

Abstract
PolyQ peptides teeter between polyproline II (PPII) and β-sheet conformations. In tandem polyQ-polyP peptides, the polyP segment tips the balance toward PPII, increasing the threshold number of Gln residues needed for fibrillation. To investigate the mechanism of cis-inhibition by flanking polyP segments on polyQ fibrillation, we examined short polyQ, polyP, and tandem polyQ-polyP peptides. These polyQ peptides have only three glutamines and cannot form β-sheet fibrils. We demonstrate that polyQ-polyP peptides form small, soluble oligomers at high concentrations (as shown by size exclusion chromatography and diffusion coefficient measurements) with PPII structure (as shown by circular dichroism spectroscopy and 3JHN-Cα constants of Gln residues from constant time correlation spectroscopy NMR). Nuclear Overhauser effect spectroscopy and molecular modeling suggest that self-association of these peptides occurs as a result of both hydrophobic and steric effects. Pro side chains present three methylenes to solvent, favoring self-association of polyP through the hydrophobic effect. Gln side chains, with two methylene groups, can adopt a conformation similar to that of Pro side chains, also permitting self-association through the hydrophobic effect. Furthermore, steric clashes between Gln and Pro side chains to the C-terminal side of the polyQ segment favor adoption of the PPII-like structure in the polyQ segment. The conformational adaptability of the polyQ segment permits the cis-inhibitory effect of polyP segments on fibrillation by the polyQ segments in proteins such as huntingtin.
Introduction
Genetic expansion of polyglutamine (polyQ) tracts is responsible for Huntington's disease and several other lethal neurodegenerative diseases (1). Other than the polyQ tract, there is no homology among the proteins encoded by the affected genes. The exact nature of the toxic species is still debated, but it may be a protein aggregate (soluble or insoluble). It is also possible that a “toxic” conformation of the monomer of the expanded polyQ region is the cause of neuronal cell death in all of these diseases (2–8).
Recent data from x-ray crystallography and solid-state NMR suggest that polyQ and other Gln-rich peptides form parallel, in-register β-sheet fibrils (9–14). Little is known, however, about the structure of fibril precursors, such as soluble oligomers and monomers of these peptides (15,16). PolyQ aggregation differs from that of other fibril-forming peptides in that in polyQ β-sheets, hydrogen bonds can form between both main-chain and side-chain amide atoms (14,17,18). This is in contrast to β-sheet forming peptides such as β-amyloids, in which stacking of β-sheets involves interactions predominantly among hydrophobic side chains (19–21).
One notable feature of polyQ expansion diseases is that there is a threshold in the number of Gln residues necessary to cause disease. For Huntington's disease, expansion of the polyQ tract beyond 35–40 residues is invariably associated with disease and protein deposits in cells, and longer polyQ tracts are associated with earlier onset and more severe disease (22). This threshold has been reproduced in models such as Caenorhabditis elegans (23), Drosophila melanogaster (24), Saccharomyces cerevisiae (25), mice (26), and zebrafish (27), as well as in nonhuman primates (28). The threshold number, however, is context-dependent (29). In contrast to Huntington's disease, the threshold for the spinocerebellar ataxia (SCA) type 6 is only 20–30 residues (30). The threshold for simpler synthetic peptides to form fibrils is even shorter, as peptides with only six glutamines can form insoluble β-sheets (31). Thus, the protein context and especially the sequences directly neighboring the polyQ tracts affect the ability of these peptides to aggregate (32,33). In huntingtin, a polyproline (polyP) tract flanks the polyQ tracts, and this polyP tract acts as a cis-inhibitor of polyQ aggregation (31,34).
Here, we investigate the structural basis of the mechanism by which the polyP tract inhibits polyQ aggregation. We present evidence that polyQ, polyP, and polyQ-polyP peptides adopt a type of polyproline II (PPII) helical structure in solution, in agreement with previous studies by our group (31) and others (35). In this work, we focus on a series of short peptides with a polyQ tract or tandem polyQ-polyP tracts in which the polyQ tracts are too short to form fibrils. We previously showed that polyQ peptides with 3–15 Gln residues teeter between a PPII-like and β-sheet structure, and the presence of an adjacent polyP tract tips the balance of tandem polyQ-polyP peptides toward the PPII conformation (31). This change in the ensemble of structures is associated with an increase in the threshold length of the polyQ segment needed for β-sheet fibril formation compared with polyQ peptides without an adjacent polyP segment. Using NMR, thermal denaturation analysis, circular dichroism (CD) spectroscopy, and size exclusion chromatography, we demonstrate that a polyP segment does not act directly as a “folding nucleus” to induce a similar structure in adjacent polyQ residues. Rather, the effect of the polyP segment on an adjacent polyQ segment results from a combination of factors, including the hydrophobicity and conformational propensities of the covalently constrained Pro residues, which permit self-association into small oligomers at high concentrations. These oligomers have a PPII-like structure, and we show, using constant time correlation spectroscopy (CT-COSY) NMR and other experiments, that the Gln residues in such oligomers themselves adopt a PPII-like structure. Furthermore, steric clashes between Gln side chains and Pro side chains to the C-terminal side of the polyQ segment favor adoption of the PPII-like structure in the polyQ segment. Thus, the effect of the polyP segment on the adjacent polyQ segment is an indirect one that results from a particular type of self-association of the hydrophobic and conformationally constrained polyP segment into oligomers with PPII structure. Finally, we will also present a model for some of the intermediates in polyQ aggregation, which includes soluble oligomers of polyQ-polyP peptides with PPII-like structure, and in which the polyQ segments permit aggregation in solution through hydrophobic contacts among side-chain atoms.
Materials and Methods
Peptide design and rationale
In this work, we utilize eight peptides with polyQ, polyP, or tandem polyQ-polyP segments modeled on the N-terminal sequence of human huntingtin protein. They can be grouped into two categories, based on sequences flanking the polyQ and/or polyP segments, as follows:
Group I
R3GQ3GY
R3GQ3P11GY
R3GP11GY
R3AQ3AY
Group II
YAQ3AR
YAQ3P11AR
YAP11AR
YGQ3GR
In all glutamine-containing peptides, the polyQ segment is only three Gln residues long, which precludes the formation of fibrils as occurs with longer polyQ peptides. In addition, this length yields peptides in which the Gln residues have distinctive chemical shifts in NMR spectroscopy. The polyP tract contains 11 Pro residues, which is the same as the polyP tract immediately flanking the C-terminus of the polyQ tract in huntingtin. Arg residues are included to increase water solubility, and Tyr is included as a chromophore. Each group has a control peptide in which the linker residue is varied, i.e., in group I, R3AQ3AY is a control for the other peptides with Gly as the linker residue, and in group II, YGQ3GR is a control for the other peptides with Ala as the linker residue. Peptide synthesis and purification were essentially as described previously (31), and are discussed in more detail in the Supporting Material.
Disaggregation procedure
Peptides underwent a disaggregation procedure to ensure complete solubilization, even though the peptides appeared to dissolve completely in water similarly to previous findings (36,37). Further specifics are given in the Supporting Material.
CD spectroscopy
CD spectra were measured using an Aviv model 202 spectropolarimeter (Lakewood, NJ) with a temperature-controlled cell holder. Data were corrected for background from buffer scans, converted to mean residue ellipticities, and then corrected for baseline deviation. For most measurements, peptides were freshly disaggregated and dissolved to yield concentrated stock solutions in buffer. The peptides were then diluted to concentrations ranging from 50 μM to a few millimolars with 10 mM sodium phosphate, pH 7.00 or 3.00.
