

Abstract
Lipoteichoic acid (LTA) is an important cell wall polymer in Gram-positive bacteria and often consists a polyglycerolphosphate backbone chain that is linked to the membrane by a glycolipid. In Listeria monocytogenes this glycolipid is Gal-Glc-DAG or Gal-Ptd-6Glc-DAG. Using a bioinformatics approach, we have identified L. monocytogenes genes predicted to be involved in glycolipid (lmo2555 and lmo2554) and LTA backbone (lmo0644 and lmo0927) synthesis. LTA and glycolipid analysis of wild-type and mutant strains confirmed the function of Lmo2555 and Lmo2554 as glycosyltransferases required for the formation of Glc-DAG and Gal-Glc-DAG. Deletion of a third gene, lmo2553, located in the same operon resulted in the production of LTA with an altered structure. lmo0927 and lmo0644 encode proteins with high similarity to the staphylococcal LTA synthase LtaS, which is responsible for polyglycerolphosphate backbone synthesis. We show that both proteins are involved in LTA synthesis. Our data support a model whereby Lmo0644 acts as an LTA primase LtaP and transfers the initial glycerolphosphate onto the glycolipid anchor, and Lmo0927 functions as LTA synthase LtaS, which extends the glycerolphosphate backbone chain. Inactivation of LtaS leads to severe growth and cell division defects, underscoring the pivotal role of LTA in this Gram-positive pathogen.
Introduction
The cell wall envelope of Gram-positive bacteria has been an area of active research for decades. By studying its assembly not only essential functions for bacterial growth and physiology but also important aspects of host pathogen interactions have been uncovered, and studies on the Gram-positive cell wall envelope have gained increased attention in the field of bacterial pathogens. A typical Gram-positive envelope is composed of peptidoglycan, proteins, often capsular polysaccharides and secondary wall polymers, which include wall teichoic acid (WTA), a polymer covalently linked to peptidoglycan, and lipoteichoic acid (LTA), a polymer tethered by a lipid anchor to the bacterial membrane (Fischer, 1988; Navarre and Schneewind, 1999). The structure of LTA varies between organisms (Fischer, 1988; Weidenmaier and Peschel, 2008); one of the best characterized structure is a polymer with an un-branched 1-3-linked glycerolphosphate chain attached to a membrane glycolipid as for instance found in Bacillus subtilis, Staphylococcus aureus, Enterococcus faecalis, Group A and B Streptococcus and Listeria monocytogenes (Fischer, 1990). Glycerolphosphate subunits can be substituted with glycosyl residues and/or d-alanine esters, which significantly contribute to cationic peptide resistance in Gram-positive bacteria (Fischer, 1990; Peschel et al., 1999). In L. monocytogenes, the polyglycerolphosphate LTA backbone is substituted with both d-alanines and α-galactosyl residues and linked to the bacterial membrane via glycolipids Gal(α1-2)Glc(α1-3)-diacylglycerol (Gal-Glc-DAG) or Gal(α1-2)Ptd-6Glc(α1-3)DAG (Gal-Ptd-6Glc-DAG), in which the glucose moiety is lipidated at position 6 with a phosphatidyl (Ptd) group (Hether and Jackson, 1983; Uchikawa et al., 1986; Fischer et al., 1990) (Fig. 1). Despite this thorough chemical analysis, the exact function of LTA is not known.
Fig. 1.

Chemical structure of L. monocytogenes LTA. L. monocytogenes LTA is a linear polyglycerolphosphate polymer attached to the membrane by the glycolipid Gal-Glc-DAG. The free hydroxyl group of the glycerolphosphate units (X1) can be esterified with d-alanine (d-Ala) or glycosylated with galactose (Gal) and the glucose moiety of Gal-Glc-DAG can be lipidated at position 6 with a phosphatidyl group (X2). The most abundant fatty acids in the glycolipid and the phosphatidyl substituent are C17 (R1) and C15 (R2) anteiso-branched fatty acids (Hether and Jackson, 1983; Uchikawa et al., 1986; Fischer et al., 1990).
