

Abstract
The phage shock protein (Psp) system found in enterobacteria is induced in response to impaired inner membrane integrity (where the Psp response is thought to help maintain the proton motive force of the cell) and is implicated in the virulence of pathogens such as Yersinia and Salmonella. We provided evidence that the two-component ArcAB system was involved in induction of the Psp response in Escherichia coli and now report that role of ArcAB is conditional. ArcAB, predominantly through the action of ArcA regulated genes, but also via a direct ArcB–Psp interaction, is required to propagate the protein IV (pIV)-dependent psp-inducing signal(s) during microaerobiosis, but not during aerobiosis or anaerobiosis. We show that ArcB directly interacts with the PspB, possibly by means of the PspB leucine zipper motif, thereby allowing cross-communication between the two systems. In addition we demonstrate that the pIV-dependent induction of psp expression in anaerobiosis is independent of PspBC, establishing that PspA and PspF can function as a minimal Psp system responsive to inner membrane stress.
Introduction
Bacteria in their native habitats encounter a plethora of stresses, which challenge their cellular integrity. Robustness of the cytoplasmic membrane is required for it to function as a barrier between the inside of the cell and the environment. Bacteria have therefore evolved a number of systems to maintain membrane integrity, one of which is the phage shock protein (Psp) response, highly conserved in Gram-negative bacteria. In Escherichia coli, Psp genes are organized in a regulon (pspF pspABCDE and pspG) that is under the control of a sigma54-dependent promoter activated by PspF, positively controlled by PspBC and negatively regulated by PspA (reviewed by Model et al., 1997; Darwin, 2005; Wigneshweraraj et al., 2008). Psp expression is induced upon inner membrane (IM) stress which dissipates the proton motive force (pmf) (potentially resulting in a reduced energy status of the cell). Psp effector proteins (e.g. PspA – effector upon stress, PspD and PspG) subsequently help to conserve pmf (Kleerebezem et al., 1996; Jovanovic et al., 2006; Kobayashi et al., 2007). Understanding the mechanism of Psp induction and its biological function is important since the Psp system is significant in protein translocation and in the growth and virulence of pathogenic enterobacteria (reviewed by Darwin, 2005; 2007; Rowley et al., 2006). The PspA homologue Vipp1 has been shown to be essential in plants and Synechocystis for thylakoid biogenesis and hence photosynthesis (Westphal et al., 2001). Further, PspA homologues have been reported in Gram-positive bacteria and archea (Bidle et al., 2008; Vrancken et al., 2008). PspA therefore appears to have a fundamental role in the three domains of life.
The nature of the stress signal and how it is detected by the Psp system remains unknown (reviewed by Darwin, 2005). Previously we observed that ArcB (the sensor kinase of the two-component ArcAB system) is required for full protein IV (pIV)-dependent psp expression in E. coli and proposed that psp-inducing stresses that dissipate pmf may activate ArcB (Jovanovic et al., 2006). The ArcAB system regulates the transition from aerobic to anaerobic respiration and fermentation (reviewed by Malpica et al., 2006) where ArcB has been shown to sense the redox state of the cell through changes in the ubiquinone : ubiquinol (UQ : UQH2) ratio (see Fig. 2A and B) (Georgellis et al., 2001; Malpica et al., 2004). Increased levels of UQH2 attenuate inhibition on ArcB kinase activity imposed by UQ, thus enabling ArcB to phosphorylate ArcA (the response regulator). Phosphorylated ArcA (ArcA-P) then represses genes involved in aerobic respiration and upregulates genes involved in anaerobic respiration and fermentation. Notably, the ArcAB system is also known to be of particular importance under microaerobic growth conditions, where oxygen is limiting rather than being completely absent (Alexeeva et al., 2003).
Fig. 2.
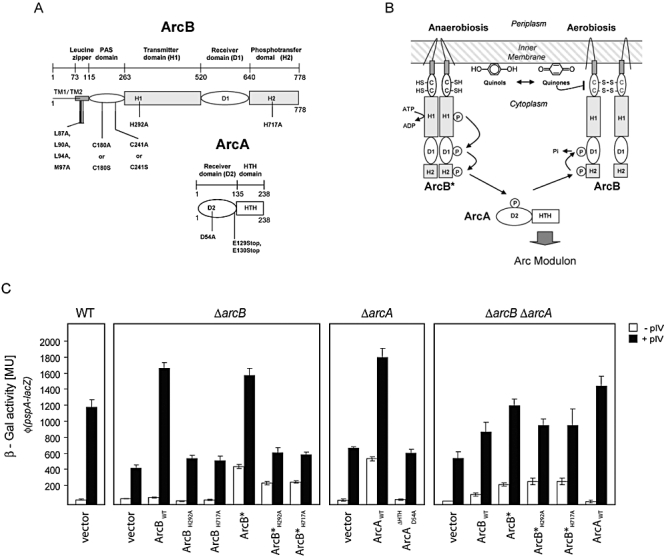
Activation of ArcAB is required for robustness of pIV-dependent induction of psp expression. A. Schematic representation of ArcB and ArcA (adapted from Malpica et al., 2004) with the locations of the amino acid substitutions used in this study. ArcB: activation mutants – C180A/S and C241A/S substitutions in PAS domain; phosphorelay mutants – H292A or H717A; leucine zipper mutant (LeuZm) – L87A, L90A, L94A and M97A. ArcA: a phosphorylation-defective mutant – D54A, and DNA-binding-deficient mutant (introduction of two stop codons, E129Stop and E130Stop) –ΔHTH. B. Regulation of the ArcAB system. ArcB is an IM-localized dimer. Its kinase activity is diminished by UQ and elevated in the presence of UQH2 leading to reduction of the ArcB Cys180 and Cys241 thiol groups (in this article functionally equivalent to ArcB* form) in PAS (Per-Arnt-Sim) domain and an increased interaction between the two ArcB TM domains. (PAS domains are signalling domains and they function as input modules in proteins that sense oxygen, redox potential, light and several other stimuli.) Reduction of ArcB is followed by autophosphorylation of H292 (H1) and phosphorelay via D576 (D1) and H717 (H2) to residue D54 (D2) within the response regulator ArcA. C. Bipartite role of ArcB activation in pIV-dependent induction of psp expression. Induction of psp expression in ΔarcB (MVA63), ΔarcA (MVA79) and a double mutant ΔarcAB (MVA94) strains expressing ArcBWT, ArcAWT and their mutants was measured using β-Gal assays under microaerobic conditions in the absence or presence of pIV. WT – wild-type strain (MVA44); vector – pCA24N.
Recently, ArcB-independent psp induction was reported (Seo et al., 2007). We therefore explored further the relationship(s) between the Psp and Arc system under different growth conditions and now report that involvement of Arc is conditional, consistent with both observations (Jovanovic et al., 2006; Seo et al., 2007). We find that ArcAB is required to propagate pIV-dependent induction of the Psp response under microaerobic, but not under aerobic or anaerobic growth conditions. The Arc system appears to be important for Psp-signal amplification, via the ArcB–ArcA phosphorelay. In addition, activated ArcB is by itself observed to elevate psp expression. Further, we have identified a direct binding interaction between ArcB and PspB that supports part of a Psp-specific signal transduction pathway used under microaerobic growth conditions. Notably in anaerobiosis, pIV-dependent psp expression is largely independent of PspBC (two positive regulators of the Psp response) and ArcAB, suggesting either that the repressive PspA–PspF complex is itself capable of recognizing inducing signals, or that PspBC and ArcAB are substituted by other gene products.