Initial CD spectra were obtained within 1 h of dilution from the concentrated stock; additional spectra were measured at various times up to 1 month, as described in the Results section and the Supporting Material. Spectra were measured using a 0.1 cm quartz cell (Starna, Atascadero, CA). Signals were collected from 260 to 190 nm at 4°C and 25°C with 0.5 nm step size, 1 nm bandwidth, and 1 s averaging time. The spectra are the result of six averaged scans per sample, and measurements were repeated on at least three replicate samples.
The methods used to assess the stability of the CD spectra over time are given in the Supporting Material. As described in the Results section, all peptides showed positive ellipticity at ≈215–230 nm, which was interpreted as being consistent with PPII helix conformation (34,36,38). This positive ellipticity decreased with increasing temperature, and this change was completely reversible. The experimental procedures used to measure thermal melting of peptides, and thermodynamically analyze that melting, are given in the Supporting Material.
NMR spectroscopy
Experiments were performed using a Varian 600 MHz NMR spectrometer (Palo Alto, CA) equipped with a triple resonance probe. Two-dimensional 1H spectra were performed to obtain residue-specific information on four peptides (R3GQ3GY, R3GQ3P11GY, YAQ3AR, and YAQ3P11AR). Total correlation spectroscopy (TOCSY) experiments were performed at ∼0°C, 25°C, and 50°C to assign resonances of each residue. Experiments were performed at multiple temperatures because chemical shifts of all residues changed with large changes in temperature, and this information was necessary for the melting experiments described below. The acquisition time was set to 0.5 s, and the mixing time was 0.06 s. Suppression of water signal was achieved using eight presaturation pulses; generally, better suppression of water signals was achieved at 5°C than at 0°C.
CT-COSY spectra were measured for the same four peptides to obtain 3JHN-Hα coupling constants at temperatures of 0–40°C, or in some cases, up to 60°C in 5°C increments. Measurements were performed twice at all temperatures—once on heating and once on recooling. For some peptides, the spectral line shapes at temperatures above 40°C became very broad at pH 7.0. Preliminary experiments showed that this broadening was due in part to rapid proton exchange of amide protons, and could be reduced by lowering the pH to 5.0 or 1.5–3.0 (see Fig. 4). 3JHN-Hα coupling constants were obtained and used to calculate torsional (ϕ) angles, as described previously (39,40) and in the Supporting Material. Values obtained for the ϕ-angles indicate that a 3JHN-Hα coupling constant of 3–4 Hz is most consistent with an α-helix, and a 3JHN-Hα coupling constant > 8 Hz suggests a β-sheet (39). As described by Shi and Kallenbach, a 3JHN-Hα coupling constant of 5–7 Hz is consistent with a PPII-like helical structure (40).
Figure 4.
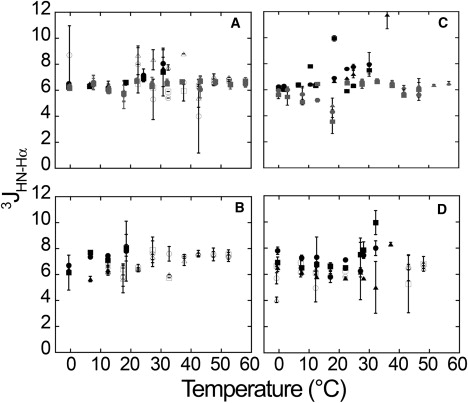
3JHN-Hα coupling constants for four peptides: (A) R3GQ3GY, (B) R3GQ3P11GY, (C) YAQ3AR, and (D) YAQ3P11AR. As described in Materials and Methods, and Results, CT-COSY spectra were obtained for these peptides at various temperatures. Because of the rapid amide proton exchange at pH 7.0 and higher temperatures, we also obtained data at lower pH values. Gln residues are numbered as they occur in the amino acid sequence. Symbols: •, •, and ○ for Gln1 at pH 7.0. 5.0, and 2.5–3.0, respectively; ■, ■, and □ for Gln2 at pH 7.0. 5.0, and 2.5–3.0, respectively; and ▴, ▴, and ▵ for Gln3 at pH 7.0. 5.0, and 2.5–3.0, respectively. Values with error bars represent the mean ± standard deviation of two measurements at the same temperature.
Nuclear Overhauser effect spectroscopy (NOESY) spectra were obtained for two peptides (R3GQ3GY and R3GQ3P11GY) at pH 7.0 (200 mM sodium phosphate) and ∼0°C and 5°C. The peptide concentrations were 3.80 and 2.68 mM for R3GQ3GY and R3GQ3P11GY, respectively. Spectra were measured with mixing times of 80 and 300 ms for R3GQ3GY, and 75 and 200 ms for R3GQ3P11GY. Better signal/noise ratios were achieved at the longer mixing times. The acquisition time was 250 ms, and the spectra are reported below. TOCSY experiments were also performed on the same samples and under the same conditions, except that the mixing time was 65 ms.
As an additional way to examine the aggregation state of polyQ and polyQ-polyP peptides, we estimated the diffusion coefficients of two peptides (R3GQ3GY and R3GQ3P11GY) using pulsed field gradient NMR, and compared the effective hydrodynamic radii of these peptides in solution with that of a protein standard, FN3s (a gift from Akiko Koide and Shohei Koide, both from the University of Chicago, Chicago, IL). FN3s, a synthetic construct for the 10th fibronectin type III domain, is a well-characterized, compact, spheroidal, globular protein with a molecular weight of 9939.23 (41). The method used was essentially that of Jones et al. (42), which is an application of an NMR method previously described by Gibbs and Johnson (43). As discussed further in the Supporting Material, the signal intensity in a pulse-gradient stimulated-echo longitudinal encode-decode sequence causes spin-lattice relaxation to appear as an exponential decay (44) in units proportional to time; thus, we obtain
| (1) |
where k = rate constant, which is proportional to the diffusion constant, D. The goal of these experiments was to compare the diffusion of our peptides with that of a reference standard, using the following relationship:
| (2) |
where RH, peptide and RH, standard are the effective hydrodynamic radii, by the Einstein-Stokes relationship, of the peptides and the reference standard, respectively.
The goal of these experiments was to compare the hydrodynamic behavior of FN3s, a protein of known dimensions, with that of R3GQ3GY and R3GQ3P11GY. NMR and x-ray crystallographic studies (44–46) indicate that fibronectin type III domains, including FN3s, are prolate ellipsoids, with dimensions of ∼43 Å × 19 Å × 17 Å. From these dimensions, we can estimate the effective spherical radius, RE, from the relationship RE = (abc)1/3, where a is the major axis, and b and c are the two minor axes, from which we calculate RE for FN3s ∼ 24.0 Å. Since this number does not take into account the hydration of the protein, the radius of gyration, Rg, may be somewhat larger. Although the structures of R3GQ3GY and R3GQ3P11GY are not known, we considered two possible theoretical values for RG, calculated from the following relationships:
| (3) |
for a symmetrical prolate ellipsoid, where R = radius and L = length, and L <<< R; and
| (4) |
for cylindrical rods (47).
