

Abstract
Factor Xa (FXa) has a prominent role in amplifying both inflammation and the coagulation cascade. In the coagulation cascade, its main role is catalyzing the proteolytic activation of prothrombin to thrombin. Efficient proteolysis is well known to require phosphatidylserine (PS)-containing membranes that are provided by platelets in vivo. However, soluble, short-chain PS also triggers efficient proteolytic activity and formation of an inactive FXa dimer in solution. In this work, we ask whether PS-containing membranes also trigger formation of an inactive FXa dimer. We determined the proteolytic activity of human FXa toward human Pre2 as a substrate both at fixed membrane concentration (increasing FXa concentration) and at fixed FXa concentration (increasing membrane concentration). Neither of these experiments showed the expected behavior of an increase in activity as FXa bound to membranes, but instead suggested the existence of a membrane-bound inactive form of FXa. We found also that the fluorescence of fluorescein attached to FXa's active site serine was depolarized in a FXa concentration-dependent fashion in the presence of membranes. The fluorescence lifetime of FXa labeled in its active sites with a dansyl fluorophore showed a similar concentration dependence. We explained all these observations in terms of a quantitative model that takes into account dimerization of FXa after binding to a membrane, which yielded estimates of the FXa dimerization constant on a membrane as well as the kinetic constants of the dimer, showing that the dimer is effectively inactive.
Introduction
The activation of prothrombin to thrombin is a crucial step in the blood coagulation process. It is accomplished by the prothrombinase complex, which consists of a serine protease, factor Xa (FXa), and a protein cofactor, factor Va (FVa), interacting on an appropriate acidic membrane surface (1,2). The prothrombinase complex sits at the intersection of the classical intrinsic and extrinsic coagulation pathways, and as such, is widely envisioned as a point of regulation of the coagulation process. Phosphatidylserine (PS) has been known for some time to be specifically required for optimal prothrombinase activity (3,4). An early article on prothrombinase showed that PS-containing membranes enhanced the kcat/KM for human prothrombin activation by FXa by ∼8000-fold (5). A more recent study put the increase in kcat/KM at roughly 1000, but also showed that PS-containing membranes made activation processive (i.e., two bonds cut without release of the substrate from the enzyme) and directed activation through one of two possible intermediates (6), both roles previously assigned to factor Va (7). However, several studies that are more recent have shown that it is not a PS-containing membrane but primarily molecular PS that is essential for prothrombinase activity. Thus, binding of a water-soluble phosphatidylserine, 1,2-dicaproyl-3sn-phosphatidylserine (C6PS) to human factor Xa also increased kcat/KM by ∼200-fold and directed activation via a single intermediate (8). Indeed, C6PS binds to FVa to assemble an FXa-FVa complex in solution that activates prothrombin by the same processive mechanism as membrane-bound prothrombinase and at a rate only a factor-of-2 less than that of the membrane complex (9). Thus, molecular PS acts as an allosteric regulator of prothrombin activation by binding to specific sites and altering the structure and function of factors Xa and Va.
The platelet membrane is asymmetric in its lipid composition (10). This asymmetry in the platelet membrane and membranes of other cells in contact with plasma is maintained by an adenosine triphosphate-driven amine-lipid translocase that moves phosphatidylserine (PS) and phosphatidylethanolamine (PE) from the outer to the inner platelet membrane leaflet (11). The expenditure of energy to maintain PS and PE asymmetry implies that the asymmetry must play a significant role in cell function. Activation of human platelets leads to appearance of discrete membranous vesicles with diameters of 80–200 nm that support the prothrombinase complex (12). Platelet-derived membranous vesicles were first described in 1967 as minute “particles”, termed platelet “dust”, found in human plasma and serum (13). These vesicles obtained from collagen-activated platelets bound factor to factor Va support blood coagulation (12). This was confirmed for complement- and thrombin-activated platelets, and it was shown that appearance of these vesicles corresponded with loss of PS asymmetry (14,15).
Both because of FXa's central role in the prothrombinase complex and because of the apparent role of PS as a signaling molecule associate with platelet activation, the effect of PS on this enzyme is of particular interest in understanding the regulation of blood coagulation. FXa, along with thrombin, has been one of the most popular targets for the development of anticoagulant/antithrombotic drugs, although this quest to date has been unsuccessful. Animal model research shows that inhibiting FXa is effective in preventing both arterial and venous thrombosis, making FXa a popular target for drug development (16). In addition to its role in blood coagulation, FXa has a prominent role in amplifying both the inflammation and coagulation cascades. In inflammation, FXa activates PAR2 (17). Protease-activated receptors are G protein-coupled receptors that function as cell-surface sensors for coagulant proteases and, by binding to effector cell protease receptor 1, participate in several cellular events that are involved in the inflammatory response (18). Clearly, FXa regulation is of considerable interest in biomedical science.
In addition to activating factors Xa and Va, C6PS binds to bovine FXa to induce Ca2+-dependent dimerization (19). Recent results showed that human FXa dimerizes in the presence of soluble PS and 5 mM Ca2+ and that the dimer is ∼107 times less active than the monomer (20). We address here the following questions:
-
1.
Does FXa form dimers on PS-containing membranes?
-
2.
If so, is the dimerization constant in a potentially physiologically significant range?
-
3.
Are FXa dimers equally inactive on PS-containing membrane as in solution?
We have recorded the florescence and fluorescence lifetime of labeled human DEGR-Xa and finally the fluorescence anisotropy of Fluorescein-Glu-Gly-Arg-chloromethylketone-modified FXa (FEGR-Xa) in response to 25% PS-containing membranes. We have also determined the proteolytic activity of human FXa toward human Pre2 as a substrate both at fixed membrane concentration (increasing FXa concentration) and at fixed FXa concentration (increasing membrane concentration). A model that takes into account FXa dimerization upon binding to a membrane successfully described all our data with surface dimer dissociation constants of 1.2 × 10−14 mole fraction for human FXa and 2.6 × 10−10 mole fraction for human DEGR-Xa or FEGR-Xa. We conclude that human FXa forms an essentially inactive dimer on PS-containing membranes even at very low membrane surface concentrations and that labeling the active site serine inhibits dimer formation.
Materials and Methods
See Experimental Procedures in the Supporting Material.