The recent identification of enzymes responsible for glycolipid and LTA backbone synthesis allowed a phenotypic characterization of strains that are deficient in LTA synthesis or produce LTA of an altered structure. The enzyme responsible for polyglycerolphosphate backbone chain formation has been discovered recently in S. aureus and named LtaS for LTA synthase (Gründling and Schneewind, 2007a). The same and two subsequent studies on S. aureus and B. subtilis revealed that LTA is important for normal growth and observed morphological alterations indicate a crucial role of LTA in the cell division process and the sporulation process in B. subtilis (Oku et al., 2009; Schirner et al., 2009).
Enzymes involved in the synthesis of glycolipids and lipid anchor for LTA have been characterized in several Gram-positive bacteria. In S. aureus and B. subtilis the enzyme YpfP (also called Ugt) is a processive glycosyltransferase, which synthesizes Glc(β1-6)Glc(β1-3)DAG (DiGlc-DAG) by the sequential addition of two glucose moieties onto diacylglycerol (DAG) using UDP-glucose as the substrate (Jorasch et al., 1998; 2000; Kiriukhin et al., 2001). On the other hand, in E. faecalis and Streptococcus agalactiae two separate enzymes are necessary for the synthesis of Glc(α1-2)Glc(β1-3)DAG (DiGlc-DAG) (Doran et al., 2005; Theilacker et al., 2009). The glycosyltransferase responsible for the addition of the second glucose moiety has been characterized in both organisms and renamed BgsA (EF2891 in strain V583) for biofilm-associated glycolipid synthesis and IagA (Gbs0682 in strain NEM316) for invasion-associated gene, respectively, to denote observed phenotypes and defects of deletion strains (Doran et al., 2005; Theilacker et al., 2009). It should be noted that phenotypes observed in strains mutated in glycolipid synthesis genes may not necessarily be due to the lack of these membrane lipids as LTA structure and production are also affected in their absence (Fedtke et al., 2007; Gründling and Schneewind, 2007b).
Little is known about LTA and glycolipid synthesis in the Gram-positive pathogen L. monocytogenes. Only the function of Dlt proteins, which incorporate d-alanines into LTA, has been investigated and it was found that this modification is important for bacterial adhesion to eukaryotic cells and virulence of L. monocytogenes in the mouse model of infection (Abachin et al., 2002). In addition, it has been reported that the L. monocytogenes internalin B protein (InlB), a non-covalently attached cell surface protein required for entry into various host cells, binds to LTA and is retained at the bacterial surface in this manner (Braun et al., 1997; 1998; Jonquieres et al., 1999). Thus, LTA directly and indirectly has important roles in bacterial physiology and virulence.
Here, we used a bioinformatics approach to identify L. monocytogenes genes required for glycolipid and LTA polyglycerolphosphate backbone synthesis. Using a combination of molecular biology and mass spectrometry approaches to characterize glycolipids and LTA synthesized in wild-type and mutant strains, we show that the previously uncharacterized L. monocytogenes genes lmo2555 and lmo2554 encode glycolipid synthesis enzymes, and renamed them LafA and LafB for LTA anchor formation proteins A and B. Two proteins, Lmo0927 and Lmo0644, with similarity to the S. aureus LTA synthase LtaS are involved in LTA backbone synthesis but they have clearly distinct enzymatic functions within the cell. Inactivation of Lmo0927 leads to the absence of LTA on the bacterial surface, a severe growth defect at elevated temperatures and morphological changes underscoring the importance of LTA for cellular functions in the Gram-positive pathogen L. monocytogenes.
