str.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 52.Penczek PA, Frank J, Spahn CMT. Conformational analysis of macromolecules analyzed by cryo-electron microscopy. In: Kotula P, Marko M, Scott JH, Gauvin R, Beniac D, Lucas G, McKernan S, Shields J, editors. Microscopy and Microanalysis 2006; Chicago. Cambridge University Press; 2006. p. CD386. [Google Scholar]
- 53.Munro JB, et al. Navigating the ribosome's metastable energy landscapes. doi: 10.1016/j.tibs.2009.04.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agirrezabala X, Lei J, Brunelle JL, Ortiz-Meoz RF, Green R, Frank J. Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol Cell. 2008;32:190–197. doi: 10.1016/j.molcel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Julian P, Konevega AL, Scheres SH, Lazaro M, Gil D, Wintermeyer W, Rodnina MV, Valle M. Structure of ratcheted ribosomes with tRNAs in hybrid states. Proc Natl Acad Sci U S A. 2008;105:16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]