Results
FXa activity in solution
The initial rate of activation of Pre2 by factor Xa in solution (50 mM Tris, 150 mM NaCl, 0.6% PEG, and 4 mM Ca2+, pH 7.4 at 37°C) was determined at different Pre2 concentrations as a function of FXa concentration (Fig. 1). For each Pre2 concentration, the initial rate of thrombin formation was proportional to the FXa concentration, with a different proportionality constant, K([Pre2]), for each Pre2 concentration. The concentrations of Pre2 used in these experiments were comparable to, and sometimes larger than, the FXa concentrations, meaning that these reactions cannot be assumed to follow pseudo second-order reaction kinetics (initial rate = kcat/KM[S][E] for [S] << KM) that derive from the steady-state (Briggs-Haldane) treatment of the Michaelis-Menten model. However, the kcat/KM for Pre2 activation to thrombin under appropriate steady-state conditions is quite small (86 ± 7 M−1 s−1) and the KM is large (KM >> 2 μM) (6), implying that, at the highest [Pre2] (250 nM), the rate was only ∼0.004 nM IIa/s and that only ∼0.5% of Pre2 was consumed in the time during which our experiments were performed. If we assume that delivery of substrate to enzyme is close to diffusion-controlled, the rate of enzyme-substrate complex formation should be substantially greater than the rate of product formation. Thus, enzyme and substrate should be in equilibrium with ES complex and the rate of product appearance should be roughly equal to the rate of ES disappearance and thus should be ≈kcat/KS, where KS is the equilibrium constant for ES formation, which is the meaning of KM when ES is in equilibrium with E and S. This conclusion is consistent with two observations. First, the time course for thrombin appearance was linear and passed through zero, consistent with steady-state conditions. Second, we plot in the inset to Fig. 1 the proportionality constant K ([Pre2]) versus Pre2 concentration, showing a linear relationship with slope K∗. This makes it clear that, for our experimental conditions, R = K∗[Pre2][Xa]. The value of K∗ obtained from the inset to Fig. 1 was 94 M−1 s−1. In addition, all five data sets were simultaneously fitted with Eq. A1 in the Supporting Material. The apparent second-order rate constant obtained from the global fit is K∗ = 94 ± 2 M−1 s−1, exactly as obtained from the inset to Fig. 1. We conclude that the initial rate of Pre2 activation under our conditions is appropriately represented as second order in enzyme and substrate concentrations. The agreement of K∗ with the kcat/KM reported previously (86 ± 7 M−1 s−1) (6) argues strongly that K∗ = kcat/KM for Pre2 activation under our conditions, and that our results can be analyzed in terms of the steady-state Michaelis-Menten formalism.
Figure 1.
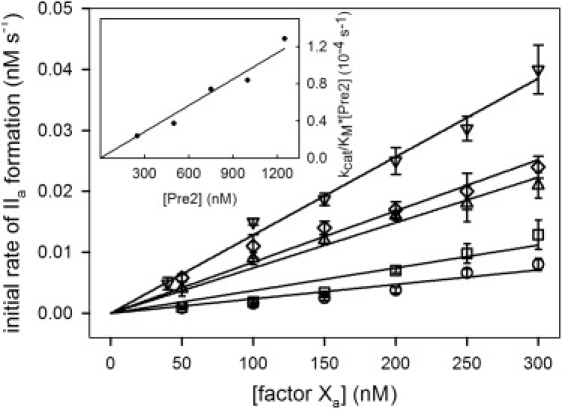
Initial rate of Pre2 activation to thrombin by FXa. The initial rate of thrombin (IIa) formation was determined at 37°C as a function of increasing concentration of FXa at different concentrations of Pre2: [Pre2] = 250 nM (○), 500 nM (□), 750 nM (▵), 1000 nM (♢), and 1250 nM (▿). Initial rates were determined in buffer A (50 mM Tris, 150 mM NaCl, and 0.6% PEG, pH 7.4) containing 4 mM CaCl2, using the substrate S-2238 as described in Experimental Procedures in the Supporting Material. The curves drawn through the symbols are straight lines, the slopes of which are R1 (as described in Appendix A in the Supporting Material) for different [Pre2]. (Inset) Linear dependence of R1 on [Pre2], which demonstrates that the reaction follows Michaelis-Menten kinetics under our conditions and gives, as the slope of this plot, kcat/KM = 94 ± 2 M−1 s−1 for free FXa. By globally fitting all five data sets to the Michaelis-Menten model (20), we obtained the same kcat/KM as well as KM = (10 ± 1) mM.
FXa activity in the presence of PS-containing membranes
It is widely reported that PS-containing membranes enhance the rate of prothrombin (5,21) or Pre2 activation (6). Fig. 2 A shows the initial rate of Pre2 conversion to thrombin (IIa) by FXa (300 nM) as a function of FXa concentration at three different concentrations of PS-containing membranes (5, 50, and 150 μM) in buffer A containing 4 mM Ca2+. At all three membrane concentrations, the initial rate did not increase proportionally with FXa concentration as one might expect it should, and the functionality was different at the three membrane concentrations. Initial rates obtained at a constant FXa concentration (300 nM) and 4 mM Ca2+ but different membrane concentrations showed that the initial rate of activation actually dropped with addition of small amounts of membranes under certain conditions and reached a minimum at membrane concentrations of ∼15–20 μM phospholipid (Fig. 2 B). Because the substrate in our experiment (Pre2) is not membrane-bound, this cannot reflect surface crowding and must mean that the membrane somehow inhibits FXa proteolytic activity at low membrane concentration, exactly the opposite of what current thought would predict. At higher membrane concentrations (>20 μM), this inhibitory effect disappeared and membranes enhanced FXa activity, as has been widely reported in previous publications (5,22,23). An experiment performed in buffer A containing 1 mM Ca2+ did not show the anomalous behavior seen in Fig. 2 B, and was well represented in terms of a simple surface-binding isotherm (line in the inset) giving an apparent Kd of 220 ± 200 nM and stoichiometry of 63 lipids per bound FXa (inset between Fig. 2, B and C).
Figure 2.
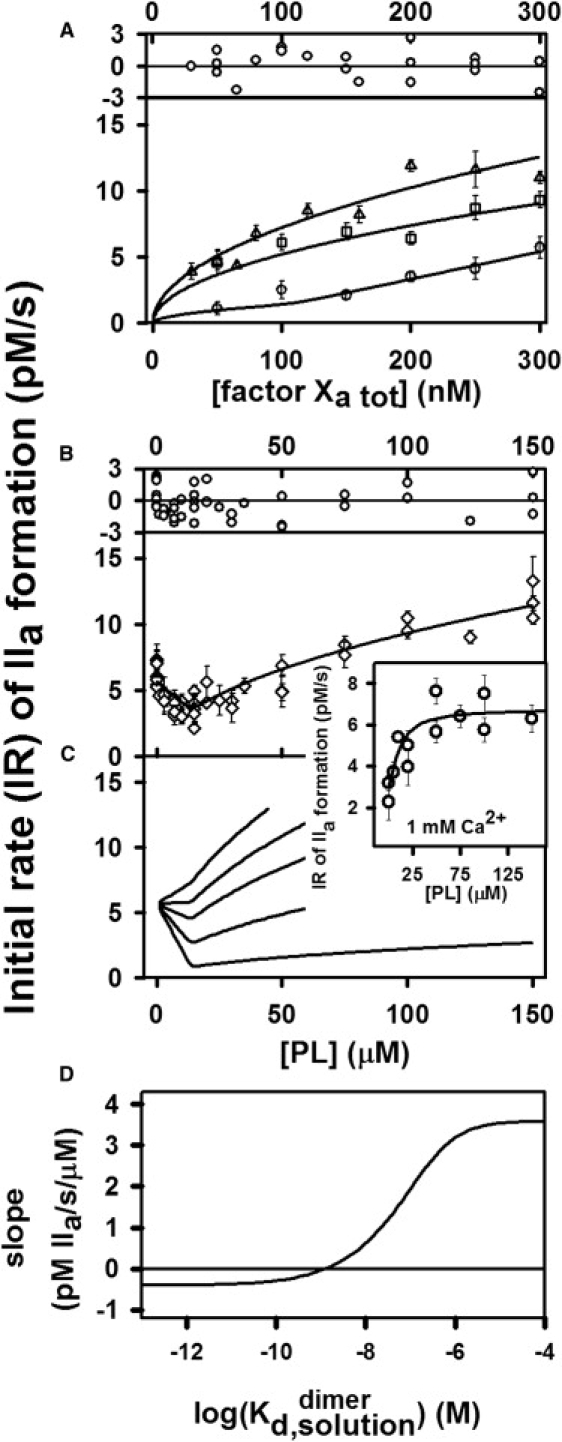
Global analysis of the initial rate of Pre2 activation by FXa in the presence of 25% PS/DOPC membranes. The initial rate of thrombin (IIa) formation was determined at 37°C using the substrate S-2238 as described in Experimental Procedures in the Supporting Material. The concentration of Pre2 was 0.5 μM in buffer A with 4 mM CaCl2. (A) The rate of activation was monitored as a function of increasing concentration of FXa at different concentrations (○, 5 μM; □, 50 μM; and ▵, 150 μM) of 25% (PS) membrane. The plot above the frame shows the deviation of experimental data from a value predicted by the dimerization model relative to the average standard error (Rexp − Rfit)/σmean. (B) The rate of Pre2 activation was monitored at 300 nM FXa in buffer A with 4 mM CaCl2 in the presence of different concentrations of 25% PS membrane. The error bar for each point is the standard error from a linear fit to six time points. The curves drawn through the symbols indicate the global fit of the data to a dimerization model as described in Appendix A in the Supporting Material, which predicts that a dimer is inactive and gives a dimer dissociation constant on a surface = (40 ± 25) × 10−15 mol/(dm)2. This corresponds to a solution dimer dissociation constant of nM calculated at [PL] = 10 μM. The plot above the frame shows the deviation of experimental