Results
Identification of potential glycolipid and LTA synthesis gene in L. monocytogenes
To begin to understand the function(s) of LTA in the Gram-positive pathogen L. monocytogenes, we used a bioinformatics approach to identify putative glycolipid and LTA synthesis enzymes in this organism. Since the LTA glycolipid anchor in L. monocytogenes consists of Gal-Glc-DAG (Hether and Jackson, 1983; Uchikawa et al., 1986; Fischer et al., 1990) presumably two distinct glycosyltransferases are required for its synthesis similar to that observed for E. faecalis and S. agalactiae. In E. faecalis and S. agalactiae the glycosyltransferases responsible for the addition of the terminal glucose moiety have been identified as IagA (Gbs0682 in strain NEM316) and BgsA (EF2891 in strain V583) and in both cases a second putative glycosyltransferase, Gbs0683 and EF2890, is encoded immediately upstream. These second proteins show high similarity to the characterized Acholeplasma laidlawii 1,2-diacylglycerol 3-glucosyltransferase (EC 2.4.1.157) (Berg et al., 2001) with E-values of 3e-53 and 2e-54. While experimental evidence is lacking, this suggests that Gbs0683 and EF2890 are responsible for Glc-DAG generation. We used the S. agalactiae proteins IagA (Gbs0682) and Gbs0683 as query sequences in blast searches (Altschul et al., 1997) against the L. monocytogenes EGD-e genome (Glaser et al., 2001). This identified the L. monocytogenes proteins Lmo2554 (E-value of 6e-20 to IagA) and Lmo2555 (E-value of 2e-76 to Gbs0683) as the closest homologues. Using the respective E. faecalis proteins in blast searches the same two L. monocytogenes proteins were identified with similar E-values of 2e-22 and 3e-83. Taken together, this suggests that Lmo2555 and Lmo02554 could encode UDP-glucose- and UDP-galactose-specific glycosyltransferases responsible for Glc-DAG and Gal-Glc-DAG synthesis respectively. The coding sequences of lmo2555 and lmo2554 overlap by eight bases and the operon is likely to contain a third gene, lmo2553, predicted to encode an integral membrane protein (Fig. 2A). The S. aureus enzyme LtaS, which is responsible for LTA polyglycerolphosphate backbone synthesis, was recently identified (Gründling and Schneewind, 2007a). Two proteins with high degree of similarity to the staphylococcal LtaS enzyme, Lmo0927 (E-value 0.0) and Lmo0644 (E-value 1e-58), are encoded in Listeria genomes (Fig. 2A).
Fig. 2.

LTA production in wild-type and mutant L. monocytogenes strains. A. Operon structure of L. monocytogenes genes involved in glycolipid and LTA formation with direction of transcription indicated by the arrows and predicted terminators shown by hairpin loops. B–E. Western blot detection of cell wall-associated LTA in wild-type, deletion and complementation strains: (B) 10403S (WT), 10403SΔlmo2553 (Δ2553), 10403SΔlmo2554 (Δ2554) and 10403SΔlmo2555 (Δ2555); (C) 10403S (WT), 10403SΔlmo0644 (Δ0644) and 10403SΔlmo0927 (Δ0927); (D) 10403S pHPL3 (WT), 10403SΔlmo2553 pHPL3-lmo2553 (Δ2553 compl.), 10403SΔlmo2553 pHPL3 (Δ2553), 10403SΔlmo2554 pHPL3-lmo2554 (Δ2554 compl.), 10403SΔlmo2554 pHPL3 (Δ2554), 10403SΔlmo2555 pPL3-lmo2555 (Δ2555 compl.) and 10403SΔlmo2555 pPL3 (Δ2555); (E) 10403S pPL3 (WT), 10403SΔlmo0644 pPL3-lmo0644 (Δ0644 compl.), 10403SΔlmo0644 pPL3 (Δ0644), 10403SΔlmo0927 pPL3-lmo0927 (Δ0927 compl.) and 10403SΔlmo0927 pPL3 (Δ0927). Positions of protein standards (in kDa) are shown on the left.
To study the requirement of lmo2555, lmo2554, lmo2553, lmo0927 and lmo0644 for glycolipid production, LTA synthesis and bacterial physiology, these genes were inactivated in the L. monocytogenes 1/2a strain 10403S. Unmarked in-frame deletions were created by allelic exchange and all gene deletions were confirmed by PCR. With the exception of strain 10403SΔlmo0927, which only grew well at 30°C (discussed below), all deletion strains had similar doubling times as compared with the parental 10403S strain (data not shown).