Size exclusion chromatography
Size exclusion chromatography of previously disaggregated peptides dissolved in 200 mM sodium phosphate, pH 7.00, was performed with the use of Superdex 75 or Superdex Peptide columns. Further details of these experiments are given in the Supporting Material.
Results
CD spectra of short polyQ, polyP, and polyQ-polyP peptides
In agreement with earlier results (31,34,38,48), the CD spectra of freshly dissolved solutions of polyQ, polyP, and polyQ-polyP peptides show a positive peak with λmax ∼ 222–230 nm, indicative of PPII or PPII-like structure (49,50). CD spectra of other polyQ, polyP, and polyQ-polyP, and variants of those peptides (group II) are shown in Fig. S1 and Fig. S2; CD spectra of the above eight peptides (groups I and II) at pH 3.0 are shown in Fig. S3. As shown in Fig. S4, these patterns were stable over time, as the CD spectra did not change significantly over the course of a month of incubation.
To examine the temperature dependence of the CD spectra of these peptides (i.e., their thermal “unfolding”), peptide solutions were heated and cooled in increments of 5°C, the CD spectra were recorded, and the results were compared with a null hypothesis, according to which there is a two-state system (see Eq. S1, Eq. S2, Eq. S3, and Eq. S4). All peptides showed completely reversible melting (Fig. 1 and Fig. S5 show data for pH 7.00), as demonstrated by the fact that the melting and cooling curves were essentially superimposable. Fig. S6 and Fig. S7 show the observed ellipticity at 229 nm, which is indicative of PPII-like helical structure, as a function of temperature for group I and group II peptides, respectively. The maximal positive ellipticity for R3GP11GY occurred at 229 nm. This spectrum was taken as that of a bona fide PPII helical peptide, and therefore its wavelength was used for all analyses. The fit of the data to Eq. S4 shows only a slight curvature, indicative of noncooperative melting (Fig. S6 and Fig. S7). The experimentally accessible part of the curve was only a small portion of the theoretical curve for unfolding, which approaches the “unfolded” state only at infinitely high temperatures. Thus, the null hypothesis was incorrect, and the melting is not indicative of a two-state system. Rather, this behavior demonstrates local unfolding or unraveling of the PPII or PPII-like helices, with loss of left-handedness, perhaps toward a more extended structure.
Figure 1.
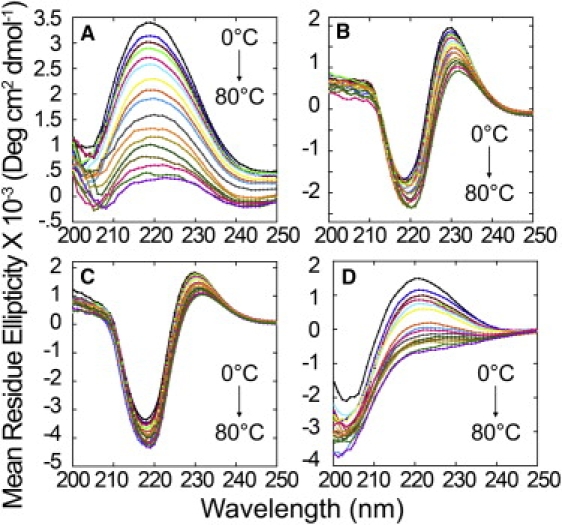
CD spectra showing thermal melting of polyQ, polyP, and polyQ-polyP peptides. Panels A–D show CD spectra of R3GQ3GY, R3GQ3P11GY, R3GP11GY, and R3AQ3AY, respectively, at temperatures from 0 to 80°C, at intervals of 5°C, and in 10 mM sodium phosphate, pH 7.00. For clarity, only the curves for heating of samples are shown; the curves obtained from cooling samples are essentially superimposable on those of the heating samples.
These results, especially the noncooperative nature of the thermal transition, underscore the difference between these PPII-like helical peptides and many α-helical peptides. Whereas the latter fold by forming folding nuclei, followed by propagation of backbone hydrogen-bond arrays between residues, no such cooperative folding or unfolding is observed for the PPII and PPII-like peptides described here.
Fig. 2 shows the CD spectra of four peptides at various concentrations and at 25°C. Similar changes also occur at 4°C (not shown). The mean residue ellipticity (MRE) changes with concentration, as shown most clearly by the two peptides containing polyP segments (R3GQ3P11GY and R3GP11GY; Fig. 2, B and C), especially for the negative MRE values at lower wavelengths. This pattern of change is consistent with the notion that the polyP segments of these peptides are constrained into a PPII helical structure even at low concentrations, but increasing the concentration induces structure in the less conformationally constrained portions of the peptide, including the polyQ segment. A similar pattern of changes in the far-ultraviolet region of the CD spectrum has been observed for other polyQ-containing peptides (34,35,51,52).
Figure 2.
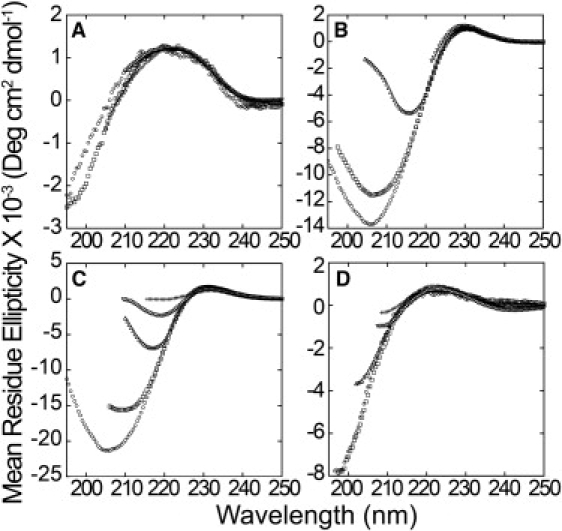
Concentration dependency of the CD spectra of peptides. Peptides were in 10 mM sodium phosphate, pH 7.00, and at 25°C. (A) R3GQ3GY at 73 (○), 95 (□), 241 (▵), 500 (▿), and 1000 (+) μM. (B) R3GQ3P11GY at 66 (○), 110 (□), 260 (▵), 500 (▿), and 1000 (+) μM. (C), R3GP11GY at 50 (○), 100 (□), 250 (▵), 500 (▿), and 1000 (+) μM. (D) R3AQ3AY at 50 (○), 100 (□), 250 (▵), 500 (▿), and 1000 (+) μM. PolyP-containing peptides show changes in spectra as concentration is increased, suggesting self-association.
Size exclusion chromatography shows oligomerization of polyQ peptides adopting a PPII conformation
The above data demonstrate that polyQ peptides, with or without a tandem polyP segment, adopt a PPII-like structure at all concentrations tested; however, the data shown in Fig. 2 suggest that a conformational change may occur with increasing concentration, and that such a change may be related to self-association. We used size exclusion chromatography to determine whether oligomers were present. As shown in Fig. 3, the polyQ-polyP peptides form small oligomers at higher concentrations (as well as in high ionic strength buffer, 200 mM sodium phosphate, pH 7.00) at room temperature. In addition, R3GQ3GY also forms oligomers, though only when higher (millimolar) concentrations of peptide are loaded onto the column.
Figure 3.