data from a value predicted by the dimerization model relative to the average standard error Rexp − Rfit)/σmean. The inset to the panel between panels B and C shows data obtained in a similar experiment but at 1 mM Ca2+. The reduced χ-square for the global fit shown in panels A and B was , and the normalized χ-square , where N is the number of observations; yi,exp is the (observed) mean dependent quantity; is variance in yi,exp; xi is the independent variable; f is the assumed relationship between xi and yi,exp (i.e., the model developed in Appendix A in the Supporting Material); f(xi) is predicted yi value; and ν = N − p − 1, where p is the number of fitting parameters. A normalized reduced χ-square close to 1 indicates that the model provides an adequate description of the data. Since ν is large (75 data points), a reduced χ-square of 1.48 corresponds to a probability for the null hypothesis (a χ-square greater than this is possible due to random variations in the data) of <0.01. (C) The rate of activation of Pre2 by 300 nM FXa as predicted with the dimerization model is plotted versus surface dimer dissociation constants (from bottom to top: ) = 1, 10, 30, 50, and 80 × 10−15 mol/(dm)2, which correspond to ≈ 0.03, 0.3, 0.8, 1.3, and 2.1 nmol calculated at [PL] = 10 μM. (D) The initial slope (in rate/membrane concentration units) of curves predicted as described in panel C[PL]=0 varied with the base 10 logarithm of the FXa dimer dissociation constant in solution units (calculated at [PL] = 10 μM). Note also that this slope is predicted to be zero at ≈ 1.3 nM = 50 × 10−15 mol/(dm)2.
Since a soluble form of phosphatidylserine (C6PS) triggers formation of an inactive FXa dimer in solution (19,20), it is reasonable to hypothesize that our current results can be explained by FXa dimerization on a PS-containing membrane. To test this interpretation, we used the model developed in Appendix A in the Supporting Material that takes into account FXa binding to a membrane followed by FXa dimerization on the membrane surface (Eqs. A1 and A4 in the Supporting Material). This model has several parameters that must be set either from previous experiments or by fitting to our experimental data. Binding of FXa to PS-containing membranes has been studied using a variety of techniques (21,24–28), which yielded varying results for the FXa membrane dissociation constant ranging from Kd = 33 nM (28) up to Kd = 2700 nM (21). If FXa does dimerize on PS-containing membranes at 4 mM Ca2+, all measured binding constants would be apparent binding constants for FXa binding as a monomer but with monomer binding linked to dimer formation. Thus, we allowed Kd,Xa to adjust as a free parameter during description of our data with the dimer model. The surface dimer dissociation constant and kcat/KM for the dimer (kcat/KM2) were also allowed to adjust. We used our previously reported stoichiometry of FXa monomer membrane binding (24), which takes into account lipids from inner and outer monolayers. Another, more recent estimate (52 lipids/ FXa) (27) is consistent with this. The stoichiometry of FXa dimer binding was also adjusted.
We used a second-order rate constant (kcat/KM) for FXa monomer in solution as obtained from Fig. 1 (94 ± 2 M−1 s−1) and used a previously published estimate (9000 M−1 s−1) (20) for a monomer bound to a PS-containing membrane. In summary, we used four adjustable parameters , to describe globally four sets of data in two types of experiments (dependence of FXa activity on FXa concentration in Fig. 1 A and titration of FXa activity by phospholipid in Fig. 1 B). The success of our dimer model in predicting both sets of experiments is illustrated by the model-generated lines through the data in Fig. 2, A and B, and by the random distribution of residuals shown above each set of data. The initial decrease in activity upon addition of PS-containing membranes was explained by the dimer model using the best-fit surface dimer dissociation constant, × 10−15 mol/(dm)2, which corresponds to a solution dimerization constant nM calculated at [PL] = 10 μM (see Eq. A10 in the Supporting Material for comparison of surface and solution dimer dissociation constant units). The fitted value for stoichiometry of a dimer was found to be . The analysis predicted that the FXa dimer was inactive (kcat/KM ∼ 0 M−1 s−1), as reported for the dimer in solution (20). In Fig. 2 C, we plot the predicted initial rate of Pre2 activation versus phospholipid concentration for several values of the surface dimer dissociation constant (from bottom to top: and 80 × 10−15 mol/(dm)2). This demonstrates that tighter dimer formation results in greater initial inhibition of activity by added phospholipid, as would be expected if the membrane-bound dimer has little or no activity. The slope of initial rate versus lipid concentration shows a sigmoidal dependence on the log of the dimer dissociation constant (Fig. 2 D), with the slope being negative below × 10−15 mol/(dm)2— corresponding to a solution dimer dissociation constant nM calculated at [PL] = 10 μM.
Response of DEGR-Xa fluorescence to titration with PS-containing membranes
DEGR-Xa fluorescence emission intensity was recorded as a function of increasing concentration of PS-containing membranes in the presence of 2 and 4 mM Ca2+. The emission intensities of DEGR-Xa measured at 2 mM Ca2+ and normalized as described in Methods in the Supporting Material are plotted in the inset to Fig. 3 A as a function of the added PS membrane concentration. These data were analyzed with a simple absorption isotherm model as described in Banerjee et al. (8), with best fit lines shown drawn through the data. The apparent dissociation constants obtained in this way at 2 mM CaCla (open symbols) decreased roughly linearly with increasing concentration of DEGR-Xa. These results are consistent with DEGR-Xa dimerization on a membrane, since the concentration of membrane-bound DEGR-Xa monomer would be depleted by dimer formation, thus shifting by mass action the binding reaction toward bound DEGR-Xa. We performed similar experiments at 4 mM CaCla (data shown in Fig. 3 B2) and attempted to analyze these with a simple adsorption isotherm (dashed lines in Fig. 3 B2). It is clear that this model will not account for the data up to high membrane concentrations. If we analyzed data only to 30 μM membranes (a concentration range in which surface dimerization of FXa should be optimized), effective Kd values in the range of 10–30 nM were obtained. These are much smaller than Kd values obtained at 2 mM Ca2+ (Fig. 3 A), consistent with our previous report that PS-stimulated bovine FXa dimer formation in solution is strongly Ca2+-dependent, with dissociation constants decreasing with increasing Ca2+ concentration (19). These results also help explain how literature estimates for the Kd for FXa binding to membranes could range from ∼30 nM to 3 μM.