Inactivation of predicted L. monocytogenes glycolipid and LTA synthases affects LTA production and structure
Initially we set out to assess if lmo2555, lmo2554, lmo2553, lmo0927 and lmo0644 contribute to LTA synthesis in L. monocytogenes by analysing LTA production in wild-type and deletion strains. In the case of S. aureus, inactivation of enzymes involved in glycolipid and LTA backbone synthesis leads to structural changes in LTA, which can be readily visualized by Western blot analysis using a polyglycerolphosphate-specific LTA antibody (Gründling and Schneewind, 2007b). Wild-type L. monocytogenes 104030S and deletion strains were grown overnight at 37°C with exception of strain 10403SΔlmo0927, which was cultivated for 2 days at 37°C, and samples were prepared for Western blot analysis of cell wall associated LTA as described under Experimental procedures. Inactivation of Lmo2554 and Lmo2555 led to a drastic reduction in the total amount of LTA produced, while deletion of lmo2553 led to the production of LTA with retarded mobility (Fig. 2B). A similar mobility shift was observed upon inactivation of Lmo0644, while no LTA-specific signal could be detected for strain 10403SΔlmo0927 (Fig. 2C). In addition, the amount of released LTA was analysed by the same Western blot method, but only minimal amounts could be detected in the culture supernatant of lmo2555, lmo2554 and lmo2553 deletion strains (data not shown). To confirm that observed phenotypes were solely due to deletion of the respective gene, complementation vectors were constructed and introduced into appropriate deletion strains. Genes lmo0644, lmo0927 and lmo2555 were cloned under their native promoter into the L. monocytogenes single-site integration vector pPL3, while lmo2553 and lmo2554 were cloned into vector pHPL3 under control of the hyper-spac promoter. As shown in Fig. 2D and E, the observed alterations in LTA production could be complemented, confirming that differences in LTA synthesis are due to inactivation of the respective gene. In summary, these results indicate that all L. monocytogenes proteins identified by our bioinformatics approach are indeed involved in LTA synthesis. The complete absence of LTA in strain 10403SΔlmo0927 suggests that Lmo0927 is responsible for the synthesis of the polyglycerolphosphate backbone chain, while Lmo0644 seems to have an accessory function.
Lmo2555 and Lmo2554 are glycosyltransferases responsible for Glc-DAG and Gal-Glc-DAG production
Alterations in glycolipid synthesis will affect LTA structure and production (Gründling and Schneewind, 2007b). To correlate observed alterations in LTA production with changes in glycolipid formation in the different L. monocytogenes deletion strains, production of these membrane lipids was further analysed. Previous studies have identified the following glycolipids in membranes of Listeria spp.: Glc-DAG, Gal-Glc-DAG and glycolipids with the proposed structure of GroP-Gal-Glc-DAG (Gal-Glc-DAG with one glycerolphosphate subunit GroP) and the d-alanine-esterified derivative D-Ala-GroP-Gal-Glc-DAG (Fischer and Leopold, 1999). Bioinformatic analysis suggested that proteins encoded in the lmo2555–lmo2553 operon are directly involved in glycolipid formation. For glycolipid analysis, wild-type, lmo2553, lmo2554 and lmo2555 deletion strains were grown overnight at 30°C, total membrane lipids isolated and separated by thin-layer chromatography (TLC). TLCs were developed with α-naphthol and sulphuric acid to visualize sugar-containing lipids. Four major glycolipid bands were detected in wild-type L. monocytogenes cells (Fig. 3A) and as shown in Fig. 3A, lmo2555–lmo2553 deletion strains showed differences in the pattern of glycolipids as compared with the wild-type strain. Using matrix-assisted laser desorption/ionization (MALDI) mass spectrometry, we were able to