Results
PIV-dependent induction of psp expression under different growth conditions
We measured pIV-dependent psp induction in strains with different complements of arc and psp genes grown under aerobic, microaerobic and anaerobic conditions (see Experimental procedures) where the ArcAB proteins are known to have differing importance (Fig. 1). We found that psp induction is dependent on ArcAB only in microaerobiosis and on PspBC (positive regulators) in microaerobiosis and aerobiosis (Fig. 1). In aerobiosis, a requirement for the ArcAB system is not apparent (Fig. 1). Under anaerobic growth, full pIV-dependent psp induction is largely independent of PspBC and the Arc system, when compared with microaerobiosis (Fig. 1). These differing dependencies on Arc are consistent with the ArcAB system functioning as a microaerobic redox regulator, required under microaerobic but not aerobic or anaerobic conditions (Alexeeva et al., 2003). Notably, under microaerobic growth conditions, the requirement for ArcB is more pronounced then for ArcA. Importantly, control reactions demonstrate that the different growth conditions per se do not affect the activity from the psp promoter since psp expression in ΔpspA cells is similar (Fig. S1A). Further since the level of psp expression in control strains ΔpspAΔarcBφ(pspA–lacZ) and ΔpspAΔarcAφ(pspA–lacZ) are unaffected by ArcB and ArcA, we infer that the Arc proteins may contribute to relieving PspA-negative regulation in microaerobiosis (Fig. S1A). In psp induction assays we used the outer membrane (OM) secretin, pIV, to induce pspA–lacZ expression. It is believed that pIV mislocalizes to the IM thereby impairing cell membrane integrity and resulting in a psp-inducing stress signal similar to that caused by PulD (Guilvout et al., 2006). To ascertain that the strains assayed were exposed to equivalent (pIV-induced) levels of ‘stress’, we tested pIV expression and determined the amount of pIV localized within the IM. As shown in Fig. 1, pIV expression did not change greatly in the different backgrounds tested or when the cells were grown under differing aeration. Additionally, the distribution of pIV between the IM and OM was similar in microaerobiosis in the wild-type (WT), arcA, arcB and pspBC deletion backgrounds (Fig. S1B).
Fig. 1.
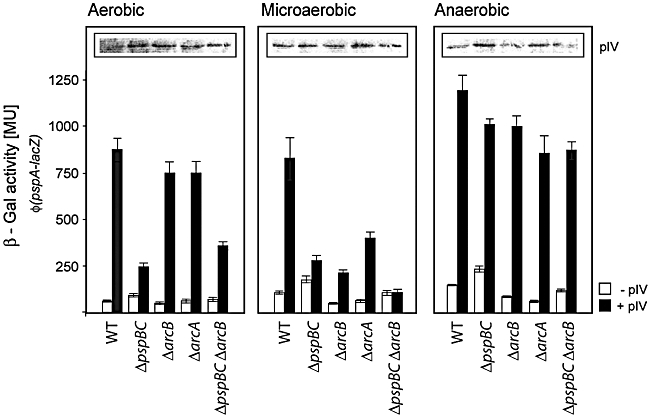
pIV-dependent induction of psp expression under different growth conditions. pIV secretin (from pGJ4) induced psp expression in E. coli[MG1655 φ(pspA–lacZ); MVA44] cells grown under aerobic, microaerobic or anaerobic conditions as measured using a β-Gal assay (see Experimental procedures). WT – wild type, MVA44. Insets: Western blot analyses of pIV expression in corresponding cells using antibodies against pIV (α-pIV) (see Experimental procedures). The relative expression levels (in arbitrary units) of pIV were determined as follows: aerobic – 1.57, 2.00, 1.93, 1.84, 2.03; microaerobic – 2.13, 2.27, 2.17, 2.32, 1.77; anaerobic – 1.71, 2.35, 1.82, 2.25, 1.77, and illustrate that the overall variability in pIV expression is ∼1.5-fold.
Since Psp effector proteins (PspA, PspD and PspG) alter expression levels of genes associated with aerobic growth (Jovanovic et al., 2006) and ArcAB regulates the switch from aerobic to anaerobic respiration and fermentation, we tested whether (i) pIV changes oxygen consumption in microaerobiosis (Fig. S1C) and (ii) other global regulators associated with aerobicity (e.g. FNR or Nar; see Fig. S1D) also affect psp induction. The results show that pIV changes oxygen consumption in an ArcB-dependent manner and that among the global regulators, the Arc system has the greatest effect on psp expression, supporting the idea of specific cross-talk between the Psp and ArcAB systems. Taken together, these results indicate a growth condition-specific requirement for ArcAB and PspBC proteins in psp induction. In addition, since under anaerobiosis induction of psp is independent of the PspBC-positive regulators we suggest that the PspA–F regulatory complex may itself be capable of perceiving a pIV-dependent inducing signal. Importantly, all subsequent experiments reported here were conducted under the microaerobic conditions where the roles of PspBC and ArcAB were most prominent.
Activation of ArcA facilitates pIV-dependent induction of psp expression
To further assess the roles of ArcAB in pIV-dependent induction of psp expression in microaerobiosis, we constructed mutants of ArcB (see Table 1) and ArcA that: (i) had a constitutively active kinase: ArcBC180A/C241A (termed ArcB*), (ii) disrupted the phosphorelay from ArcB to ArcA: ArcBH292A, ArcBH717A, ArcB*H292A and ArcB*H717A, and (iii) inactivated ArcA (by disrupting phosphorylation and preventing binding to target promoters): 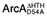 (Fig. 2A). Initially we determined that WT and mutant forms of ArcB proteins were similarly expressed (Fig. S2Ai and data not shown) and appropriately localized within the IM (Fig. S2Aii and data not shown). As a further measure of functionality, we showed that the WT and certain mutant ArcB and ArcA proteins (only those with intact phosphorelay systems) were able to activate ArcA-P-dependent pfl transcription (Fig. S2B). We then assayed the ability of Arc mutants to complement ΔarcB or ΔarcA mutations and support pIV-dependent induction of psp expression. As shown in Fig. 2C, ArcBWT and ArcB* or ArcAWT fully complemented ΔarcB or ΔarcA mutations, respectively, for pIV-dependent induction of psp expression. None of the phosphorelay mutants in ArcB or the inactive ArcA variant fully supported pIV-dependent induction of psp expression (Fig. 2C), indicating that ArcA-P has a role in promoting pIV-dependent induction. Importantly, control reactions demonstrated that similar levels of pIV secretin were present in all the samples tested (Fig. S2C and data not shown). Overexpression of ArcB proteins in a ΔarcAB strain showed that in the absence of ArcAWT, the ArcBWT and ArcB* similarly to ArcB* phosphorelay mutants cannot fully support pIV-dependent induction of psp expression (compared with results obtained with a ΔarcB) and that ArcB* and its phosphorelay mutants promote an ArcA- and pIV-independent low-level induction of psp (Fig. 2C). These data indicate that ArcB has an activity with respect to Psp that is partially ArcA-independent. In contrast, overexpression of ArcAWT in the ΔarcAB strain led to strong pIV-dependent induction of psp (Fig. 2C), indicating that there are two levels of Arc involvement in psp expression, one mediated by ArcB independent of ArcA, and one directed by ArcA.
(Fig. 2A). Initially we determined that WT and mutant forms of ArcB proteins were similarly expressed (Fig. S2Ai and data not shown) and appropriately localized within the IM (Fig. S2Aii and data not shown). As a further measure of functionality, we showed that the WT and certain mutant ArcB and ArcA proteins (only those with intact phosphorelay systems) were able to activate ArcA-P-dependent pfl transcription (Fig. S2B). We then assayed the ability of Arc mutants to complement ΔarcB or ΔarcA mutations and support pIV-dependent induction of psp expression. As shown in Fig. 2C, ArcBWT and ArcB* or ArcAWT fully complemented ΔarcB or ΔarcA mutations, respectively, for pIV-dependent induction of psp expression. None of the phosphorelay mutants in ArcB or the inactive ArcA variant fully supported pIV-dependent induction of psp expression (Fig. 2C), indicating that ArcA-P has a role in promoting pIV-dependent induction. Importantly, control reactions demonstrated that similar levels of pIV secretin were present in all the samples tested (Fig. S2C and data not shown). Overexpression of ArcB proteins in a ΔarcAB strain showed that in the absence of ArcAWT, the ArcBWT and ArcB* similarly to ArcB* phosphorelay mutants cannot fully support pIV-dependent induction of psp expression (compared with results obtained with a ΔarcB) and that ArcB* and its phosphorelay mutants promote an ArcA- and pIV-independent low-level induction of psp (Fig. 2C). These data indicate that ArcB has an activity with respect to Psp that is partially ArcA-independent. In contrast, overexpression of ArcAWT in the ΔarcAB strain led to strong pIV-dependent induction of psp (Fig. 2C), indicating that there are two levels of Arc involvement in psp expression, one mediated by ArcB independent of ArcA, and one directed by ArcA.
Table 1.