Figure 3.
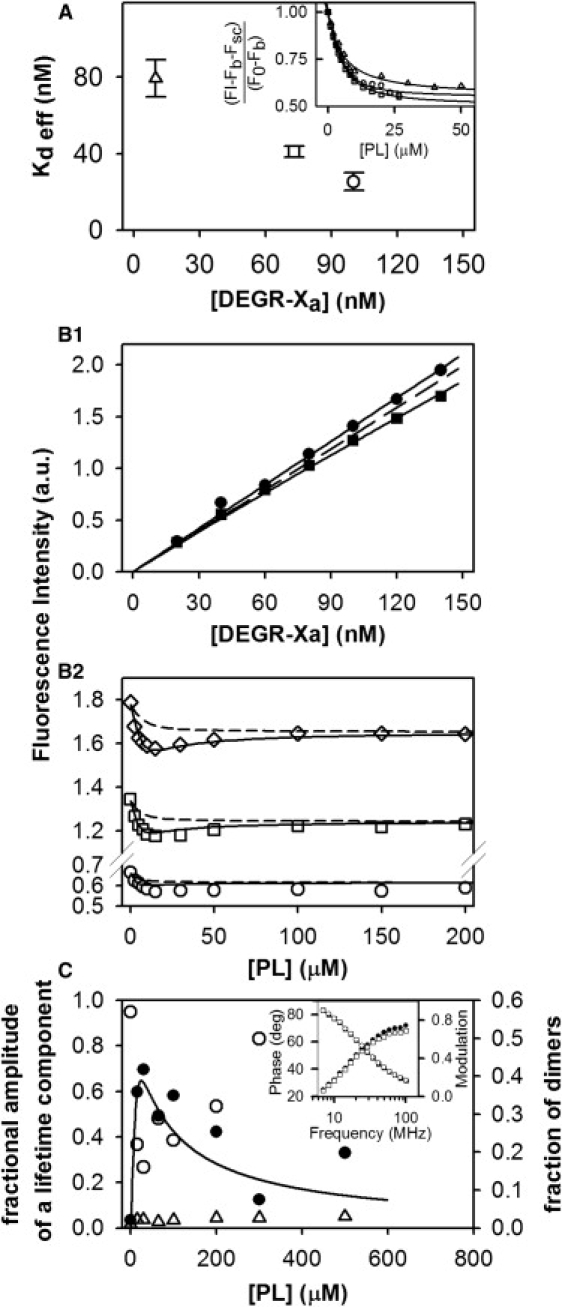
Fluorescence intensity studies of DEGR-Xa. (A) DEGR-Xa binding to membranes depends on DEGR-Xa concentration. (Inset) DEGR-Xa binding to 25% PS/DOPC membranes ([PL]) was monitored at 23°C as a relative fluorescence change as PL was added at different concentrations of DEGR-Xa (▵, 10 nM; □, 73 nM; and ○, 100 nM) in buffer A containing 2 mM Ca2+. (Main plot) Open symbols show apparent dissociation constants for DEGR-Xa binding obtained from the data shown in the inset fit (inset symbols are consistent with main plot) to a simple binding model (8). (B1 and B2) Global analysis of DEGR-Xa membrane binding using either (B1) DEGR-Xa or (B2) membranes as titrant. (B1) Fluorescence intensity of DEGR-Xa (scattering and background subtracted) in Buffer B containing 4 mM CaCl2 at 23°C as a function of increasing DEGR-Xa concentration at different concentrations (●, 0 μM; and ■, 15 μM) of 25% PS/DOPC membranes. (B2) Fluorescence intensity of DEGR-Xa (○, 50 nM; □, 100 nM; and ♢, 150 nM) at increasing concentrations of 25% PS/DOPC membranes. Data in both frames were fitted globally to the dimerization model (Appendices A and B in the Supporting Material) (solid lines), yielding a surface dimer dissociation constant × 10−9 mol/(dm)2 for DEGR-Xa, which corresponds to a solution dimer dissociation constant μM calculated at [PL] = 10 μM. Dotted lines show fits to a model that assumes that DEGR-Xa can bind to membranes only in its monomer form. Reduced χ-square for the global fit equals , and normalized χ-square , The p value for the null hypothesis is <0.02. (C) DEGR-Xa fluorescence lifetime during titration with 25:75 PS/DOPC membranes. DEGR-Xa (300 nM) was titrated with 25% PS/DOPC membranes in buffer B containing 4 mM Ca2+ at 23°C, and its fluorescence lifetime was resolved into three lifetime components using software supplied with the Spex FluoroLog-3 phase and modulation spectrofluorometer (HORIBA Jobin Yvon, Edison, NJ). Fractional amplitudes of lifetime components with lifetime τ ≈ 7.4 ns (●), 13 ns (○), and 0.5 ns (▵) are plotted, along with the relative proportion of DEGR-Xa in dimer form (solid line) as predicted by the dimer model using the dimerization constant obtained from the global fit shown in panel B. (Inset) Phase and modulation of the signal measured at low lipid concentration [PL] = 15 μM (●, phase; ■, modulation) and high lipid concentration [PL] = 300 μM (○, modulation; □, phase).
The titrations in Fig. 3 B2 alone were insufficient to estimate the DEGR-Xa dimerization constant on the membrane surface. To obtain this estimate, we needed the intensities of dimer and monomer fluorescence. As a result, the experiments in Fig. 3 B1 were performed, in which DEGR-Xa fluorescence intensity was recorded with increasing concentration of DEGR-Xa in the absence (squares) and presence of 15 μM PS/DOPC (25:75) vesicles. Global analysis of the results shown in Fig. 3, B1 and B2, using the dimer model described in Appendix B in the Supporting Material, provided a good description of the data at 100 nM (squares) and 150 nM DEGR-Xa (diamonds), but only an approximate description of the data at 50 nM DEGR-Xa (circles), as shown by the solid lines through data points. Clearly, a simple monomer-binding model cannot describe data obtained at 100 nM and 150 nM DEGR-Xa concentrations, whereas the dimer model developed in Appendix A in the Supporting Material can. However, the dimer model showed significant residuals at low lipid concentration at 50 nM DEGR-Xa, whereas the monomer-binding model had more randomly distributed residuals at this concentration of DEGR-Xa. It should be noted that both the fluorescence intensity and the contribution from dimers were quite low at 50 nM DEGR-Xa, making meaningful analysis more difficult at this low concentration.
The dimer model analysis yielded a surface dimerization constant of nmol/dm2 for DEGR-Xa compared to fmol/dm2 for FXa, which was obtained from our analysis of activity data (Fig. 2). The DEGR-Xa surface dimer dissociation constant corresponds to a solution dimer dissociation constant of μM (also calculated at 10 μM PS membrane), which compares to nM for FXa. Clearly, DEGR-Xa dimers form with roughly 105-fold lower dimerization affinity than seen for FXa dimer formation, presumably because the active site is involved directly in or linked to dimer contact sites. Analysis of the data in Fig. 3, B1 and B2, also yielded estimates of the fluorescence intensities (relative to solution monomer fluorescence, Ffree) of membrane-bound monomer and dimer (∼0 but constrained to be ≥0) DEGR-Xa, thus accounting for the decrease followed by an increase of fluorescence seen for lipid titration of 150 nM DEGR-Xa (Fig. 3 B1, triangles).