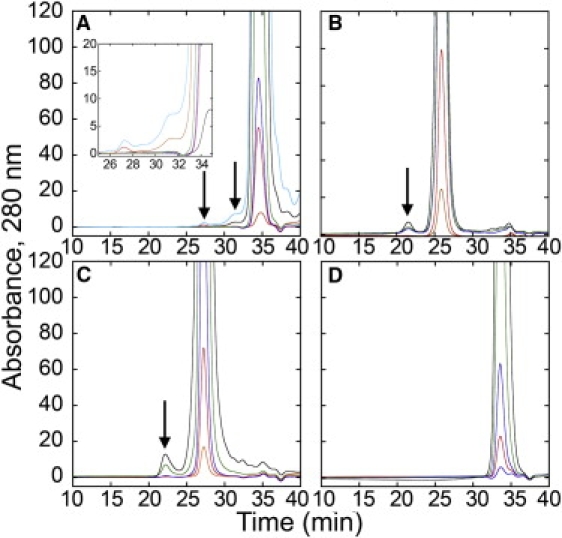
Concentration dependency of the size exclusion chromatographs of peptides. Peptides were eluted from the column in 200 mM sodium phosphate, pH 7.00, and at room temperature (∼22°C), and were loaded onto the Superdex Peptide column at various concentrations. (A) R3GQ3GY at 10 μM (orange), 60 μM (red), 100 μM (blue), 455 μM (green), 3.03 mM (black), and 5.88 mM (cyan). The inset shows a magnified view of the region of the chromatograph in which the oligomers elute. (B) R3GQ3P11GY at 10 μM (orange), 60 μM (red), 100 μM (blue), 455 μM (blue), and 2.75 mM (black). (C) R3GP11GY at 10 μM (orange), 45 μM (red), 100 μM (blue), 455 μM (green) and 2.25 mM (black). (D) R3AQ3AY at 10 μM (orange), 37 μM (red), 100 μM (blue), 455 μM (green), and 1.87 mM (black). Flow rate was 0.5 mL/min, void volume was at 15.4 min, and total volume was at 35.9 min.
In size exclusion chromatography, peptides become diluted as they migrate through the column; hence, the peptide that is loaded onto the column is ≈10 times more concentrated than when it emerges from the column. Thus, while oligomer peaks are apparent in higher-concentration samples (≥100 μM), they are present at significantly lower concentrations when the oligomers elute from the column. Thus, we calculate from the extinction coefficient that when 100 μM of R3GQ3P11GY are injected into the column, the peak concentration of monomer eluting from the column is ∼15 μM.
TOCSY and CT-COSY NMR spectra of oligomerizing polyQ and polyQ-polyP peptides indicate PPII conformation over a range of temperatures
To obtain residue-specific indicators of the conformations of the Gln residues in the polyQ and polyQ-polyP peptides discussed above, we measured TOCSY and CT-COSY spectra at a variety of temperatures from 0 to 60°C and a pH range of 1.5–7.0. TOCSY spectra (see examples in Fig. S8) were acquired at temperatures of 0°C, 25°C, and 50°C to obtain chemical shift information as a function of temperature, since the location of the chemical shifts changes over this temperature range.
From the two-dimensional CT-COSY spectra, we extracted one-dimensional F2 traces at the Hα for each Q residue. Such traces through the HN-Hα crosspeak appear as antiphase doublets, with one positive and one negative peak, separated by the HN-Hα coupling constant, 3JHN-Hα. 3JHN-Hα coupling constants were then used to compute backbone torsional (ϕ) angles of these residues using a Karplus relationship. The solvent had the same ionic strength as that used for size exclusion chromatography, which indicated peptide oligomerization, i.e., 200 mM sodium phosphate. Peptide concentration was well into the millimolar range, above the 455 μM concentration at which self-association was observed by size exclusion chromatography and/or CD spectroscopy. To obtain a more objective estimate of the coupling constants, the curves from one-dimensional slices of the spectrum were fitted to the equation of a difference of two Lorentzian functions, as described in Materials and Methods and in Eq. S5 (see the Supporting Material for further details). Sample CT-COSY spectra and examples of fits of the data to this equation are shown in Fig. S9 and Fig. S10, respectively. As shown in Fig. 4, all three Gln residues of both polyQ and polyQ-polyP peptides maintained coupling constants that fell within a narrow range of ≈6–8 Hz, at all temperatures tested from 0°C to 60°C and in the pH range of 1.5–7.0. From the Karplus relationship (see Materials and Methods and the Supporting Material), this suggests values of ϕ ≈ −75–90°, which is consistent with a PPII helical conformation.
In addition, we examined TOCSY spectra of the Pro residues at multiple temperatures from 0 to 60°C, and found only minor variations of the chemical shifts of Cβ, Cγ, and Cδ protons, all of which remained in the range of 2.2–3.65 ppm (data not shown). These chemical shifts are consistent with a trans peptide bond (39), and suggest that the secondary structure of the Pro residues does not change within this temperature range.
Diffusion of R3GQ3GY and R3GQ3P11GY indicates formation of soluble oligomers
As described in Materials and Methods, we used pulsed field gradient NMR spectroscopy to compare the diffusion coefficients of two peptides (R3GQ3GY and R3GQ3P11GY) with that of a reference standard protein, FN3s, a synthetic fibronectin type III domain (41). Fig. 5, A–C, show the decay of signal intensity with time for the dominant (backbone amide) peaks associated with these two peptides and protein, respectively, with all of the signals normalized to an initial value of one; the nonnormalized signal decays are shown in Fig. S11. Fig. 5 D shows the t1/2 values calculated for decay of the signals from the amide proton peaks of R3GQ3GY and R3GQ3P11GY, and for 20 representative peaks from the FN3s. The mean value for the first-order rate constant for the decay of T2 of R3GQ3GY, R3GQ3P11GY, and FN3s were 7.6 × 10−5, 6.1 × 10−5, and 5.2 × 10−5, respectively. Using a value of 24.0 Å for the minimal hydrodynamic radius for FN3s, we calculate RH = 16.4 and 20.5 Å for R3GQ3GY and R3GQ3P11GY, respectively.
Figure 5.