Biochemical phenotypes of arcB alleles.
| Protein | Motif(s) mutation | ArcB activationa | ArcB constitutively activeb | ArcA activationc |
|---|---|---|---|---|
| ArcBWT | Wild type | + | − | + |
| ArcBH292A | H292A | + | − | − |
| ArcBH717A | H717A | + | − | − |
| ArcB* | C180A C241A | − | + | + |
| ArcB*H292A | C180A C241A H292A | − | + | − |
| ArcB*H717A | C180A C241A H717A | − | + | − |
| ArcBLeuZm | L87A L90A L94A M97A | + | + | + |
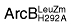 |
L87A L90A L94A M97A H292A | + | + | − |
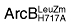 |
L87A L90A L94A M97A H717A | + | + | − |
Kinase ‘on’, wild-type C180 and C241 (apo form).
Kinase constitutively ‘on’.
Phosphorelay ‘on’.
An active form of ArcB promotes low-level induction of psp expression independent of pIV
In the absence of pIV (white bars in Fig. 2C) we observed that overexpression of either ArcBWT or the ArcB phosphorelay mutants (ArcBH292A or ArcBH717A) did not significantly induce psp expression. However, overexpression of either activated ArcB (ArcB*) or the ArcB* phosphorelay mutants (e.g. ArcB*H717A) were able to induce a low level of psp expression (compared with ArcBWT). When ArcB* is overexpressed in the ΔarcAB strain, a similar low level of pIV-independent psp expression was observed (Fig. 2C). We reasoned that the low-level induction of psp expression caused by the ArcB* phosphorelay mutants appears to be due to an effect of activated ArcB*, which is completely independent of ArcA or ArcA-P action.
Overexpression of ArcAWT in a ΔarcA strain in the absence of pIV results in induction of psp expression (Fig. 2C). Notably, this elevation of psp expression by overexpression of ArcAWT is ArcB-dependent (compare ΔarcA and ΔarcAB strains) (Fig. 2C). However in the presence of pIV, overexpression of ArcAWT in the ΔarcAB strain results in strong psp expression even in the absence of ArcB (although we note that this activity could be due to ArcA-P formation arising from cross-talk or acetyl phosphate). Together with the results implying that low-level induction of psp expression can occur in an ArcA-independent manner in the presence of the ArcB* phosphorelay mutants, these data again strongly suggest that activation of the Arc system results in two distinct outcomes (one via ArcA-P, the other via an active form of ArcB) that generate the Arc-dependences in the signalling pathway for pIV-dependent induction of psp expression.
Activation of ArcAB results in decreased electron potential (Δψ)
To address the nature of the signal that is responsible for low-level induction of psp expression by ArcB* and to potentially discriminate between roles of ArcB* and ArcA-P, we measured the electron potential (Δψ) of strains expressing either WT or mutant ArcB or ArcA proteins in the presence and absence of pIV (Fig. 3).
Fig. 3.
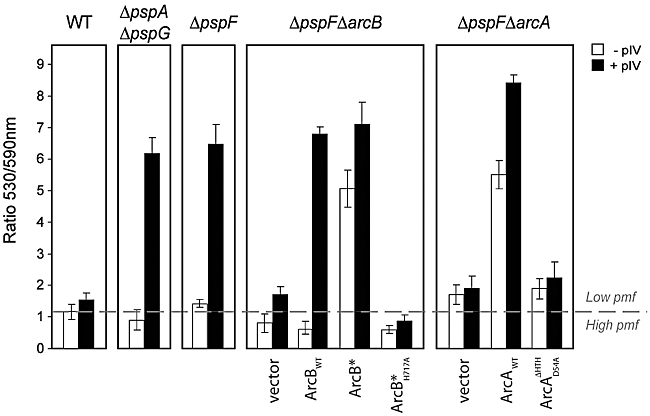
Activation of ArcAB reduces the electron potential (Δψ). Cells were grown microaerobically, treated with JC-1 dye, and the change in Δψ calculated as the 530/590 nm (green/red) ratio. ArcBWT, ArcAWT and key variants were expressed in the absence or presence of pIV. An increase in the 530/590 ratio indicates a decrease in Δψ compared with WT (E. coli MG1655) with vector plasmid pCA24N.
In the absence of a functioning Psp system (i.e. no effectors present, ΔpspAG; or no Psp proteins present, ΔpspF), a clear decrease in Δψ under pIV stress is observed (Fig. 3) that appears to be ArcB-dependent (Jovanovic et al., 2006). As shown in Fig. 3, overexpression of ArcBWT, ArcB* or ArcAWT in ΔpspFΔarcB or ΔpspFΔarcA strains resulted in a clear drop in Δψ under pIV stress conditions. Interestingly, when we analysed the phosphorelay ArcB and ArcA mutants we no longer observed a drop in Δψ, suggesting that phosphorylation of ArcA could therefore be a prerequisite for the pIV-dependent drop in pmf.
Notably, in the absence of pIV, overexpression of ArcB* or ArcAWT (which also results in psp expression; Fig. 2C) was also sufficient to significantly reduce Δψ (Fig. 3). In contrast, overexpression of ArcBWT, ArcB* phosphorelay mutants (e.g. ArcB*H717A) or inactive ArcA (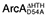 ) did not reduce Δψ in the absence of stress (Fig. 3).
) did not reduce Δψ in the absence of stress (Fig. 3).
ArcB and PspB interact in vivo
To understand how ArcB*-dependent signalling might be integrated into the PspBC-specific signal transduction pathway, we used an adenylate cyclase (Cya)-based bacterial two-hybrid system (BACTH) to study potential Arc–Psp protein–protein interactions in vivo (Karimova et al., 1998; 2005; and see Experimental procedures). We constructed ArcBWT, ArcB*, PspA, PspB, PspC and PspF fusion proteins with Cya T18, T18C and T25 fragments (see Experimental procedures).
As a control we first analysed the ability of ArcB and ArcB* to self-associate (since ArcB forms dimers; reviewed by Malpica et al., 2006) and show that the ArcBWT and ArcB* fusion proteins are capable of self-associating (Table 2). Next, we tested whether ArcBWT or ArcB* could interact with several key members of the Psp regulon (PspA, B, C, F). Additionally we confirmed that known interactions between PspA–PspF, PspA–PspC and PspB–PspC (Table 2 and data not shown) were detected with our fusion proteins (reviewed by Darwin, 2005). We demonstrated that a clear interaction between either ArcBWT or ArcB* (T25-fused protein) occurs with the C-terminal PspB fusion protein (PspB–T18) (Table 2). A Triton X-100 fractionation and Western blotting method was used to demonstrate that the interaction between ArcB, ArcB* and PspB occurs in the IM (using antibodies against Cya T25, Fig. S3Bi or PspB, Fig. S3Bii). As a control, an N-terminal T18C (T18C–PspB) PspB fusion (where T18C is now embedded in the IM) did not detectably interact with ArcB (data not shown). Using an in vivo cross-linking assay, we demonstrate that a higher-molecular-weight cross-linked ‘Psp complex’ (labelled cross-linked species in Fig. S3C) can be detected with PspB antibodies in WT and ΔarcA strains but not in a ΔarcB deletion strain (Fig. S3C), providing further evidence for an interaction between ArcB and PspB.
Table 2.
Interactions of Psp and ArcB proteins in vivo.
| T25 fusions |
||||
|---|---|---|---|---|
| T18 fusions | T25–ArcBWT | T25–ArcB* | T25–ArcBLeuZm | T25–PspC |
| T18C–ArcBWT | +/− | ND | ND | +/− |
| T18C–ArcB* | ND | + | ND | +/− |
| PspB–T18 | ++ | ++ | + | ++ |
| PspBLeuZm–T18 | − | − | − | ++ |
| T18C–PspA | +/− | +/− | +/− | + |
| T18C–PspC | +/− | +/− | +/− | − |
| T18C–ArcBLeuZm | ND | ND | +/− | +/− |
| T18C–PspF | − | − | − | − |
The BACTH system was used to detect the protein–protein interactions between the Psp and ArcB proteins. Negative control: BTH101/pKT25+pUT18C vectors alone (74 ± 4 MU); positive control: BTH101/pKT25-zip+pUT18C-zip (1017 ± 34 MU); interaction estimates including SD were as follows: (+/−) weak interaction (235–350 MU); (+) interaction (350–500 MU); (++) strong interaction (>700 MU); (−) no interaction (≤ 234 MU); ND, not determined. For construction of fusion proteins, growth conditions and mean values with SD, see Experimental procedures.