Response of DEGR-Xa fluorescence lifetime to titration with PS-containing membranes
To obtain additional evidence for DEGR-Xa dimerization on PS-containing membranes, 300 nM DEGR-Xa was titrated with 25:75 PS/DOPC membranes in buffer B containing 4 mM Ca2+ at 23°C. Fluorescence lifetime data were obtained as described in Methods (see the Supporting Material) using the PhaseMax fluorometer from HORIBA Jobin Yvon (Edison, NJ) and the Lifetime Modeling Application software developed by the same company. At each lipid concentration, three lifetime components were resolved: τ1 = 0.5 ± 0.3 ns, τ2 = 7.4 ± 2.4 ns, and τ3 = 13.2 ± 1.4 ns. Although the signal was measured at 16 different frequencies that span a considerable range, we were unable to resolve more than three lifetime components. The fractional contributions of each component are plotted in Fig. 3 C as a function of lipid concentration (τ1, open triangles; τ2, solid circles; and τ3, open circles). At zero lipid, the data were explained in terms of only two components. The shortest-lifetime component had fractional amplitude that was roughly constant across all lipid concentrations, whereas that of the long-lifetime component (τ3 = 13.2 ns) decreased and subsequently increased in amplitude as lipid concentration increased. The second component (τ2 = 7.4 ns) appeared only in the presence of membranes, and its fractional amplitude increased and subsequently decreased with increasing membrane concentration (i.e., its behavior was approximately opposite to the behavior of component 3). These results show that two DEGR-Xa species coexist in the presence of membranes. This offers direct evidence that two forms of DEGR-Xa coexist and further supports the monomer/dimer equilibrium in terms of which we have interpreted the data in Figs. 1–3. In this interpretation, the lifetime of the monomer should correspond to component 3 (τ3 = 13.2 ns), which is one of the two components present in the absence of membranes. A small amount of component 1 (τ1 ≈ 0.5 ns) is present under all conditions and presumably is not sensitive to the state of oligomerization of DEGR-Xa. Component 2 (τ2 = 74 ns) should correspond to dimers, as its contribution increases as DEGR-Xa binds to membranes and dimerizes and then decreases as dimers dissociate with increasing available surface. To test this interpretation, we plot as a solid line in Fig. 3 C the fraction of DEGR-Xa predicted to exist as dimer under the conditions of this experiment as derived using the dimer model (Appendix A in the Supporting Material) and the obtained from global fitting of the data in Fig. 3, B1 and B2. It is clear that the predicted fraction of dimer correlates well with the behavior of component 2 (τ2 = 74 ns) from the lifetime analysis. In the dimer interpretation, this component should decrease at sufficiently high membrane concentration because dimer dissociates into monomer as the surface concentration of DEGR-Xa decreases, as predicted by the dimer model (solid line).
FEGR-Xa fluorescence anisotropy during titration with 25/75 PS/DOPC membranes
Although the results presented thus far offer strong evidence in favor of surface dimerization of FXa and DEGR-Xa, they do not provide direct physical evidence for close approach of two FXa molecules. To demonstrate this, we used a form of fluorescence resonance energy transfer in which one fluorescein chromophore transfers the energy of its excited state to another nearby fluorescein, a process termed nonradiative homotransfer (29). As for all fluorescence resonance energy transfer processes, homotransfer depolarizes fluorescence emission, since orientational information is not preserved during the transfer process. In this way, depolarization of fluorescein emission can serve as a useful tool for detecting protein oligomerization. Fig. 4 shows the fluorescence anisotropy (Fig. 4 A) and fluorescence intensity (Fig. 4 B) of 150 nM FEGR-Xa titrated with 25:75 PS/DOPC membranes in buffer B containing 4 mM Ca2+ at 23°C. Fluorescence anisotropy (solid circles) decreased after addition of a small amount of PS-containing membranes. As more membranes were added, the fluorescence anisotropy increased and ultimately reached the anisotropy of free FEGR-Xa in solution (i.e., at [PL] = 0 μM). The decrease in fluorescence anisotropy indicates close juxtaposition of fluorescein moieties on different FXa molecules and provides direct evidence of FEGR-Xa self-associated upon binding to PS-containing membranes. The return of anisotropy to that of free FEGR-Xa at high membrane concentration indicates that self-association reverses upon dilution of FEGR-Xa on the membrane surface. After addition of detergent to the membrane-rich sample (solid diamond at [PL] = 500 μM), the anisotropy was that of free FEGR-Xa in solution (horizontal dashed line), confirming that FEGR-Xa behaves like a solution monomer at sufficiently high dilution on a membrane surface. Fluorescein anisotropy also remained constant when FEGR-Xa was titrated with 25:75 PS/DOPC membranes in buffer B containing 1 mM Ca2+ (open squares), a condition under which FEGR-Xa binds to membranes but does not dimerize. This again confirms that binding of FEGR-Xa to membranes does not change fluorescein fluorescence anisotropy. We have also plotted in Fig. 4 B the FEGR-Xa fluorescence intensity as a function of added PS/DOPC membrane concentration.
Figure 4.
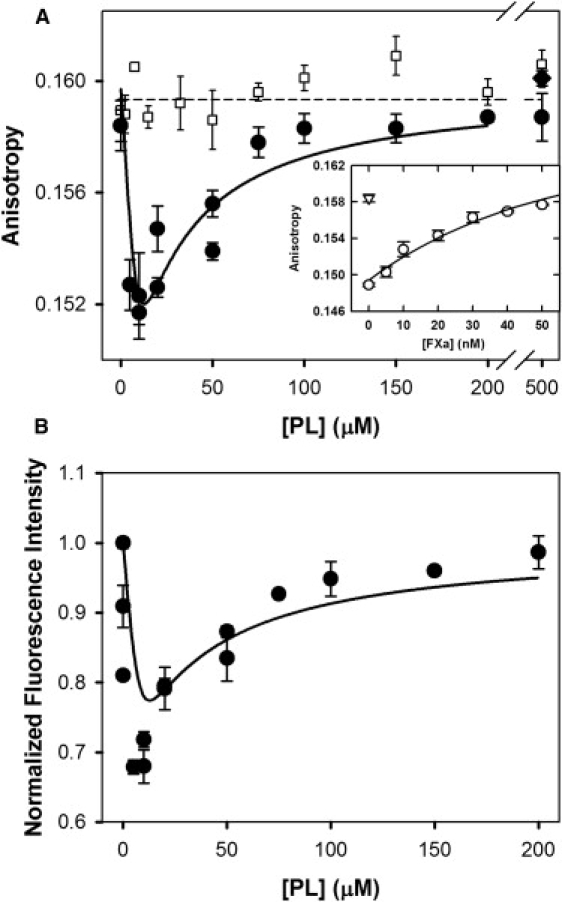
FEGR-Xa fluorescence during titration with 25:75 PS/DOPC membranes. (A) FEGR-Xa (150 nM) in buffer B containing 1 mM Ca2+ (□) or 4 mM Ca2+ (●) was titrated with 25% PS/DOPC membranes, and its fluorescence anisotropy is plotted versus PS-containing membrane concentration ([PL]) before (□ and ●) and after (♦) addition of detergent. (Inset) Fluorescence anisotropy increase of FEGR-Xa after addition of increasing amounts of FXa in the presence of 10 μM 25% PS/DOPC membrane. Anisotropy was fitted using a model similar to the one in Appendix B in the Supporting Material with the addition of competition between FXa and FEGR-Xa. The data were fitted with only one parameter: for FEGRXa-FXa dimer, which was similar to the dimer dissociation constant for FXa dimer; all other parameters were used as obtained from previously described experiments. Obtained reduced χ-square is 2.98, and normalized χ-square is 1.73. The p value for the null hypothesis is <0.01. (B) Fluorescence intensity (relative to intensity at [PL] = 0) of FEGR-Xa is plotted versus PS-containing membrane concentration ([PL]) in buffer containing 4 mM Ca2+ (●). Solid lines through the data in both frames were obtained from the best global fit of the dimer model to the data, as described in Appendix C in the Supporting Material. The dashed line in panel A shows the fraction of FEGR-Xa present as monomer plus free species (i.e., 1-Xdimer), as derived using × 10−9 mol/(dm)2 as obtained from the global fit. Reduced χ-square for the global fit equals , and normalized χ-square . The p value for the null hypothesis is <0.02.