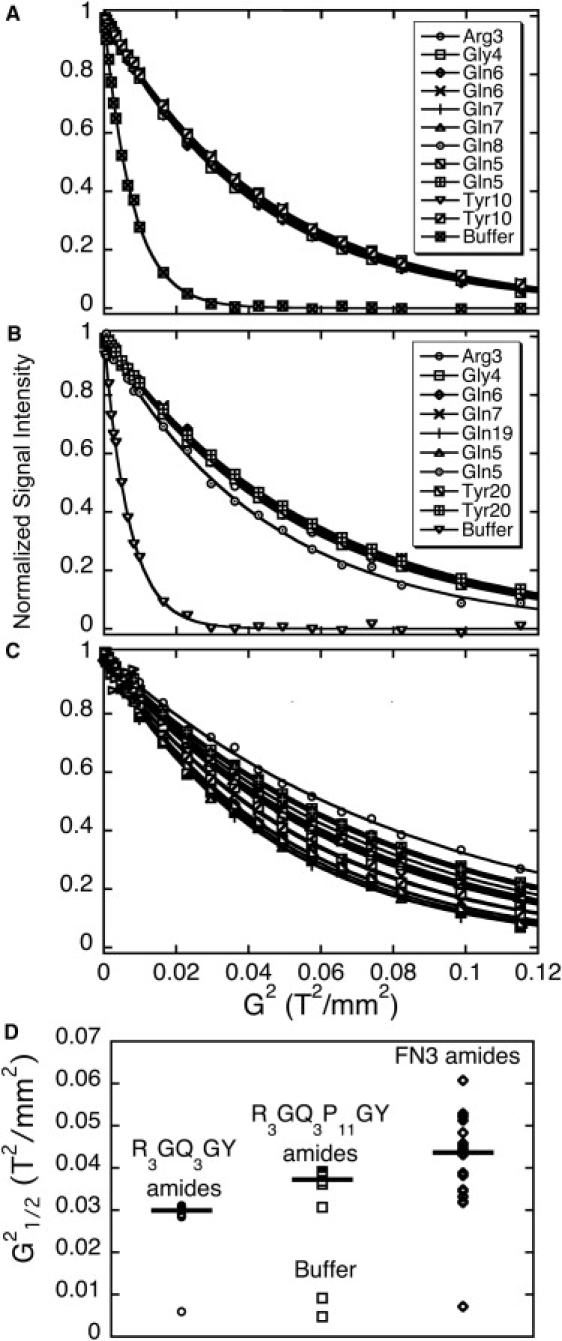
Estimation of diffusion coefficients of R3GQ3GY and R3GQ3P11GY by comparison with a protein standard, FN3s (42,43). Data are shown for the backbone amides of two peptides ((A) R3GQ3GY and (B) R3GQ3P11GY) and for (C) the protein standard FN3s, a small protein of known dimensions. Points are experimental data; lines are fits to the equation of a monoexponential decay (Eq. 1). The figure also includes several peaks derived from buffer instead of the peptides.
Thus, the relative diffusion coefficients estimated for R3GQ3GY and R3GQ3P11GY are larger than would be expected for a monomer, which is consistent with the view that both of these peptides form small oligomers in solution. The observed values for RH are close to that of FN3s. Although the shapes of the small oligomers of R3GQ3GY and R3GQ3P11GY are not known, the monomers would have to have an extremely high axial ratio to explain these high numbers entirely in terms of asymmetry. To take an extreme example, consider R3GQ3GY and R3GQ3P11GY depicted as completely rigid, rod-shaped molecules with a PPII conformation. From molecular modeling of these peptides as PPII helices (see Fig. 7), this corresponds to lengths of ∼25.5 and ∼56.7 Å for R3GQ3GY and R3GQ3P11GY, and a diameter of ∼7.6 Å for both peptides. Even given the extreme case of a completely rigid rod, we calculate radii of gyration of 7.8 and 16.5 Å, respectively. These values are both smaller than the observed values for RH. Although the difference is not definitive, the assumption of a completely rigid rod for polyP peptides is an exaggeration, as previously shown experimentally by fluorescence resonance energy transfer (FRET) measurements (53–55) that revealed the flexibility of polyP peptides. Thus, our data suggest that R3GQ3P11GY and possibly even R3GQ3GY form small soluble oligomers, with ∼2–4 molecules per aggregate, and with the dominant NMR signals coming from the aggregated species.
Figure 7.
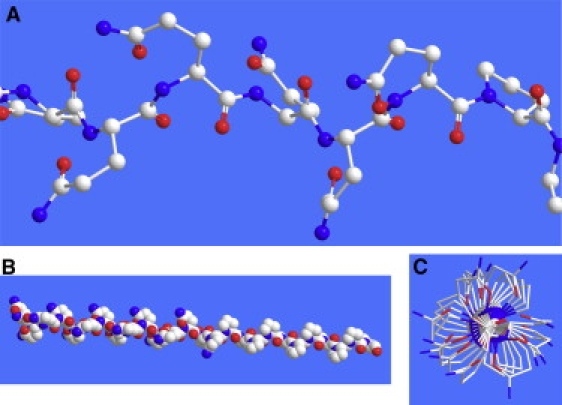
PolyQ-polyP peptide (Q15P11) arrayed as a PPII helix (ϕ = −75°, ψ = 150°). (A) Close-up view showing the side chains of Gln and Pro in similar conformations. (B and C) Full length of the peptide in side view and end-on view, respectively, showing the PPII helical conformation with “folded-over” side chains.
NOESY NMR spectra of polyQ and polyQ-polyP peptides support a PPII-like helical conformation and suggest modes of stabilization of the PPII-like structure
NOESY spectra were obtained for R3GQ3GY and R3GQ3P11GY at peptide concentrations of 3.80 and 2.68 mM, respectively, and for both in 200 mM sodium phosphate, pH 7.00, at 5°C. At these concentrations of peptide and salt, both peptides were shown (by CD spectroscopy, size exclusion chromatography, and diffusion coefficient measurements) to form soluble oligomers in which the peptide assumed a PPII helical structure, and CT-COSY spectra indicated that the Gln residues, in particular, were among those that adopted the PPII helical structure. In the NOESY experiments, of particular interest was the possibility that NOEs can exist between either backbone NH or Cα protons of the three Gln residues, and the Cβ, Cγ, or Nδ protons of the same residue.
Fig. 6 A shows the HN-Cα region of the NOESY spectrum of R3GQ3GY (300 ms mixing time), and Fig. 6 C shows a similar region of the spectrum of R3GQ3P11GY (200 ms mixing time). Close-ups of the regions containing the Cα and side-chain protons of the three Gln residues are shown in Fig. 6, B and D, for R3GQ3GY and R3GQ3P11GY. Both peptides show clear crosspeaks between the Cα protons and the Cβ and Cγ protons of the same residue, and the intensities of these crosspeaks increase with increasing mixing time. These data indicate that the side chain of the Gln residues forms a compact structure, with all atoms in close proximity to the peptide backbone, and the side chain oriented toward the N-terminus of the peptide. These findings are anticipated for a PPII-like helical structure with a “folded-back” side chain, but not for an extended β-strand structure in which the side chain would be more fully extended above and below the peptide backbone, and the Cγ protons would be too distant from the backbone to show easily observable NOEs. Our findings suggest that glutamines can adopt this “folded-over conformation”, as previously seen in crystal structures (56,57).
Figure 6.
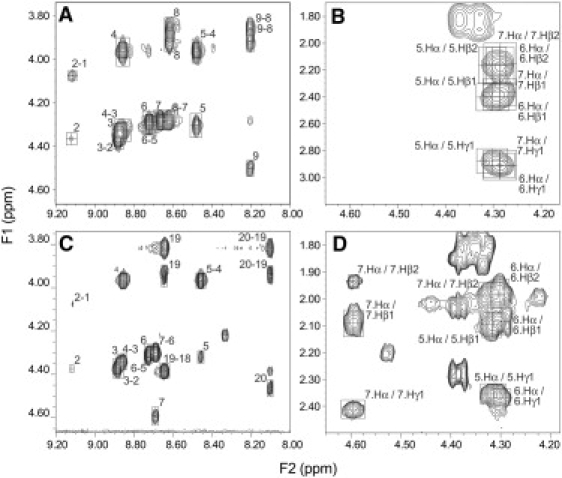
NOESY spectra of R3GQ3GY (A and B), and R3GQ3P11GY (C and D). R3GQ3GY concentration is 3.80 mM and R3GQ3P11GY concentration is 2.68 mM, both samples in 200 mM sodium phosphate (with 0.1% NaN3, w:v), pH 7.00, 5°C. A and C show the “footprint” (HN-Hα) region of the NOESY spectra of these peptides. B and D highlight NOEs between Cα protons of each of the three Gln residues and both Cβ and Cγ protons of the same residue. Numbers in the figure refer to the residue numbers in the two peptides.