We also found evidence for a weak, but detectable, interaction between ArcBWT or ArcB* and N-terminal T18 fusions to either PspA or PspC (Table 2), but no interaction between ArcB, ArcB* and PspF was detected (Table 2). Since interactions between PspF, PspA, PspB, PspC have previously been established (Adams et al., 2003; Elderkin et al., 2005; Maxson and Darwin, 2006), we conclude that (potentially regardless of its state) ArcB could interact with members of a Psp[FABC] complex via PspB.
Since both ArcB and PspB contain putative leucine zipper motifs that are most likely positioned at the IM/cytoplasm interface (see Fig. 2A and Fig. S3A), we reasoned that the interaction observed between ArcB and PspB may be occurring directly through these sequences. To determine whether this was indeed the case, we mutated the leucine zipper motifs (LeuZm) of both ArcB (ArcBLeuZm; L87A, L90A, L94A, M97A) and PspB (PspBLeuZm; L10A L15A L18A) and fused these mutants to T25– and –T18 respectively (see Experimental procedures and Table S1). Importantly, the PspBLeuZm–T18 fusion protein remained localized within the IM (Fig. S3Bii); however, PspBLeuZm is no longer able to interact with either ArcBWT or ArcB* (Table 2). Interestingly, PspBLeuZm retained the ability to interact with PspC (Table 2), indicating that the interaction with PspC is not dependent on the leucine zipper sequences. These results support a physical interaction between ArcB and PspB mediated by the PspB leucine zipper motif.
Like PspBLeuZm–T18, the T25–ArcBLeuZm fusion protein remained localized within the IM (Fig. S4Aii) and importantly retained the ability to activate pfl transcription (Fig. S4B), similar to T25–ArcB*. However, T25–ArcBLeuZm showed reduced pIV-independent and pIV-dependent induction of psp expression compared with T25–ArcB* (Fig. S4B). Interestingly, the ArcBLeuZm mutant was still able to interact with PspB (albeit not as strongly, Table 2), implying that the site of PspB interaction on ArcB does not solely require the integrity of the putative ArcB leucine zipper motif. Like ArcBWT, ArcBLeuZm self-associates (Table 2), suggesting that the ArcB leucine zipper motif does not play a major role in self-association. Finally, ArcBLeuZm retained a very weak interaction with PspA and PspC (Table 2), at a similar level to those observed with ArcBWT or ArcB*, suggesting no role for the ArcB leucine zipper in these interactions.
ArcBLeuZm is in an active kinase form
Since the primary role of the putative LeuZm in ArcB is not in binding to PspB, we decided to investigate the role of this sequence. Initially, we tested the activity of ArcBLeuZm in a ΔarcB strain and found that, in contrast to ArcBWT, ArcBLeuZm is able to significantly upregulate the pfl promoter (Fig. 4A) as does ArcB* (see also Fig. S2B). Importantly, upregulation of the pfl promoter by ArcBLeuZm is dependent on the phosphorelay residues H292 and H717; the 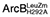 and
and 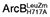 (see Experimental procedures, Table S1) do not show elevated pfl–lacZ activities (Fig. 4A), but are expressed at similar levels (Fig. S4Ai and data not shown). Overexpression of ArcBLeuZm in a ΔarcB strain resulted in elevated psp expression in the absence of pIV and full induction in the presence of pIV (Fig. 4A), similar to ArcB* (Fig. 2C). Consistent with the pfl–lacZ findings, phosphorelay in the ArcBLeuZm (Fig. 4A, compare ArcBLeuZm with
(see Experimental procedures, Table S1) do not show elevated pfl–lacZ activities (Fig. 4A), but are expressed at similar levels (Fig. S4Ai and data not shown). Overexpression of ArcBLeuZm in a ΔarcB strain resulted in elevated psp expression in the absence of pIV and full induction in the presence of pIV (Fig. 4A), similar to ArcB* (Fig. 2C). Consistent with the pfl–lacZ findings, phosphorelay in the ArcBLeuZm (Fig. 4A, compare ArcBLeuZm with 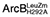 and
and 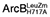 ) appears critically important for pIV-dependent induction of psp expression, but less so for pIV-independent elevation of psp expression. Overexpression of ArcBLeuZm in a ΔpspFΔarcB strain (Fig. 4B) complemented the arcB mutation and reduced Δψ in the presence and absence of pIV (similar to ArcB*). However as shown in Fig. 4B, overexpression of
) appears critically important for pIV-dependent induction of psp expression, but less so for pIV-independent elevation of psp expression. Overexpression of ArcBLeuZm in a ΔpspFΔarcB strain (Fig. 4B) complemented the arcB mutation and reduced Δψ in the presence and absence of pIV (similar to ArcB*). However as shown in Fig. 4B, overexpression of 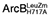 neither complemented the arcB mutation in the presence of pIV, nor reduced Δψ in the absence of pIV, consistent with the inability of
neither complemented the arcB mutation in the presence of pIV, nor reduced Δψ in the absence of pIV, consistent with the inability of 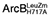 to upregulate pfl transcription or stimulates psp expression (Fig. 4A).
to upregulate pfl transcription or stimulates psp expression (Fig. 4A).
Fig. 4.
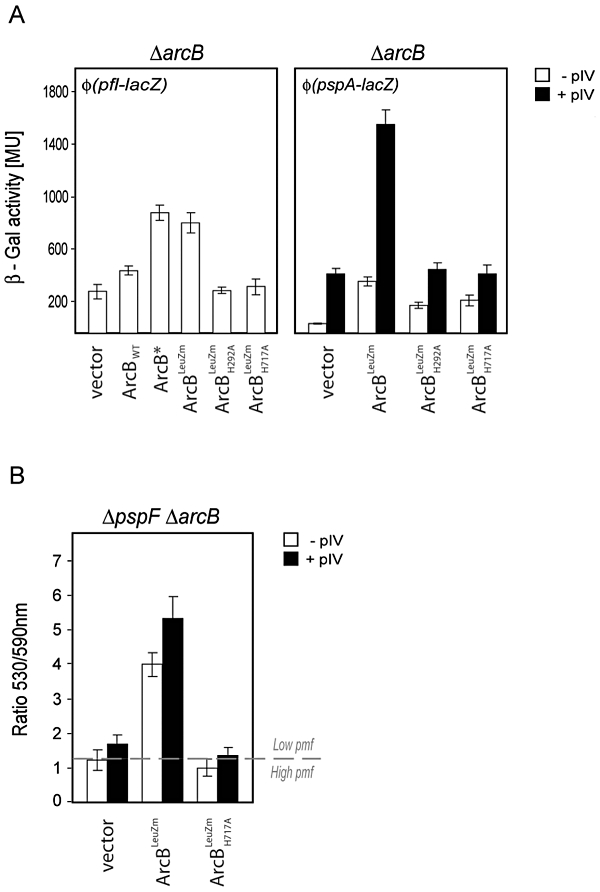
Activities of the ArcBLeuZm protein. A. ArcB LeuZm has an ‘on’ kinase activity and supports induction of psp expression. Activities of overexpressed ArcBLeuZm and its corresponding phosphorelay mutants were assessed in a microaerobically grown ΔarcB strain (MVA59) also carrying the pfl–lacZ (pGJ46) transcriptional fusion construct (left panel). Activities of ArcBLeuZm and its variants were compared with ArcBWT and ArcB*. pIV-independent and -dependent induction of psp expression after complementation of the ΔarcB mutation (MVA63) with ArcBLeuZm and its variants is presented in the right panel. pfl or psp expression was measured using β-Gal assays. Vector control was pCA24N. B. ArcBLeuZm reduces cells' Δψ. E. coliΔpspFΔarcB cells (MVA61) expressing ArcBLeuZm and its variants in the presence and absence of pIV were treated with JC-1 dye and changes in Δψ calculated. An increase in the 530/590 ratio indicates a decrease in Δψ compared with ΔpspFΔarcB strain with the vector plasmid pCA24N.