Based on these data, it appears that the fluorescence intensity of the membrane-bound FEGR-Xa monomer is the same as that of FEGR-Xa in solution (normalized to 1). As described in Appendix C in the Supporting Material, these intensity data are required to analyze our FEGR-Xa anisotropy data in terms of the dimerization model developed in Appendix A in the Supporting Material. The curves resulting from a global fit of the data in Fig. 4, A and B, are shown as solid lines in Fig. 4, A and B. This fit was obtained using rmonomer = rfree ≈ 0.1595 (horizontal line in Fig. 4 A) and varying rdimer from 0 to 2 × rfree and varying and Fdimer/Ffree to minimize the sum of mean-squared deviations of the data in panels A and B from values calculated via the dimer model (χ2). The value of χ2 decreased steeply as rdimer/rfree decreased but was insensitive to its value for rdimer/rfree ≤ 0.87, which corresponds to R/R0 ≤ 0.75 (see Eq. C1 in the Supporting Material). Thus, we can say only that the two active-site located flouresceins in the FEGR-Xa dimer are separated by <∼37 Å. The value of Fdimer/Ffree varied in this range from 0.34 to 0.25, with lower values corresponding to lower rdimer/rfree. The surface dimer dissociation constant for FEGR-Xa did not vary significantly in this range, with nmol/dm2, which compares to μM, calculated at [PL] = 10 μM. These results convincingly demonstrate that FEGR-Xa self-associates on a PS-containing membrane and strongly suggest that the aggregate form is a dimer, as we have demonstrated unambiguously for FXa in solution (19).
Discussion
Factor Xa dimerizes in the presence of water-soluble C6PS (19) to dimers that are nearly inactive in solution. Based on ellipsometry binding stoichiometry data, Hathcock et al. (27) proposed that FXa but not FX might dimerize on a membrane. In addition, Husten et al. (30) suggested from fluorescence data that DEGR-Xa might form oligomers on binding to PS-containing membranes. However, these claims were not taken seriously since these reports offered no direct evidence for dimer formation or quantitative analysis of observations in terms of a dimerization model. This study provides both. We present five independent lines of evidence for dimer formation, each of which is quantitatively consistent with a dimerization model:
-
1.
Activity decrease upon binding to membranes (Fig. 2).
-
2.
Effective Kd for membrane binding as a function of membrane concentration (Fig. 3 A).
-
3.
Anomalous binding isotherms for DEGR-Xa (Fig. 3, B1 and B2).
-
4.
Two DEGR-Xa fluorescence lifetime components vary with membrane concentration (Fig. 3 C).
-
5.
FEGR-Xa anisotropy decrease due to nonradiative homotransfer (Fig. 4).
In trying to explain our observations, we used a dimer model, both because this is the simplest possible oligomer model and because FXa forms a dimer in solution when bound to soluble C6PS (19,20). Furthermore, the same surface dimerization model (Appendices A–C in the Supporting Material) explains all these anomalous behaviors. We conclude that FXa (and even its active-site derivatized forms) form dimers on PS-containing membranes.
One of the most interesting aspects of this phenomenon is that addition of PS-containing membranes to FXa at near physiological concentrations leads to a drop, not an increase, in its proteolytic activity. To demonstrate this, we used a substrate that does not bind to membranes and does not block the membrane surface (evidenced by proteolytic rate unaffected by membrane concentration at low FXa concentration (8)), Pre2. The rate of Pre2 conversion by FXa at 10 mM calcium was shown some time ago to be only slightly increased (approximately twofold) by the presence of phospholipid membranes (1:1 dioleoylphosphatidylcholine/dioleoylphosphatidylglycerol) as compared to the absence of membranes (31). Since soluble PG does not stimulate FXa activity against prothrombin (32) but soluble PS produces a 500-fold increase (8), the small effect previously reported must have been a nonspecific change in the protein due to absorption to a PG-containing membrane surface (24). However, in the experiments summarized in Fig. 2 B, addition of PS-containing membranes leads to a twofold decrease in activity rather than the 500-fold increase we have reported previously (6). This also contrasts with reports from other groups (e.g., (5,22)) that PS-containing membranes increase FXa activity by anywhere between 20- and 7000-fold, depending on the experimental conditions. This discrepancy results from the fact that the studies presented here were performed with high concentrations of FXa, akin to those that might exist in a platelet plug (potentially 150–200 nM if all FX is activated), whereas most of the earlier studies were carried out with 1–10 nM FXa.
Now that we have demonstrated the existence of a membrane-assembled FXa dimer, it is reasonable to ask whether it may be similar to the C6PS-assembled species. At 4 mM Ca2+, the dissociation constant for the solution-phase human FXa dimer is ∼80 nM (20), whereas the surface dissociation constant obtained from fitting the data in Fig. 2 = (40 ± 25) × 10−15 mol/(dm)2 corresponds to a solution dissociation constant of 10, 1, or 0.1 nM at 1, 10, or 100 μM membranes. Expressed in the same units (nM), the solution and surface dimer dissociation constants are comparable, except that the membrane-associated constant is smaller, as would be expected if localization of the monomer to a membrane reduces the unfavorable entropy of self-association. In addition, both the membrane- and C6PS-assembled dimers are essentially inactive toward prethrombin 2 as a substrate. Based on these observations, it seems that the solution and membrane-bound dimers are comparable. Since head-to-head or head-to-tail dimers would be difficult to imagine on a membrane, it is likely that both are side-by-side complexes. Based on this conclusion, on our fluorescein nonradiative homotransfer results, and on lysine acetylation results, it is concluded elsewhere that the structure of FXa in the dimer must be significantly different from monomer structures proposed based on crystallographic data (20).
Our results also make it clear that the dimer interface must either involve the enzyme active site or be linked to the active site, since labeling of this site results in a significantly higher surface dimerization dissociation constant (0.7 and 5 nmol/dm2 for FEGR-Xa and DEGR-Xa, respectively) than appears to apply to unlabeled FXa dimerization (0.04 nmol/dm2). This is consistent with the fact that C6PS-triggered DEGR-Xa dimerization is barely detectable in solution (20). In the dimer, fluorescein depolarization suggests that fluorescein moieties linked to the active site are within 37 Å of each other (Fig. 4). The active site is not blocked in the dimer, since it remains active against a synthetic tri-peptide substrate (20). However, the membrane-bound dimer is essentially inactive toward a protein substrate, prethrombin 2, exactly as was observed for a soluble dimer assembled in solution through binding of soluble C6PS, suggesting that dimer formation interferes with a substrate exosite rather than with direct interaction with the active site (20).