provide structural information for glycolipids within three main bands labelled top, middle and bottom in Fig. 3. Obtained masses were consistent with expected masses for sodium adducts of Glc-DAG (753.6 m/z – top band; Fig. 4A), Gal-Glc-DAG (915.7 m/z – middle band; Fig. 4C) and GroP-Gal-Glc-DAG (1069.5 m/z – bottom band; Fig. 4D) with C17 and C15 fatty acid side-chains. Calculated absolute masses for glycolipids and experimentally observed masses are summarized in Table 1. The predominant masses for lipids with C17 and C15 fatty acid chains are consistent with previous findings that anteiso-branched C17 and C15 fatty acid at C1 and C2 positions, respectively, are the most abundant fatty acids in Listeria lipids (Kosaric and Carroll, 1971; Fischer and Leopold, 1999). In addition, we also observed masses, which are consistent with glycolipids with C15/C15 or C17/C17 fatty acid chains or disodium adducts (replacement of H+ with Na+) (Table 1). Only small amounts of Glc-DAG accumulated in wild-type cells (Fig. 3A) and as seen in Fig. 4A the observed mass signal of 753.6 m/z for this lipid was weak as compared with background signals but the signal was specific (background peaks were separated from each other by 44 mass units). Deletion of lmo2553, predicted to encode an integral membrane protein, resulted in small but reproducible changes in the glycolipid profile with reduction of a glycolipid of unknown structure, indicated by an asterisk in Fig. 3A. Deletion of lmo2555 led to a complete absence of glycolipids and deletion of lmo2554 led to the accumulation of Glc-DAG (Fig. 3A). Glycolipid structure as well as presence and absence of Gal-Glc-DAG (middle band) and GroP-Gal-Glc-DAG (bottom band) was confirmed by MALDI mass spectrometry (Fig. 4B and E–J, Table 1). We had difficulties detecting a signal above background for the top glycolipid band. Only in strain 10403SΔlmo2554, in which this lipid accumulated to significant levels, a clear m/z signal of 753.5 was obtained as expected for a lipid with the structure Glc-DAG (Fig. 4B). These results are consistent with a function of Lmo2555 and Lmo2554 as glycosyltransferase responsible for the formation of Glc-DAG and Gal-Glc-DAG respectively. The exact function of Lmo2553 remains unknown; nevertheless we propose to rename proteins encoded in the lmo2555–lmo2553 operon, LafA (Lmo2555), LafB (Lmo2554) and LafC (Lmo2553) for LTA anchor formation proteins A to C.
Fig. 3.
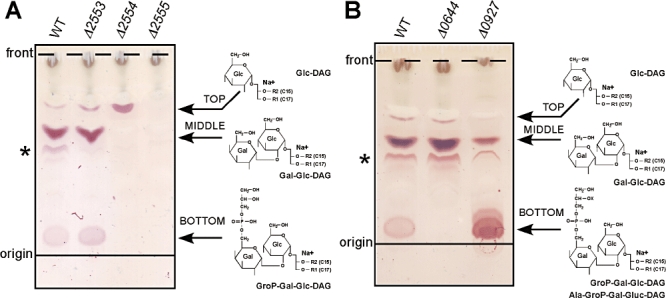
Glycolipid production in wild-type and mutant L. monocytogenes strains. Total membrane lipids were isolated from wild-type and deletion strains grown overnight at 30°C. Five hundred micrograms of lipids were separated by TLC and glycolipids visualized with α-naphthol/sulphuric acid. (A) 10403S (WT), 10403SΔlmo2553 (Δ2553), 10403SΔlmo2554 (Δ2554) and 10403SΔlmo2555 (Δ2555); (B) 10403S (WT), 10403SΔlmo0644 (Δ0644) and 10403SΔlmo0927 (Δ0927). Solid and dashed lines indicate the positions of origin and solvent front respectively. Top, middle and bottom glycolipid bands are indicated by arrows and structures, as identified in this study, are shown on the right. The glycolipid of unknown structure is marked with an asterisk.
Fig. 4.

MALDI-TOF analysis of glycolipids produced by wild-type and mutant L. monocytogenes strains. Lipids from areas containing glycolipids labelled top, middle and bottom in Fig. 3 were further purified and subjected to MALDI-TOF mass spectrometry. Spectra are shown for (A) WT top band, (B) Δ2554 top band, (C and D) WT, (E and F) Δ2553, (G and H) Δ2554, (I and J) Δ2555, (K and L) Δ0644 and (M and N) Δ0927 middle and bottom bands respectively. m/z signals on the x-axes are given in percentage (y-axes) of maximal value shown on the top left. Observed masses corresponding to predicted masses of glycolipids are highlighted in red.