It seems that ArcBLeuZm resembles ArcB* (an active ArcB form) for the upregulation of pfl transcription (compare Fig. S2B and Fig. 4A) and induction of psp expression (compare Figs 2C and 4A), as well as its effect on Δψ (compare Figs 3 and 4B). Introduction of the H292A or H717A mutations in ArcBLeuZm led to the same reduction in activity seen for ArcB*, demonstrating the importance of an intact phosphorelay system for ArcBLeuZm activity.
PspBC propagates the signal onwards from the Arc system
Since a specific binding interaction between ArcB and PspB is evident, we next explored the relationship between ArcB and PspB in the context of a PspBC-specific signal transduction pathway resulting in psp induction.
In the absence of PspB (ΔpspB) neither pIV nor overexpression of ArcB* or ArcAWT significantly induced psp expression (Fig. 5). Overexpression of ArcB* in the presence of pIV in ΔpspB cells failed to induce psp expression (Fig. 5). PspBWT but not PspBLeuZm complemented ΔpspB for pIV-dependent induction of psp expression (Fig. 5), consistent with the inability of PspBLeuZm to interact with ArcB (Table 2). Importantly, both PspBWT and PspBLeuZm are stably expressed and localized within the IM (Fig. S5). Further, in the absence of PspC (ΔpspC), psp induction by pIV and by ArcB* was also abolished (Fig. 5). Considering that pIV-dependent induction of psp expression involves ArcB and ArcA activities, we suggest that the Arc and Psp systems are integrated via the PspB leucine zipper motif and as such, Arc conditionally forms part of the PspBC-specific signal transduction pathway.
Fig. 5.
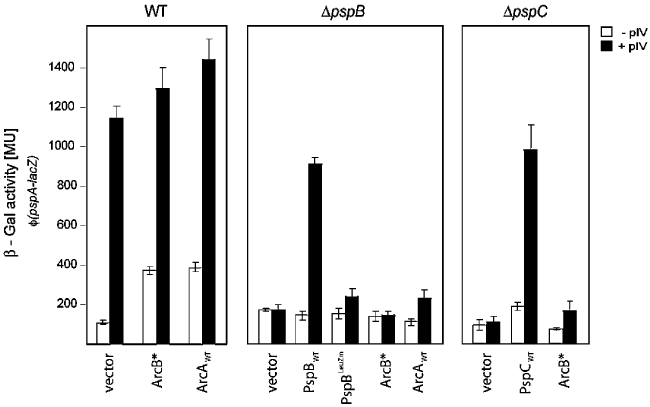
Roles of PspB and PspC in transducing the psp-inducing signal(s). psp expression was induced by either pIV, ArcB* or ArcAWT. Cells were grown under microaerobic conditions and expression of psp was measured using β-Gal assays. The ΔpspB mutation was complemented with either PspB (pAJM1) or PspBLeuZm (pGJ48), and ΔpspC mutation was complemented by PspC (pAJM2). WT – wild-type strain [MC1061 φ(pspA–lacZ); MVA4]; vector (WT panel) – pCA24N; vector (ΔpspB/C panels) – pBAD18-cam.
Discussion
Conditional requirement for Arc activation in pIV-dependent induction of psp expression
In this work, several lines of evidence demonstrate that the ArcAB system in microaerobiosis is required to facilitate pIV-dependent induction of psp expression and may potentially contribute to maintaining pmf (via the Psp response). ArcB activation is clearly essential for pIV-dependent induction of psp expression as is the integrity of the phosphorelay signalling cascade (which results in phosphorylation of ArcA) and ArcA itself. However, we doubt that the Arc system directly controls expression of the psp genes, since neither of the two E. coli psp promoters contains recognizable ArcA binding sites and the absence of Arc proteins does not affect transcription of psp genes in ΔpspA cells (i.e. in the absence of negative control).
In microaerobiosis, Arc-dependent induction of psp expression in the presence of pIV correlates with a decrease in Δψ (pmf), which is dependent both on the phosphorelay activity of ArcB and on ArcA itself. Under pIV stress conditions, the Psp response is not able to conserve pmf when the Arc proteins are absent, therefore we suggest that activation of the ArcAB system helps with generating and/or propagating a pIV-dependent psp-inducing signal threshold needed to mount the Psp response. Since the Psp response helps to maintain the cells pmf (and energy status) and is implicated in the growth and virulence of enterobacteria (reviewed by Darwin, 2005; Rowley et al., 2006; Darwin, 2007), the cross-talk between the Arc and Psp systems is of physiological relevance given that changes in oxygen availability often prevail during pathogenesis.
The requirement for ArcAB activation in pIV-dependent induction of psp expression significantly decreases in aerobiosis and anaerobiosis, consistent with the proposed major importance of the ArcAB regulators in microaerobic growth and in managing transitions between aerobic and anaerobic conditions (Alexeeva et al., 2003; Partridge et al., 2007). Conditional use of ArcAB can explain results in which involvement of Arc in psp induction was not evident under aerobic growth conditions (Seo et al., 2007).
We speculate that in anaerobiosis, the more reducing environment in the cytoplasm will decrease the psp-inducing signal threshold resulting in increasingly Arc-independent pIV-dependent psp expression, consistent with the cellular redox state (as defined by the NADH/NAD ratio) favouring a reducing environment in anaerobiosis versus an oxidizing environment in aerobiosis (de Graef et al., 1999). In addition, under anaerobic conditions, the contribution of UQ to the respiratory chain is significantly lower (and is replaced by menaquinone) (Gennis and Stewart, 1996) and so may contribute to a decreased requirement for Arc in pIV-dependent induction of psp expression. Hence, Arc-dependent induction of psp expression under pIV stress depends on specific growth conditions. Other examples of conditional effects of Arc on certain genes are known (Mika and Hengge, 2005).
ArcB communicates with PspB
There appear to be two separate actions of the Arc system in pIV-dependent induction of psp expression. One is related to activation of ArcA (and subsequent control of ArcA-P target genes) required for full psp induction under pIV stress. The other action relates to a more direct effect of activated ArcB that promotes low-level induction of psp expression, where ArcB*, in the absence of pIV, can induce low-level psp expression in a ΔarcA strain as can ArcB* phosphorelay mutants in ΔarcB cells. We demonstrated that ArcB interacts with PspB and that this interaction is dependent on the integrity of the PspB leucine zipper motif. Further, mutational analysis revealed that the PspB leucine zipper is important for pIV-dependent induction of psp expression, but not required for direct binding interactions between PspB and PspC, suggesting that PspB may be directly involved in receiving (or transducing) an inducing signal from activated ArcB.
The linker region of ArcB connects the trans-membrane (TM) domain with the catalytic domain of ArcB (see Fig. 2A and B) and contains a putative leucine zipper motif (conserved leucine residues at positions 73, 80, 87 and 94) and a PAS domain, the former of unknown function for ArcB activity. It has been suggested that Leu87 functions in intramolecular signal propagation in ArcB kinase activation (reviewed by Malpica et al., 2006). Here we show that the ArcB leucine zipper motif is functionally important in intramolecular activation of ArcB. Our results indicate that the ArcB leucine zipper mutant (ArcBLeuZm) is in a constitutive ‘kinase on’ state, capable of inducing the pfl promoter, but not if the phosphorelay residues H292 or H717 are compromised (e.g. 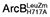 ). ArcBLeuZm also facilitates pIV-dependent and increases pIV-independent induction of psp expression. However, induction of psp expression by
). ArcBLeuZm also facilitates pIV-dependent and increases pIV-independent induction of psp expression. However, induction of psp expression by 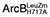 (for example) is significantly lower when compared with ArcB*H717A. In addition, the ArcBLeuZm mutant exhibits a decreased affinity for PspB, further suggesting that under specific psp-inducing conditions, the leucine zipper motifs of both ArcB and PspB are involved in ArcB–PspB communication.
(for example) is significantly lower when compared with ArcB*H717A. In addition, the ArcBLeuZm mutant exhibits a decreased affinity for PspB, further suggesting that under specific psp-inducing conditions, the leucine zipper motifs of both ArcB and PspB are involved in ArcB–PspB communication.
The PAS domain of ArcB may allow ArcB to respond to a number of factors related to changes in, e.g. oxygen supply and associated metabolic processes. To date only the interaction between ArcB and UQ has been reported (reviewed by Malpica et al., 2006). Since ArcB specifically interacts with PspB, in principle this would allow PspB to respond to signals received by ArcB, and vice versa. A similar relationship between signalling and control components has been described for other systems (e.g. for PhoP and PmrD) (Kato et al., 2007) where communication often occurs through membrane interactions (Szurmant et al., 2008).