Since it is now established that PS binding triggers formation of inactive dimers both in solution and on membranes, it is reasonable to speculate as to the possible physiological significance of dimer formation. Human FXa functions in vivo either on PS-exposing membranous vesicles derived from activated platelets (12,14,33,34) or on platelet membranes having PS exposed on their surfaces (35). The concentration of human FX in plasma is ∼150–200 nM, and ∼100% is activated to FXa in the platelet plug at the site of injury when platelets are exposed to both collagen and thrombin (36). The activity of human FXa in solution is very low (kcat/KM for prothrombin active site formation ≅ 130 M−1 s−1 at 91 nM FXa), but increases by roughly 500-fold (kcat/KM ≅ 7 × 104 M−1 s−1 at 50 μM PS/PC and 5 nM FXa) when bound to PS-containing membranes (6). Membrane binding produces this rate enhancement by roughly a 250-fold increase in the rate of the active intermediate (meizothrombin) formation and by channeling intermediates directly to thrombin without release from the enzyme (∼250-fold) (6). A maximal rate of prothrombin activation is not obtained until FXa binds to factor Va to form the prothrombinase complex (kcat/KM ∼1.2 × 108 M−1 s−1 (23)), roughly another 1800-fold rate enhancement. Recent work has shown that FXa dimerization and binding to factor Va2 in solution are competitive events (R. Majumder and B. R. Lentz, unpublished). Thus, it may be that FXa dimerization and prothrombinase complex formation are competitive events on a PS-containing membrane, a possibility that we are currently investigating. The local concentration of platelet-derived vesicles in the platelet plug is not known, but it is likely to be high in a mature plug, probably above the 15–20 μM minimum seen in Fig. 2 B. However, in the initial wound, before thrombin triggers a burst of platelet activation, the platelet vesicle concentration could be low. The concentration of FXa in Fig. 2 B was high (300 nM), but comparable to the concentration of FX in plasma (∼150–200 nM). Under these conditions, FXa dimerization on membranes should be possible, depending on the local free Ca2+ concentration. The free Ca2+ concentration in the experiments recorded in Fig. 2 B was 4 mM, well above the free Ca2+ concentration in plasma (∼1–1.2 mM). We performed one experiment at 1 mM Ca2+ (inset between Fig. 2, B and C) and did not observe the anomalous behavior seen in Fig. 2 B, indicating that FXa did not dimerize under these conditions. Since FXa dimerization in solution is quite sensitive to Ca2+ concentration (19,20), dimerization on a PS-containing membrane is likely to be as well, a possibility that we are investigating. Depending on the details of the Ca2+-dependence of FXa dimerization on membranes, FXa in the early platelet mass could exist as an inactive dimer, an active monomer, or in a super-active form complexed with FVa as platelet activation proceeds. Since Ca2+ concentration in the early wound (platelet plug) drops to ∼2/3 that in plasma (37), the balance between these different forms of FXa could play an important regulatory role in coagulation. In this way, PS-containing membranes could turn out to downregulate FXa at higher Ca2+ and upregulate it at lower Ca2+—a fascinating possibility that demands further investigation.
Supporting Material
Experimental Procedures section as well as three appendices with 18 equations are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01306-X.
Supporting Material
Acknowledgments
We gratefully acknowledge the help of Jim Mattheis, HORIBA Jobin Yvon, with fluorescence lifetime measurements, and are thankful for the opportunity to work with the pre-release version of FluoroMax-4.
This work was supported by United States Public Health Service grant No. HL 45916 and by the National Heart, Lung and Blood Institute grant No. HL 072827 (to B.R.L.).
Footnotes
Tilen Koklic and Rinku Majumder contributed equally to this work.
Tilen Koklic's present address is Laboratory of Biophysics (EPR Center), Jozef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia.
Abbreviations used: see the Supporting Material.
References
- 1.Papahadjopoulos D., Hougie C., Hanahan D. Influence of surface change of phospholipids on their clot-promoting activity. Proc. Soc. Exp. Biol. Med. 1962;111:413–416. [Google Scholar]
- 2.Jackson C.M., Nemerson Y. Blood coagulation. Annu. Rev. Biochem. 1980;49:765–811. doi: 10.1146/annurev.bi.49.070180.004001. [DOI] [PubMed] [Google Scholar]
- 3.Jones M.E., Lentz B.R., Dombrose F.A., Sandberg H. Comparison of the abilities of synthetic and platelet-derived membranes to enhance thrombin formation. Thromb. Res. 1985;39:711–724. doi: 10.1016/0049-3848(85)90255-5. [DOI] [PubMed] [Google Scholar]
- 4.Gerads I., Govers-Riemslag J.W., Tans G., Zwaal R.F., Rosing J. Prothrombin activation on membranes with anionic lipids containing phosphate, sulfate, and/or carboxyl groups. Biochemistry. 1990;29:7967–7974. doi: 10.1021/bi00486a027. [DOI] [PubMed] [Google Scholar]
- 5.Rosing J., Tans G., Govers-Riemslag J.W., Zwaal R.F., Hemker H.C. The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 1980;255:274–283. [PubMed] [Google Scholar]
- 6.Wu J.R., Zhou C., Majumder R., Powers D.D., Weinreb G. Role of procoagulant lipids in human prothrombin activation. 1. Prothrombin activation by factor Xa in the absence of factor Va and in the absence and presence of membranes. Biochemistry. 2002;41:935–949. doi: 10.1021/bi0116893. [DOI] [PubMed] [Google Scholar]
- 7.Boskovic D.S., Giles A.R., Nesheim M.E. Studies of the role of factor Va in the factor Xa-catalyzed activation of prothrombin, fragment 1.2-prethrombin-2, and dansyl-L-glutamyl-glycyl-L-arginine-meizothrombin in the absence of phospholipid. J. Biol. Chem. 1990;265:10497–10505. [PubMed] [Google Scholar]
- 8.Banerjee M., Majumder R., Weinreb G., Wang J., Lentz B.R. Role of procoagulant lipids in human prothrombin activation. 2. Soluble phosphatidylserine upregulates and directs factor Xa to appropriate peptide bonds in prothrombin. Biochemistry. 2002;41:950–957. doi: 10.1021/bi0116902. [DOI] [PubMed] [Google Scholar]
- 9.Majumder R., Weinreb G., Lentz B.R. Efficient thrombin generation requires molecular phosphatidylserine, not a membrane surface. Biochemistry. 2005;44:16998–17006. doi: 10.1021/bi051469f. [DOI] [PubMed] [Google Scholar]
- 10.Schick P.K., Kurica K.B., Chacko G.K. Location of phosphatidylethanolamine and phosphatidylserine in the human platelet plasma membrane. J. Clin. Invest. 1976;57:1221–1226. doi: 10.1172/JCI108390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devaux P.F. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 1991;30:1163–1173. doi: 10.1021/bi00219a001. [DOI] [PubMed] [Google Scholar]