Molecular modeling of polyQ and polyQ-polyP peptides suggests that the hydrophobic effect mediates self-association of PPII helical peptides
As shown above, polyQ and polyQ-polyP peptides adopt a stable PPII-like helical structure (Figs. 1, 2, and 4, and Fig. S1, Fig. S2, and Fig. S3). Furthermore, in this conformation, these peptides may self-associate into small soluble oligomers, as suggested by CD spectroscopy (Fig. 2), size exclusion chromatography (Fig. 3), and diffusion coefficient measurement (Fig. 5 and Fig. S11), all of which were performed at fairly high ionic strengths to enhance the hydrophobic effect (58,59). Finally, NOESY NMR experiments (Fig. 6) indicated that under conditions in which these peptides formed oligomers, the Gln residues were part of a PPII-like helix with side chains in a compact or “folded-over” conformation. We used molecular modeling to investigate the mechanism by which polyQ and polyQ-polyP peptides might self-associate when they form a PPII-like helix.
Fig. 7 shows polyQ and polyQ-polyP peptides arrayed in PPII helical conformations using ϕ = −75° and ψ = 150°. The overall shape of these peptides, arrayed in this fashion, is rod-like. In this conformation, the peptides form a solvent-exposed hydrophobic surface consisting of the entire side chain of Pro residues, and the β- and γ-methylene carbon atoms of Gln residues. Yoon et al. (60) examined polyP peptides and proposed that self-association of these peptides could result from interactions between these hydrophobic ridges. As shown in Fig. 7, a similar hydrophobic ridge can also arise in polyQ peptides when they adopt a PPII-like conformation and the side chains are “folded over” toward the backbone. Thus, a polyQ segment appears prone to self-associate through hydrophobic interactions when it adopts a PPII-like helix with this compact side-chain structure that resembles that of Pro side chains.
Discussion
In this work, we have shown that short polyQ, polyP, and polyQ-polyP tandem peptides adopt a stable PPII or PPII-like structure in solution, and that soluble oligomers of these peptides display that conformation. The polyP and polyQ-polyP peptides that contained the relatively hydrophobic polyP segment had a greater tendency to self-associate than peptides that contained only polyQ segments. Self-association was demonstrated by the change in the pattern of the CD spectrum (MRE) with peptide concentration, by size exclusion chromatography, and by diffusion coefficient measurements using pulsed field gradient NMR.
For all of the polyQ-containing peptides, the polyQ segment was only three residues long, and thus too short to form β-sheet fibrils. Especially in the context of an adjacent polyP segment, the polyQ-containing peptides self-associated as PPII-like helical peptides. As demonstrated in Fig. 7, polyP peptides are constrained to present a hydrophobic edge, formed by the methylene groups in their side chains, toward the solvent (60), which favors self-association through the hydrophobic effect. For simplicity, these models assume an all-trans conformation in the polyP segment; however, as shown by Eaton and co-workers (53,54), internal cis prolines can introduce kinks into the chain, and are primarily responsible for the higher than expected observed FRET efficiency in polyP peptides. This caveat notwithstanding, the majority of Pro residues have trans peptide bonds, as depicted in Fig. 7 and evaluated by TOCSY experiments. Fig. 7 also shows the Gln residues arrayed in a similar conformation, with side-chain structures strikingly similar to those of the Pro residues, i.e., with two methylene groups oriented toward the solvent, and the side-chain amide in proximity to the backbone, possibly also including the formation of a hydrogen bond between side-chain and backbone amide atoms. NOESY spectra (Fig. 6) demonstrate that our peptides do indeed adopt a conformation of this type: the Gln side chains form a compact structure close to the peptide backbone, with the potential for hydrophobic self-association.
For disease-causing polyQ proteins, such as huntingtin, genetic expansion of the polyQ tract beyond a sharp threshold is associated with disease, and longer polyQ tracts are associated with more severe and earlier-onset disease. This threshold, however, varies with the “context” (i.e., the protein in which the polyQ occurs) (22,29,32,33). In huntingtin, polyQ domains of >35–40 residues lead to disease, whereas 20–30 Gln residues in the α1A-voltage-dependent calcium channel subunit lead to SCA type 6 (30). For short polyQ peptides, the threshold for fibril formation is much shorter: ∼6 Gln residues (31). Presumably, the large globular domains of huntingtin inhibit efficient packing of some of the polyQ domain. In addition, huntingtin contains a polyP domain adjacent to the polyQ domain, and the polyP domain inhibits aggregation in vitro. Similarly, an adjacent polyP domain inhibits fibril formation by synthetic polyQ peptides, and increases the threshold from 6 to 9–12 Gln residues (31,34). We previously showed that this effect occurs because the polyP tract tends to induce a PPII-like conformation in the adjacent polyQ tract. A polyQ tract can readily adopt either a PPII-like or β-sheet conformation, and the polyP tract tips the balance toward the PPII-like conformation (31). It is also noteworthy that the polyP tract has this cis-inhibitory effect on fibrillation only if it is placed to the C-terminal side of the polyQ tract; the effect does not occur if it is placed to the N-terminal side (34).
The results presented here can help to explain both the threshold phenomenon and the requirement that the polyP segment must be placed to the C-terminal side of the polyQ tract. Indeed, it was perplexing that the polyP tract should be able to “induce” a PPII-like conformation in an adjacent polyQ tract at all, since the PPII-helix (and similar structures formed by polyQ peptides) showed no cooperativity of folding, in contrast to α-helices (61–63), where a nucleation event initiated helix formation followed by cooperative folding of the remainder of the helix. We observed completely noncooperative thermal “unfolding” (see, for example, Fig. 1 and Fig. S5, Fig. S6, and Fig. S7), perhaps to a somewhat more extended structure, of polyQ, polyP, and polyQ-polyP peptides.
As shown in Fig. 7, the “induction” of a PPII-like structure in the polyQ segment of polyQ-polyP peptides could be attributable in part to the self-association of the polyP segment (47,60). In the setting of an oligomer, where one hydrophobic edge of Pro residues associates with another, the Gln residues could assume a conformation similar to that of the Pro residues. Since the Pro side chain is covalently constrained, the Gln residues could follow suit and adopt a similar structure only if the Gln side chains are oriented toward the N-terminus of the peptide, as also depicted in Fig. 7.
Such self-association was apparent at high peptide concentrations. Even allowing for dilution of peptides as they migrate through size exclusion chromatography columns, the oligomer peak accounted for a small percentage of the total peptide. It is unlikely, therefore, that self-association through the hydrophobic effect alone accounts for cis-inhibition of polyQ aggregation by an adjacent polyP segment. In addition to this effect, at least three other effects may account for cis-inhibition.
First, as suggested by NMR and modeling, a Pro residue to the C-terminal side of a Gln residue occludes much of the conformational freedom of the Gln side chain, and thereby favors folding of the Gln side chain into a compact structure resembling the Pro side chain. Our results help to explain the fact that polyP inhibits polyQ fibrillation only when the polyP segment is to the C-terminal side of the polyQ segment (34).