Integration of the Arc system in the PspB/C-specific signalling pathway
In vivo it has been shown that PspB, PspC and PspA and PspA and PspF interactions occur under non-stress conditions (reviewed by Darwin, 2005). Here, we show that ArcB and PspB interact, potentially within one or more ArcB–Psp[B/C/A/F] regulatory complexes or subcomplexes. Stress-induced activation of ArcB may result in conformational changes in either PspB or PspC (or both) and presumably through altered protein–protein interactions releases the PspA-imposed negative regulation of PspF.
The precise nature of the signal(s) that induces psp expression remains to be determined. It is possible that under stress conditions, besides changes in the integrity of the IM and/or Δψ (pmf), the redox state of respiratory chain components specifically quinones, may also contribute to Arc-dependent induction of psp expression. One possibility is that alongside Arc-independent signalling, psp-inducing stimuli reduce UQ and, following an increased diffusion of H+ into the cytosol, the consequent dissipation of the pmf, may then activate ArcB. This view is supported by the observation that both UQ and ArcAB are required to detect a drop in Δψ in the presence of pIV, although UQ may be required for the structural integrity ArcB (Jovanovic et al., 2006; this study). Notably induction of psp expression and a significant downregulation of aerobic respiration and pmf occur simultaneously in cells undergoing contact-dependent growth inhibition (Aoki et al., 2009).
In microaerobiosis, activation of the Arc system greatly facilitates pIV-dependent induction of psp expression in a PspBC-dependent manner. However, since part of the induction process is Arc-independent but still PspBC-dependent (Fig. 1) it seems likely that one (or both) of the psp encoded sensors (PspB and/or PspC) can receive a signal. Therefore, a ‘double check point’ potentially exists to regulate pIV-dependent induction of psp expression. This arrangement of sensors would help the Psp system maintain specificity to stimuli, as described for unorthodox two-component systems which maintain their robustness to noises (Kim and Cho, 2006).
PspA–PspF is a minimal Psp system
This study clearly shows that in anaerobiosis, pIV-dependent induction of psp expression is PspBC-independent, consistent with the observation that some psp-inducing stimuli are PspBC-independent (e.g. extreme heat shock, CCCP) or only partially PspBC-dependent (ethanol treatment and hyperosmotic shock) [(reviewed by Model et al., 1997; Darwin, 2005); Jovanovic et al., 2006]. These results strongly suggest either (i) that PspA (and/or PspF) can directly receive the inducing signal and relieve the PspA-imposed negative regulation or (ii) that other factors substitute for PspBC and ArcAB. That a minimal PspA–PspF system is capable of responding to extracytoplasmic stimulus is consistent with a regulated PspA stress response in Gram-positive bacteria and archebacteria where PspBC are absent [(reviewed by Darwin, 2005); Bidle et al., 2008; Vrancken et al., 2008]. PspA homologues are found among Gram-positive bacteria, cyanobacteria, archea and higher plants [(reviewed by Darwin, 2005); Bidle et al., 2008; Vrancken et al., 2008]. In all these organisms, PspA homologues often respond to stress conditions similar to those described for inducing the Psp response (e.g. via extreme heat shock, ethanol treatment, hyperosmotic shock, CCCP) in a partially PspBC-dependent or -independent manner in enterobacteria.
Experimental procedures
Bacterial strains and growth conditions
The bacterial strains used in this study are listed in Table S1. Strains were constructed by transduction using the P1vir bacteriophage (Miller, 1992) (see Table S1) and were routinely grown in Luria–Bertani (LB) broth or on LB agar plates at 37°C (Miller, 1992). For BACTH assays, strains were grown in LB at 30°C. For in vivo cross-linking experiments, strains were grown in minimal medium [50 mM MOPS (pH 7), 2 mM MgSO4, 0.5% Glucose (w/v), 10 mM NH4Cl, 0.75 mM Na2SO4, 1.2 mM NH4NO3, 0.5 mM KH2PO4] at 30°C. For aerobic growth, overnight cultures of cells were diluted 100-fold into 5 ml of LB in a universal tube with loose fitting caps and shaken at 200 r.p.m. For microaerobic growth, overnight cultures of cells were diluted 100-fold into 5 ml of LB and shaken at 100 r.p.m. For an anaerobic growth, overnight cultures grown at 37°C in a universal tube with tightly closed caps without shaking were transferred (100-fold dilution) into a fully LB-filled suba-sealed universal tube (to avoid any air space) and incubated overnight at 37°C without shaking. The cells were then taken by syringe for β-galactosidase (β-Gal) assays. The pIV secretin was constitutively expressed from pGJ4. For induction of the pBAD ara promoter, 0.001% final arabinose (Ara) was added for 1 h. For induction of the lac promoter, 0.1 mM isopropyl-β-d-galactopyranoside (IPTG) was added for 1 h. For scoring the lacZ+ colonies, indicator plates containing 40 µl of 20 mg ml−1 stock solution of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) and 0.5 mM IPTG were used. Antibiotics were routinely used at the following concentrations: ampicillin (Amp; 100 µg ml−1), kanamycin (Kan; 25 µg ml−1), chloramphenicol (Cam; 30 µg ml−1) and tetracycline (Tet; 10 µg ml−1).
Construction of the ΔarcB59 mutant
To eliminate the Kan cassette from a ΔarcB::Kan mutant (strain MVA59) and construct the marker-less arcB59 variant (strain MVA92), we used plasmid pCP20 and the method described by Cherepanov and Wackernagel (1995). The resulting strain MVA92 had lost the FRT-flanked Kan resistance gene and the FLP helper plasmid (pCP20) was used to construct the strains MVA93 and MVA94 (see Table S1). The PCR and ParcB/elbB pair of primers were used to verify that arcB59 mutant in MVA92–94 strains had the correct structure (see Fig. S2D). PCR reactions were carried out either with isolated chromosomal DNA (Qiagen, DNeasy) or with a freshly grown colony suspended in 15 µl of water.
Measurement of oxygen consumption in bacterial cultures
Oxygen content in bacterial cultures was measured using a non-invasive OxySense 4000B system (Air Monitors). Bacterial cultures grown either in aerobiosis, microaerobiosis or anaerobiosis (see below) were prepared as described above, except that the tubes for dilution of overnight cultures contained an O2xyDot sensor (OxySense, Dallas, TX, USA) to detect O2 concentrations. The output fluorescence was measured by an OxySense Reader Pen (through the growth tube) and analysed using the OxySense 4000B system expressed either as a percentage or p.p.b. (1000 p.p.b. = 1 mg of O2 l−1). For oxygen consumption experiments, autocalibrated O2xyDot sensors were used and control measurements of ambient air (20.446 ± 1.005%) and the LB medium under either aerobic (8.845 ± 0.965%), microaerobic (4.901 ± 0.181%; air above the medium 17.147 ± 0.624%) or anaerobic conditions (0.075 ± 0.010%) were taken. An optimal signal-to-noise ratio was achieved by measuring in reduced ambient light conditions. Mean values of the oxygen consumption were calculated from 10 measurements with 100 signals captured per sample taken from technical duplicates of three independently grown cultures of each strain.
DNA manipulations
Plasmids used in this study are shown in Table S1. Using PCR-based site-specific mutagenesis (Quickchange mutagenesis kit, Stratagene) of the plasmid templates pJW5536(−) (ArcB), pJW4364(−) (ArcA) and pAJM1, we constructed plasmids pGJ21-23/27/29-31/33, pGJ45 and pGJ48 respectively (see Table S1). The positions of substituted residues in ArcB and ArcA and the mutant proteins activities are shown in Fig. 2A and Table 1. Plasmid pGJ46 that carries the φ(pfl–lacZ) transcription fusion was constructed as shown in Table S1. A DNA fragment carrying the pfl promoter region was chosen according to Drapal and Sawers (1995). All constructs were verified by DNA sequencing.