Table 1.
Summary of TLC and MALDI-TOF data of glycolipids produced by wild-type and mutant L. monocytogenes strains.
| Lipid (fatty acid chain length) Formula – calculated mass | WT | Δ2553 | Δ2554 | Δ2555 | Δ0644 | Δ0927 |
|---|---|---|---|---|---|---|
| Top band | X | X | X | Not observed | X | X |
| Glc-DAG (C17, C15) | ||||||
| C41H78Na1O10 – 753.5 | 753.6 | 753.5 | ||||
| Glc-DAG (C15, C15) | ||||||
| C39H74Na1O10 – 725.5 | 725.4 | |||||
| Middle band | X | X | Not observed | Not observed | X | X |
| Gal-Glc-DAG (C17, C15) | ||||||
| C47H88Na1O15 – 915.6 | 915.7 | 915.6 | 915.6 | 915.7 | ||
| Gal-Glc-DAG (C15, C15) | ||||||
| C45H84Na1O15 – 887.6 | 887.6 | 887.6 | 887.6 | 887.6 | ||
| Gal-Glc-DAG (C17, C17) | ||||||
| C49H92Na1O15 – 943.6 | 943.8 | 943.6 | 943.6 | 943.7 | ||
| Bottom band | X | X | Not observed | Not observed | Not observed | X |
| GroP-Gal-Glc-DAG (C17, C15) | ||||||
| C50H95Na1O20P1 – 1069.6 | 1069.5 | 1069.5 | 1069.8 | |||
| GroP-Gal-Glc-DAG (C17, C15) | ||||||
| C50H94Na2O20P1 – 1091.6 (disodium adduct) | 1091.5 | 1091.5 | 1091.8 | |||
| Ala-GroP-Gal-Glc-DAG (C17, C15) | Not observed | Not observed | Not observed | Not observed | Not observed | X |
| C53H100 N1Na1O21P1 – 1140.6 | 1140.8 | |||||
| Ala-GroP-Gal-Glc-DAG (C17, C15) | ||||||
| C53H99 N1Na2O21P1 – 1162.6 (disodium adduct) | 1162.8 |
Presence or absence of glycolipids in different L. monocytogenes strains shown on top is denoted with ‘X’ when present and ‘Not observed’ when absent. Abbreviations for glycolipids in top, middle and bottom bands as indicated in Fig. 3 are shown in the left column with fatty acid chain length given in parenthesis along with molecular formula and calculated absolute mass of sodium adducts or disodium adducts (minus one proton).
Lmo0644 functions as an LTA primase
Based on the accumulation of glycolipids with a single glycerolphosphate in some Gram-positive bacteria such as GroP-Gal-Glc-DAG in L. monocytogenes, a two-enzyme system for LTA synthesis has been proposed, whereby an LTA primase initiates LTA synthesis by the transfer of the first glycerolphosphate subunit onto the glycolipid and a second enzyme, an LTA synthase, extends the chain to form the polyglycerolphosphate backbone (Fischer, 1990). Lmo0927 is likely to be the LTA synthase, as in its absence no LTA could be detected on the bacterial surface (Fig. 2C). In the presence of an LTA primase, one would expect that even in the absence of the LTA synthase (Lmo0927) production of GroP-Gal-Glc-DAG should occur. To test if Lmo0644 could serve as LTA primase, we determined the glycolipid profile of lmo0644 and lmo0927 deletion strains. In the absence of Lmo0644 no GroP-Gal-Glc-DAG (bottom band) could be detected, while in the absence of Lmo0927 GroP-Gal-Glc-DAG (bottom band) was produced and accumulated to higher levels as compared with a wild-type strain (Fig. 3B). MALDI mass spectrometry analysis confirmed the structure and presence or absence of Gal-Glc-DAG and GroP-Gal-Glc-DAG in respective deletion strains (Fig. 4K–N; Table 1). The m/z signal of 1069.8, expected for a sodium adduct of GroP-Gal-Glc-DAG with a calculated mass of 1069.6, was only observed in the presence of Lmo0644. In addition, upon deletion of lmo0927, which resulted in the accumulation of the bottom glycolipid band, an additional mass signal of 1040.8 was observed, which is consistent with the expected mass of 1040.6 for d-Ala-GroP-Gal-Glc-DAG, a d-Ala esterified derivative of GroP-Gal-Glc-DAG. These results are consistent with a model whereby Lmo0644 serves as LTA primase and Lmo0927 functions as LTA synthase and we propose to rename Lmo0644, LtaP, for LTA primase and Lmo0927, LtaS, for LTA synthase.