Second, the rod-like polyP segment may act as a rigid strut, limiting the conformational freedom of the polyQ segment. Since the polyQ segment itself has a high tendency to adopt a PPII-like structure, it does so when conformationally constrained against adopting another structure, such as a β-sheet.
A third effect is suggested by our recent experiments on side-chain N-methylated polyQ peptides, which act as trans-inhibitors of polyQ aggregation (J. D. Lanning, unpublished results). Although the affinity of inhibitors (e.g., one designated 5QMe2) for a fibril-forming target polyQ peptide, YAQ12A, is only moderate (Kd ∼ 1 μM), complexes form in a chaperone-like manner, with very rapid binding and desorption of inhibitor and target. Thus, such inhibitors are active even at significantly substoichiometric inhibitor concentrations (e.g., 1:10 = inhibitor/target). In a similar way, even low concentrations of small oligomers of QnPm peptides with a PPII-like structure could inhibit fibril formation. In both cases (the N-methylated inhibitor and small oligomers of QnPm peptides), inhibition of fibrillation may stem from the fact that nucleation is slow and results from a rare conformational change. Wetzel and co-workers have argued that the nucleus for polyQ fibril formation could even be a monomer of altered conformation (64). Thus, fibrillation and its inhibition can be represented be the following scheme:
Formation of the nucleus is unfavorable for both polyQ and polyQ-polyP peptides, but apparently it is more so for the latter. We infer from our observations that either the self-association of a polyQ-polyP peptide (depicted as QnPm in the above scheme) into an oligomer with PPII-like structure, or the formation of a complex with an inhibitor peptide such as 5QMe2, in effect “resets the clock”. Upon dissociation, the polyQ-polyP peptide is again a monomer with a PPII-like structure, and the slow process of nucleation begins anew.
In addition to the P11 segment adjacent to the polyQ domain of huntingtin, there are two additional Pro-rich segments of similar length. The codons for Gln differ by only one nucleotide from two of the codons for Pro. In view of the tendency of polyQ domains to expand and lead to protein aggregation diseases, it is possible that polyP domains arose adjacent to the polyQ domains as a protection against this tendency (65). An understanding of these effects could help lead to the future development of reagents that can affect the tendency of polyQ-containing proteins to self-associate. Indeed, recently developed inhibitors have been shown to act by forming a heterodimeric complex that stabilizes the PPII-like state of polyQ peptides (J. D. Lanning, unpublished results).
Supporting Material
Additional materials and methods, equations, references, and figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01312-5.
Supporting Material
Acknowledgments
We thank Elena Solomaha (University of Chicago Biophysics Core Facility), Giridher Reddy (Peptide/Protein Facility for ESI-Mass Spectrometry), and Shohei Koide (University of Chicago Biomolecular NMR Facility) for assistance in the NMR experiments, and Jen Lanning for helpful discussions.
This work was funded by a grant from the National Institutes of Health (NS042852 to S.C.M.) and a National Institutes of Health Molecular and Cell Biology Training Grant (T32GM07183 to G.D.).
References
- 1.Gusella J.F., MacDonald M.E. Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem. Sci. 2006;31:533–540. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Iuchi S., Hoffner G., Verbeke P., Djian P., Green H. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc. Natl. Acad. Sci. USA. 2003;100:2409–2414. doi: 10.1073/pnas.0437660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borrell-Pages M., Zala D., Humbert S., Saudou F. Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 2006;63:2642–2660. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gidalevitz T., Ben-Zvi A., Ho K.H., Brignull H.R., Morimoto R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez I., Mahlke C., Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–379. doi: 10.1038/nature01301. [DOI] [PubMed] [Google Scholar]
- 6.Nagai Y., Mahlke C., Yuan J. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 7.Glabe C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 8.Glabe C.G., Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66(Suppl. 1):S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- 9.Shewmaker F., Ross E.D., Tycko R., Wickner R.B. Amyloids of shuffled prion domains that form prions have a parallel in-register β-sheet structure. Biochemistry. 2008;47:4000–4007. doi: 10.1021/bi7024589. [DOI] [PubMed] [Google Scholar]
- 10.Shewmaker F., Wickner R.B., Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc. Natl. Acad. Sci. USA. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson R., Eisenberg D. Structural models of amyloid-like fibrils. Adv. Protein Chem. 2006;73:235–282. doi: 10.1016/S0065-3233(06)73008-X. [DOI] [PubMed] [Google Scholar]
- 12.Nelson R., Eisenberg D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006;16:260–265. doi: 10.1016/j.sbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Chan J.C., Oyler N.A., Yau W.M., Tycko R. Parallel β-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669–10680. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baxa U., Wickner R.B., Steven A.C., Anderson D.E., Marekov L.N. Characterization of β-sheet structure in Ure2p1–89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry. 2007;46:13149–13162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 15.Crick S.L., Jayaraman M., Frieden C., Wetzel R., Pappu R.V. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein F.A., Pastore A., Masino L., Zeder-Lutz G., Nierengarten H. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: towards a length-dependent toxicity gradient. J. Mol. Biol. 2007;371:235–244. doi: 10.1016/j.jmb.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 17.Perutz M. Polar zippers: their role in human disease. Protein Sci. 1994;3:1629–1637. doi: 10.1002/pro.5560031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng J., Ma B., Nussinov R. Consensus features in amyloid fibrils: sheet-sheet recognition via a (polar or nonpolar) zipper structure. Phys. Biol. 2006;3:P1–P4. doi: 10.1088/1478-3975/3/3/P01. [DOI] [PubMed] [Google Scholar]
- 19.Kim W., Hecht M.H. Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer's Aβ42 peptide. Proc. Natl. Acad. Sci. USA. 2006;103:15824–15829. doi: 10.1073/pnas.0605629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hills R.D., Brooks C.L. Hydrophobic cooperativity as a mechanism for amyloid nucleation. J. Mol. Biol. 2007;368:894–901. doi: 10.1016/j.jmb.2007.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balbach J.J., Ishii Y., Antzutkin O.N., Leapman R.D., Rizzo N.W. Amyloid fibril formation by Aβ 16–22, a seven-residue fragment of the Alzheimer's β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. doi: 10.1021/bi0011330. [DOI] [PubMed] [Google Scholar]
- 22.Djousse L., Knowlton B., Hayden M., Almqvist E.W., Brinkman R. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am. J. Med. Genet. A. 2003;119A:279–282. doi: 10.1002/ajmg.a.20190. [DOI] [PubMed] [Google Scholar]
- 23.Brignull H.R., Morley J.F., Morimoto R.I. The stress of misfolded proteins: C. elegans models for neurodegenerative disease and aging. Adv. Exp. Med. Biol. 2007;594:167–189. doi: 10.1007/978-0-387-39975-1_15. [DOI] [PubMed] [Google Scholar]