- 12.Bode A.P., Sandberg H., Dombrose F.A., Lentz B.R. Association of factor V activity with membranous vesicles released from human platelets: requirement for platelet stimulation. Thromb. Res. 1985;39:49–61. doi: 10.1016/0049-3848(85)90121-5. [DOI] [PubMed] [Google Scholar]
- 13.Wolf P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967;13:269–288. doi: 10.1111/j.1365-2141.1967.tb08741.x. [DOI] [PubMed] [Google Scholar]
- 14.Sims P.J., Faioni E.M., Wiedmer T., Shattil S.J. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J. Biol. Chem. 1988;263:18205–18212. [PubMed] [Google Scholar]
- 15.Comfurius P., Senden J.M., Tilly R.H., Schroit A.J., Bevers E.M. Loss of membrane phospholipid asymmetry in platelets and red cells may be associated with calcium-induced shedding of plasma membrane and inhibition of aminophospholipid translocase. Biochim. Biophys. Acta. 1990;1026:153–160. doi: 10.1016/0005-2736(90)90058-v. [DOI] [PubMed] [Google Scholar]
- 16.Wong P.C., Crain E.J., Nguan O., Watson C.A., Racanelli A. Antithrombotic actions of selective inhibitors of blood coagulation factor Xa in rat models of thrombosis. Thromb. Res. 1996;83:117–126. doi: 10.1016/0049-3848(96)00112-0. [DOI] [PubMed] [Google Scholar]
- 17.Borensztajn K., Stiekema J., Nijmeijer S., Reitsma P.H., Peppelenbosch M.P. Factor Xa stimulates proinflammatory and profibrotic responses in fibroblasts via protease-activated receptor-2 activation. Am. J. Pathol. 2008;172:309–320. doi: 10.2353/ajpath.2008.070347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ambrosini G., Altieri D.C. Molecular dissection of effector cell protease receptor-1 recognition of factor Xa. Assignment of critical residues involved in antibody reactivity and ligand binding. J. Biol. Chem. 1996;271:1243–1248. doi: 10.1074/jbc.271.2.1243. [DOI] [PubMed] [Google Scholar]
- 19.Majumder R., Wang J., Lentz B.R. Effects of water soluble phosphatidylserine on bovine factor Xa: functional and structural changes plus dimerization. Biophys. J. 2003;84:1238–1251. doi: 10.1016/S0006-3495(03)74939-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chattopadhyay R., Iacob R., Majumder R., Sen S., Tomer K. Functional and structural characterization of factor Xa dimer in solution. Biophys. J. 2009;96:974–986. doi: 10.1016/j.bpj.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nesheim M.E., Kettner C., Shaw E., Mann K.G. Cofactor dependence of factor Xa incorporation into the prothrombinase complex. J. Biol. Chem. 1981;256:6537–6540. [PubMed] [Google Scholar]
- 22.Nesheim M.E., Taswell J.B., Mann K.G. The contribution of bovine factor V and factor Va to the activity of prothrombinase. J. Biol. Chem. 1979;254:10952–10962. [PubMed] [Google Scholar]
- 23.Weinreb G.E., Mukhopadhyay K., Majumder R., Lentz B.R. Cooperative roles of factor Va and phosphatidylserine-containing membranes as cofactors in prothrombin activation. J. Biol. Chem. 2003;278:5679–5684. doi: 10.1074/jbc.M208423200. [DOI] [PubMed] [Google Scholar]
- 24.Cutsforth G.A., Whitaker R.N., Hermans J., Lentz B.R. A new model to describe extrinsic protein binding to phospholipid membranes of varying composition: application to human coagulation proteins. Biochemistry. 1989;28:7453–7461. doi: 10.1021/bi00444a045. [DOI] [PubMed] [Google Scholar]
- 25.Erb E.M., Stenflo J., Drakenberg T. Interaction of bovine coagulation factor X and its glutamic-acid-containing fragments with phospholipid membranes—a surface plasmon resonance study. Eur. J. Biochem. 2002;269:3041–3046. doi: 10.1046/j.1432-1033.2002.02981.x. [DOI] [PubMed] [Google Scholar]
- 26.Giesen P., Willems G., Hermens W. Production of thrombin by the prothrombinase complex is regulated by membrane-mediated transport of prothrombin. J. Biol. Chem. 1991;266:1379–1382. [PubMed] [Google Scholar]
- 27.Hathcock J.J., Rusinova E., Gentry R.D., Andree H., Nemerson Y. Phospholipid regulates the activation of factor X by tissue factor/factor VIIa (TF/VIIa) via substrate and product interactions. Biochemistry. 2005;44:8187–8197. doi: 10.1021/bi050338b. [DOI] [PubMed] [Google Scholar]
- 28.Andree H.A., Contino P.B., Repke D., Gentry R., Nemerson Y. Transport rate limited catalysis on macroscopic surfaces: the activation of factor X in a continuous flow enzyme reactor. Biochemistry. 1994;33:4368–4374. doi: 10.1021/bi00180a034. [DOI] [PubMed] [Google Scholar]
- 29.Chen L., Yao Q., Brungardt K., Squier T., Bigelow D. Changes in spatial arrangement between individual Ca-ATPase polypeptide chains in response to phospholamban phosphorylation. Ann. NY Acad. Sci. 1998;853:264–266. doi: 10.1111/j.1749-6632.1998.tb08275.x. [DOI] [PubMed] [Google Scholar]
- 30.Husten E.J., Esmon C.T., Johnson A.E. The active site of blood coagulation factor Xa. Its distance from the phospholipid surface and its conformational sensitivity to components of the prothrombinase complex. J. Biol. Chem. 1987;262:12953–12961. [PubMed] [Google Scholar]
- 31.Esmon C.T., Owen W.G., Jackson C.M. The conversion of prothrombin to thrombin. V. The activation of prothrombin by factor Xa in the presence of phospholipid. J. Biol. Chem. 1974;249:7798–7807. [PubMed] [Google Scholar]
- 32.Banerjee M., Drummond D.C., Srivastava A., Daleke D., Lentz B.R. Specificity of soluble phospholipid binding sites on human factor Xa. Biochemistry. 2002;41:7751–7762. doi: 10.1021/bi020017p. [DOI] [PubMed] [Google Scholar]
- 33.Sandberg H., Bode A.P., Dombrose F.A., Hoechli M., Lentz B.R. Expression of coagulant activity in human platelets: release of membranous vesicles providing platelet factor 1 and platelet factor 3. Thromb. Res. 1985;39:63–79. doi: 10.1016/0049-3848(85)90122-7. [DOI] [PubMed] [Google Scholar]
- 34.Bevers E.M., Comfurius P., Tilly H.J., Zwaal R.F.A. Transbilayer movement of procoagulant phospholipids during platelet microvesicle formation. Throm. Haemostasis. 1991;65:688a. [Google Scholar]
- 35.Solum N.O. Procoagulant expression in platelets and defects leading to clinical disorders. Arterioscler. Thromb. Vasc. Biol. 1999;19:2841–2846. doi: 10.1161/01.atv.19.12.2841. [DOI] [PubMed] [Google Scholar]
- 36.Kempton C.L., Hoffman M., Roberts H.R., Monroe D.M. Platelet heterogeneity: variation in coagulation complexes on platelet subpopulations. Arterioscler. Thromb. Vasc. Biol. 2005;25:861–866. doi: 10.1161/01.ATV.0000155987.26583.9b. [DOI] [PubMed] [Google Scholar]
- 37.Grzesiak J.J., Pierschbacher M.D. Shifts in the concentrations of magnesium and calcium in early porcine and rat wound fluids activate the cell migratory response. J. Clin. Invest. 1995;95:227–233. doi: 10.1172/JCI117644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.