For the BACTH system experiments, the ArcB, ArcB* or ArcBLeuZm proteins were fused to either T25 (plasmid pKT25), T18C (pUT18C) or T18 (pUT18) Cya domains. The corresponding genes were amplified from plasmids pJW5536(−), pGJ23 and pGJ22 using primers that introduce either XbaI–KpnI, HindIII–KpnI or XbaI–EcoRI restriction sites, cloned in pGEM-T Easy (Promega), and then subcloned in frame into multiple cloning sites (MCS) of pKT25, pUT18 and pUT18C respectively. The genes for PspA, PspB, PspC and PspF were amplified by PCR using primers that introduce XbaI–KpnI restriction sites, cloned in pGEM-T Easy and then subcloned in frame into MCS of pKT25, pUT18 and pUT18C. The PspBLeuZm mutant was amplified from plasmid pGJ48 using primers that introduce HindIII–KpnI restriction sites, cloned in pGEM-T Easy and then subcloned in frame into MCS of pUT18. All constructs were verified by DNA sequencing. The production of fusion proteins, following induction by 0.5 mM IPTG at 30°C, was verified using Western blots (see below) with antibodies corresponding to either the fused proteins or Cya T25 (for pKT25 constructs). Transformation of bacteria was performed as described by Miller (1992).
In vivo BACTH
The Cya-based BACTH allows detection of protein–protein interactions in vivo and is particularly appropriate for studying interactions among membrane proteins (Karimova et al., 1998; 2005). Proteins of interest are fused to the T18 and T25 fragments (which do not function when physically separated) of the catalytic domain of Cya, and co-transformed into the host strain BTH101 (cya-). Upon interactions of fused proteins, cAMP synthesis is restored leading to transcriptional activation of CAP/cAMP-dependent genes, such as the lac operon on the chromosome of the host reporter strain.
The topologies of ArcB (Fig. 2A) (reviewed by Malpica et al., 2006), PspA, PspB and PspC (Fig. S3A) (reviewed by Darwin, 2005) were taken into account for construction of the fusion proteins and analysis of results (e.g. a Cya domain fused to the PspB region embedded in IM could interfere with localization of PspB). As a negative control we used pKT25 and pUT18C vectors in the absence of fusion proteins, as a positive control we used pKT25-zip and pUT18C-zip plasmids carrying fused GCN4 leucine-zipper sequence (Karimova et al., 1998). After co-transformation of the BTH101 strain with the two plasmids expressing the fusion proteins, selection plates (Kan, Amp, X-gal and IPTG) were incubated at room temperature for 72 h. The levels of the interactions were quantified by β-Gal activity in liquid cultures (see below). LacZ expression threefold above the negative control value was considered a positive interaction signal. For this measurement bacteria were grown in LB medium containing 100 µg ml−1 Amp and 50 µg ml−1 Kan at 30°C for 16 h, then cultures were diluted 1:25 and grown until the OD600 reached ∼0.3, then 0.5 mM IPTG was added and the cells incubated for a further 1 h at 30°C.
β-Gal assays
Activity from a single-copy chromosomal φ(pspA–lacZ) transcriptional fusion was assayed to gauge the level of psp expression. The φ(pspA–lacZ) transcriptional reporter fusion was introduced as a single copy into the chromosomal att site to retain the native pspA locus and permit the WT psp response. Activity from the plasmid-derived φ(pfl–lacZ) transcriptional fusion (pGJ46) was assayed to gauge activity of ArcA-P. Cells were grown overnight at 37°C in LB broth containing the appropriate antibiotic and then diluted 100-fold (initial OD600 ∼ 0.025) into the same medium (5 ml). Following incubation to OD600 ∼ 0.2–0.3, cultures were induced with Ara or IPTG for 1 h (where appropriate), and then assayed for β-Gal activity as described by Miller (1992). For anaerobically grown cells, β-Gal activity was measured following overnight growth at 37°C with no shaking. For all β-Gal assays, mean values of six independent assays taken from technical duplicates of three independently grown cultures of each strain were used to calculate activity. The data are shown as a mean values with SD error bars.
Bacterial cell fractionation
The bacterial cultures (5 ml) were separated into soluble and membrane fractions by a lysozyme-EDTA-osmotic shock protocol and IM and OM were selectively extracted with Triton X-100 (Russel and Kaźmierczak, 1993). All the samples were air-dried before adding 30 µl of 4% SDS and 30 µl of Laemlli buffer (Sigma). Samples were analysed by Western blotting (see below).
Western blot analysis
For the Western blot analysis, bacterial cells were collected (1 ml) at OD600 ∼ 0.5. Proteins were separated on either 10%, 12.5% or 15% (SDS)-PAGE and transferred onto PVDF membranes using a semi-dry transblot system (Bio-Rad). Western blotting was performed as described (Elderkin et al., 2002) using antibodies to PspF (1:4000 with anti-mouse; Ammersham), PspA (1:10 000 with anti-mouse) (Jones et al., 2003), PspB (a gift from P. Model) (1:5000 with anti-rabbit; Ammersham), PspC (a gift from P. Model) (1:5000 with anti-rabbit), pIV (a gift from M. Russel) (1:10 000 with anti-rabbit), ArcB (a gift from D. Georgellis) (1:10 000 with anti-rabbit) and Cya T25 (a gift from D. Ladant) (1:5000 with anti-rabbit). The proteins were detected using ECL plus Western Blotting Detection Kit according to manufacturer's guidelines (GE Healthcare). Images were captured in a FujiFilm – intelligent Dark Box by an image analyser with a charge-coupled device camera (LAS-3000). Densitometry analysis was performed with MultiGauge 3.0 software (FujiFilm USA, Valhalla, NY, USA) and quantification (results expressed in arbitrary units) performed using the AIDA software.
In vivo cross-linking
In vivo cross-linking was performed as described (Adams et al., 2003) using the thiol-reactive cross-linking reagent dithiobis (succinimydylpropionate) DSP (PIERCE). Cells were grown in minimal medium at 30°C to an OD600 ∼ 1.0, harvested and washed in 0.9% NaCl. Proteins were exposed to the cross-linker [100 µM DSP in 125 mM HEPES (pH 7.3)] for 30 min at 25°C. To quench the reaction, 50 µl of 1 M Tris-HCl (pH 8.0) was added to the reaction mix and the samples then transferred to ice for 5 min. Proteins were separated on a 7.5% SDS-PAGE (run at 200 V for 50 min) and the cross-linked species detected using antibodies specific to PspB (α-PspB) as described above.
Confocal fluorescence microscopy to assess membrane electron potential (Δψ)
The Δψ component of pmf was measured as described by Becker et al. (2005), with the following modifications. Cells from an overnight LB culture were subcultured into fresh LB to an initial OD600 ∼ 0.025 and grown to an OD600 of 0.8. Bacterial culture (1 ml) was centrifuged and then re-suspended in 1 ml of permeabilization buffer (10 mM Tris pH 7.5, 1 mM EDTA, 10 mM glucose). Then 2 µl of 5 mg ml−1 1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodine (JC-1) (Molecular Probes) was added and the preparation incubated for 30 min at room temperature. Cells were then centrifuged and re-suspended in 500 µl of permeabilization buffer. Microscope slides were prepared as follows: 80 µl of molten 0.8% agarose was pipetted onto a clean microscope slide, covered with a coverglass pre-treated with sigmacote (Sigma) and left to dry at room temperature for 30 min, prior to removing the coverglass. To obtain a thin layer of agarose, the microscope slide was left to dry overnight at 37°C. Fluorescent bacteria were examined using a Leica TCS-NT confocal microscope (Leica Microsystems, Germany) equipped with a krypton/argon laser with an excitation wavelength of 485 nm. Leica confocal software (Leica Microsystems, Germany) was used to calculate the green/red fluorescence emission ratio from 100 individual cells taken from technical duplicates of three independently grown cultures of each strain. The threshold to distinguish low pmf from high pmf was chosen based on the Δψ (pmf under given experimental conditions) of the WT strain under normal (non-stress) growth conditions.