- 24.Jung J., Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 25.Krobitsch S., Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White J.K., Auerbach W., Duyao M.P., Vonsattel J.P., Gusella J.F. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 27.Schiffer N.W., Broadley S.A., Hirschberger T., Tavan P., Kretzschmar H.A. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J. Biol. Chem. 2007;282:9195–9203. doi: 10.1074/jbc.M607865200. [DOI] [PubMed] [Google Scholar]
- 28.Yang S.H., Cheng P.H., Banta H., Piotrowska-Nitsche K., Yang J.J. Towards a transgenic model of Huntington's disease in a non-human primate. Nature. 2008;453:921–924. doi: 10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.La Spada A.R., Taylor J.P. Polyglutamines placed into context. Neuron. 2003;38:681–684. doi: 10.1016/s0896-6273(03)00328-3. [DOI] [PubMed] [Google Scholar]
- 30.Shizuka M., Watanabe M., Ikeda Y., Mizushima K., Okamoto K. Molecular analysis of a de novo mutation for spinocerebellar ataxia type 6 and (CAG)n repeat units in normal elder controls. J. Neurol. Sci. 1998;161:85–87. doi: 10.1016/s0022-510x(98)00270-6. [DOI] [PubMed] [Google Scholar]
- 31.Darnell G., Orgel J.P., Pahl R., Meredith S.C. Flanking polyproline sequences inhibit β-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 2007;374:688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Ignatova Z., Thakur A.K., Wetzel R., Gierasch L.M. In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. J. Biol. Chem. 2007;282:36736–36743. doi: 10.1074/jbc.M703682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nozaki K., Onodera O., Takano H., Tsuji S. Amino acid sequences flanking polyglutamine stretches influence their potential for aggregate formation. Neuroreport. 2001;12:3357–3364. doi: 10.1097/00001756-200110290-00042. [DOI] [PubMed] [Google Scholar]
- 34.Bhattacharyya A., Thakur A.K., Chellgren V.M., Thiagarajan G., Williams A.D. Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 2006;355:524–535. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 35.Chen S., Berthelier V., Yang W., Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 36.O'Nuallain B., Thakur A.K., Williams A.D., Bhattacharyya A.M., Chen S. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- 37.Chen S.M., Wetzel R. Solubilization and disaggregation of polyglutamine peptides. Protein Sci. 2001;10:887–891. doi: 10.1110/ps.42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chellgren B.W., Miller A.F., Creamer T.P. Evidence for polyproline II helical structure in short polyglutamine tracts. J. Mol. Biol. 2006;361:362–371. doi: 10.1016/j.jmb.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 39.Wuthrich K. Wiley; New York: 1986. NMR of Proteins and Nucleic Acids. 292. [Google Scholar]
- 40.Shi Z., Olson C.A., Rose G.D., Baldwin R.L., Kallenbach N.R. Polyproline II structure in a sequence of seven alanine residues. Proc. Natl. Acad. Sci. USA. 2002;99:9190–9195. doi: 10.1073/pnas.112193999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koide A., Bailey C.W., Huang X., Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 42.Jones J.A., Wilkins D.K., Smith L.J., Dobson C.M. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR. 1997;10:199–203. [Google Scholar]
- 43.Gibbs S.J., Johnson C.S. A PFG NMR experiment for accurate diffusion and flow studies in the presence of eddy currents. J. Magn. Reson. 1991;93:395–402. [Google Scholar]
- 44.Sklenar V., Torchia D., Bax A. Measurement of C-13 longitudinal relaxation using H-1 detection. J. Magn. Reson. 1987;73:375–379. [Google Scholar]
- 45.Copie V., Tomita Y., Akiyama S.K., Aota S., Yamada K.M. Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regions: comparison with the human fibronectin crystal structure. J. Mol. Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- 46.Dickinson C.D., Veerapandian B., Dai X.P., Hamlin R.C., Xuong N.H. Crystal-structure of the 10th type-III cell-adhesion module of human fibronectin. J. Mol. Biol. 1994;236:1079–1092. doi: 10.1016/0022-2836(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 47.Burke M.G., Woscholski R., Yaliraki S.N. Differential hydrophobicity drives self-assembly in Huntington's disease. Proc. Natl. Acad. Sci. USA. 2003;100:13928–13933. doi: 10.1073/pnas.1936025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Z.S., Chen K., Liu Z.G., Sosnick T.R., Kallenbach N.R. PII structure in the model peptides for unfolded proteins: studies on ubiquitin fragments and several alanine-rich peptides containing QQQ, SSS, FFF, and VVV. Proteins. 2006;63:312–321. doi: 10.1002/prot.20788. [DOI] [PubMed] [Google Scholar]
- 49.Rucker A.L., Creamer T.P. Polyproline II helical structure in protein unfolded states: lysine peptides revisited. Protein Sci. 2002;11:980–985. doi: 10.1110/ps.4550102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rucker A.L., Pager C.T., Campbell M.N., Qualls J.E., Creamer T.P. Host-guest scale of left-handed polyproline II helix formation. Proteins. 2003;53:68–75. doi: 10.1002/prot.10477. [DOI] [PubMed] [Google Scholar]
- 51.Chen S.M., Berthelier V., Hamilton J.B., O'Nuallain B., Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 52.Chen S.M., Ferrone F.A., Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuler B., Lipman E.A., Steinbach P.J., Kumke M., Eaton W.A. Polyproline and the “spectroscopic ruler” revisited with single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2005;102:2754–2759. doi: 10.1073/pnas.0408164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Best R.B., Merchant K.A., Gopich I.V., Schuler B., Bax A. Effect of flexibility and cis residues in single-molecule FRET studies of polyproline. Proc. Natl. Acad. Sci. USA. 2007;104:18964–18969. doi: 10.1073/pnas.0709567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merchant K.A., Best R.B., Louis J.M., Gopich I.V., Eaton W.A. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc. Natl. Acad. Sci. USA. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly M.A., Chellgren B.W., Rucker A.L., Troutman J.M., Fried M.G. Host-guest study of left-handed polyproline II helix formation. Biochemistry. 2001;40:14376–14383. doi: 10.1021/bi011043a. [DOI] [PubMed] [Google Scholar]
- 57.Stapley B.J., Creamer T.P. A survey of left-handed polyproline II helices. Protein Sci. 1999;8:587–595. doi: 10.1110/ps.8.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zangi R., Berne B.J. Aggregation and dispersion of small hydrophobic particles in aqueous electrolyte solutions. J. Phys. Chem. B. 2006;110:22736–22741. doi: 10.1021/jp064475+. [DOI] [PubMed] [Google Scholar]
- 59.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–647. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 60.Yoon Y.R., Lim Y.B., Lee E., Lee M. Self-assembly of a peptide rod-coil: a polyproline rod and a cell-penetrating peptide Tat coil. Chem. Commun. (Camb.) 2008;16:1892–1894. doi: 10.1039/b719868j. [DOI] [PubMed] [Google Scholar]
- 61.Pokarowski P., Droste K., Kolinski A. A minimal proteinlike lattice model: an α-helix motif. J. Chem. Phys. 2005;122:214915. doi: 10.1063/1.1924601. [DOI] [PubMed] [Google Scholar]
- 62.Jacobs D.J., Wood G.G. Understanding the α-helix to coil transition in polypeptides using network rigidity: predicting heat and cold denaturation in mixed solvent conditions. Biopolymers. 2004;75:1–31. doi: 10.1002/bip.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang A.S., Honig B. Free energy determinants of secondary structure formation: I. α-helices. J. Mol. Biol. 1995;252:351–365. doi: 10.1006/jmbi.1995.0502. [DOI] [PubMed] [Google Scholar]
- 64.Bhattacharyya A.M., Thakur A.K., Wetzel R. Polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction. Proc. Natl. Acad. Sci. USA. 2005;102:15400–15405. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duennwald M.L., Jagadish S., Muchowski P.J., Lindquist S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. USA. 2006;103:11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.