Acknowledgments
This work was supported by funding from the Wellcome Trust. We acknowledge P. Model, M. Russel, D. Ladant, D. Georgellis, J. Beckwith, J. Brown, H. Mori and A. Mayhew for providing strains, plasmids and antibodies, and S. Amartey for help with anaerobic work. We especially would like to thank Dr P. Burrows, Dr N. Joly and Dr J. Schumacher for their constructive help with the manuscript. We also thank other laboratory members for helpful discussions, comments on the manuscript and support.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adams H, Teertstra W, Demmers J, Boesten R, Tommassen J. Interactions between phage-shock proteins in Escherichia coli. J Bacteriol. 2003;185:1174–1180. doi: 10.1128/JB.185.4.1174-1180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeeva S, Hellingwerf KJ, Teixeira de Mattos MJ. Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J Bacteriol. 2003;185:204–209. doi: 10.1128/JB.185.1.204-209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki SK, Webb JS, Braaten BA, Low DA. Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. J Bacteriol. 2009;191:1777–1786. doi: 10.1128/JB.01437-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker LA, Bang L-S, Crouch M-L, Fang FC. Compensatory role of PspA, a member of the phage shock protein operon, in rpoE mutant Salmonella enterica serovar Typhimurium. Mol Microbiol. 2005;56:1004–1016. doi: 10.1111/j.1365-2958.2005.04604.x. [DOI] [PubMed] [Google Scholar]
- Bidle KA, Kirkland PA, Nannen JL, Maupin-Furlow JA. Proteomic analysis of Haloferax volcanii reveals salinity-mediated regulation of the stress response protein PspA. Microbiology. 2008;154:1436–1443. doi: 10.1099/mic.0.2007/015586-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Darwin AJ. The phage-shock-protein response. Mol Microbiol. 2005;57:621–628. doi: 10.1111/j.1365-2958.2005.04694.x. [DOI] [PubMed] [Google Scholar]
- Darwin AJ. Regulation of the phage-shock-protein stress response in Yersinia enterocolitica. In: Perry RD, Fetherston JD, editors. The Genus Yersinia. From Genomics to Function. New York: Springer; 2007. pp. 167–177. Advances in Experimental Medicine Biology, Vol. 603. [DOI] [PubMed] [Google Scholar]
- Drapal N, Sawers G. Promoter 7 of the Escherichia coli pfl operon is a major determinant in the anaerobic regulation of expression by ArcA. J Bacteriol. 1995;177:5338–5341. doi: 10.1128/jb.177.18.5338-5341.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elderkin S, Jones S, Schumacher J, Studholme D, Buck M. Mechanism of action of the Escherichia coli phage shock protein PspA in repression of the AAA family transcription factor PspF. J Mol Biol. 2002;320:23–37. doi: 10.1016/S0022-2836(02)00404-7. [DOI] [PubMed] [Google Scholar]
- Elderkin S, Bordes P, Jones S, Rappas M, Buck M. Molecular determinants for PspA-mediated repression of the AAA transcriptional activator PspF. J Bacteriol. 2005;187:3238–3248. doi: 10.1128/JB.187.9.3238-3248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennis RB, Stewart V. Respiration. In: Neidhardt FC, Ingraham JL, Lin EC, Low KB, Magasanik B, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. Washington, DC: American Society for Microbiology Press; 1996. pp. 217–261. [Google Scholar]
- Georgellis D, Kwon O, Lin EC. Quinones as the redox signal for the arc two-component system of bacteria. Science. 2001;292:2314–2316. doi: 10.1126/science.1059361. [DOI] [PubMed] [Google Scholar]
- de Graef M, Alexeeva S, Snoep JL, Mattos MJT. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J Bacteriol. 1999;181:2351–2357. doi: 10.1128/jb.181.8.2351-2357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilvout I, Chami M, Engel A, Pugsley AP, Bayan N. Bacterial outer membrane secretin PulD assembles and inserts into the inner membrane in the absence of its pilotin. EMBO J. 2006;25:5241–5249. doi: 10.1038/sj.emboj.7601402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE, Lloyd LJ, Tan KK, Buck M. Secretion defects that activate the phage shock response of Escherichia coli. J Bacteriol. 2003;185:6707–6711. doi: 10.1128/JB.185.22.6707-6711.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic G, Lloyd LJ, Stumpf MPH, Mayhew AJ, Buck M. Induction and function of the phage shock protein extracytoplasmic stress response in Escherichia coli. J Biol Chem. 2006;281:21147–21161. doi: 10.1074/jbc.M602323200. [DOI] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Mitrophanov AY, Groisman EA. A connector of two-component regulatory systems promotes signal amplification and persistence of expression. Proc Natl Acad Sci USA. 2007;104:12063–12068. doi: 10.1073/pnas.0704462104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-R, Cho K-H. The multi-step phosphorelay mechanism of unortodox two-component systems in E. coli realizes ultrasensitivity to stimuli while maintaining robustness to noises. Comput Biol Chem. 2006;30:438–444. doi: 10.1016/j.compbiolchem.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Kleerebezem M, Crielaard W, Tommassen J. Involvement of stress protein PspA (phage shock protein A) of Escherichia coli in maintenance of the protonmotive force under stress conditions. EMBO J. 1996;15:162–171. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi R, Suzuki T, Yoshida M. Escherichia coli phage-shock protein A (PspA) binds to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol Microbiol. 2007;66:100–109. doi: 10.1111/j.1365-2958.2007.05893.x. [DOI] [PubMed] [Google Scholar]
- Malpica R, Franco B, Rodriguez C, Kwon O, Georgellis D. Identification of a quinine-sensitive redox switch in the ArcB sensor kinase. Proc Natl Acad Sci USA. 2004;101:13318–13323. doi: 10.1073/pnas.0403064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpica R, Peña Sandoval GR, Rodríguez C, Franco B, Georgellis D. Signaling by the Arc two-component system provides a link between the redox state of the quinone pool and gene expression. Antioxid Redox Signal. 2006;8:781–795. doi: 10.1089/ars.2006.8.781. [DOI] [PubMed] [Google Scholar]
- Maxson ME, Darwin AJ. PspB and PspC of Yersinia enterocolitica are dual function proteins: regulators and effectors of the phage-shock-protein response. Mol Microbiol. 2006;59:1610–1623. doi: 10.1111/j.1365-2958.2006.05047.x. [DOI] [PubMed] [Google Scholar]
- Mika F, Hengge R. A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of sigmaS (RpoS) in E. coli. Genes Dev. 2005;19:2770–2781. doi: 10.1101/gad.353705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Model P, Jovanovic G, Dworkin J. The Escherichia coli phage-shock-protein (psp) operon. Mol Microbiol. 1997;24:255–261. doi: 10.1046/j.1365-2958.1997.3481712.x. [DOI] [PubMed] [Google Scholar]
- Partridge JD, Sanguinetti G, Dibden DP, Roberts RE, Poole RK, Green J. Transition of Escherichia coli from aerobic to micro-aerobic conditions involves fast and slow reacting regulatory components. J Biol Chem. 2007;282:11230–11237. doi: 10.1074/jbc.M700728200. [DOI] [PubMed] [Google Scholar]
- Rowley G, Spector M, Kormanec J, Roberts M. Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol. 2006;4:383–394. doi: 10.1038/nrmicro1394. [DOI] [PubMed] [Google Scholar]
- Russel M, Kaźmierczak B. Analysis of the structure and subcellular location of filamentous phage pIV. J Bacteriol. 1993;175:3998–4007. doi: 10.1128/jb.175.13.3998-4007.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J, Savitzky DC, Ford E, Darwin AJ. Global analysis of tolerance to secretin-induced stress in Yersinia enterocolitica suggests that the phage-shock-protein system may be a remarkably self-contained stress response. Mol Microbiol. 2007;65:714–727. doi: 10.1111/j.1365-2958.2007.05821.x. [DOI] [PubMed] [Google Scholar]
- Szurmant H, Bu L, Brooks CL, 3rd, Hoch JA. An essential sensor histidine kinase controlled by transmembrane helix interactions with its auxiliary proteins. Proc Natl Acad Sci USA. 2008;105:5891–5896. doi: 10.1073/pnas.0800247105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrancken K, Van Mellaert L, Anné J. Characterization of the Streptomyces lividans PspA response. J Bacteriol. 2008;190:3475–3481. doi: 10.1128/JB.01966-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal S, Heins L, Soll J, Vothknecht UC. Vipp1 deletion mutant of Synechocystis: a connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci USA. 2001;98:4238–4242. doi: 10.1073/pnas.061501198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigneshweraraj S, Bose D, Burrows PC, Joly N, Schumacher J, Rappas M, et al. Modus operandi of the bacterial RNA polymerasecontaining the sigma54 promoter-specificity factor. Mol Microbiol. 2008;68:538–546. doi: 10.1111/j.1365-2958.2008.06181.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.