LtaP (Lmo0644) and LtaS (Lmo0927) are processed in L. monocytogenes
In S. aureus, LtaS is efficiently processed and the extracellular enzymatic domain can be found in the culture supernatant as well as in the cell wall fraction (Lu et al., 2009). To test if L. monocytogenes LtaP (Lmo0644) and LtaS (Lmo0927) are processed similarly, localization and cleavage of these proteins was analysed. To this end, plasmids pPL3-lmo644His6 and pPL3-lmo927His6 were constructed for expression of C-terminally His-tagged LtaP and LtaS proteins under their native promoter and integrated into the chromosome of strain 10403S. We also introduce as controls the empty vector pPL3 and pPL3-ltaSSAHis6, which encodes a C-terminally tagged version of the S. aureus LtaS protein under the control of its native promoter. Resulting strains 10403S pPL3, 10403S pPL3-lmo644His6, 10403S pPL3-lmo927His6 and 10403S pPL3-ltaSSAHis6 were grown overnight at 37°C and cell-associated and supernatant protein samples were prepared for Western blot analysis as described in the Experimental procedure section and tagged proteins were detected with a His-tag-specific antibody. As can be seen in Fig. 5A, L. monocytogenes LtaP and LtaS, and the S. aureus LtaSSA protein could be detected in culture supernatant, indicating that all proteins were cleaved in L. monocytogenes. Processed forms of the L. monocytogenes LTA synthase (Lmo927) and the S. aureus control protein were also detected in the cell wall-associated fraction (Fig. 5B). We were not able to detect any full-length proteins in L. monocytogenes, while our previously published results have shown that small amounts of full-length LtaSSA protein can be detected in S. aureus using an identical sample preparation method (Lu et al., 2009). Taken together, these results show that both proteins, LtaP and LtaS, are processed in L. monocytogenes and suggest that simply a difference in enzyme processing cannot explain the difference in enzyme function between an LTA primase and an LTA synthase.
Fig. 5.

Production and processing of L. monocytogenes proteins Lmo0644 and Lmo0927. L. monocytogenes strains expressing C-terminally His-tagged versions of Lmo0644, Lmo0927 and the staphylococcal LtaSSA control protein were grown overnight at 37°C and samples prepared for Western blot analysis as described under Experimental procedures. (A) Supernatant and (B) cell wall-associated protein samples were separated on 10% SDS polyacrylamide gels and tagged proteins detected with a His-tag-specific antibody. Samples were obtained from L. monocytogenes strains 10403S pPL3 (pPL3), 10403S pPL3-lmo0644His6 (pPL3-0644His6), 10403S pPL3-lmo0927His6 (pPL3-0927His6) and 10403S pPL3-ltaSSAHis6 (pPL3-ltaSSAHis6). The positions of protein standards (in kDa) are indicated on the left. Predicted masses for his-tagged full-length and cleaved proteins are 75.3 and 50.2 kDa for LtaSSA, 75.6 and 49.5 kDa for Lmo0927, and 70.1 and approximately 48 kDa for Lmo0644.
Inactivation of the LTA synthase LtaS (Lmo0927) severely affects growth and morphology of L. monocytogenes
LtaS is required for growth of S. aureus under standard laboratory growth conditions (tryptic soya broth medium at 37°C) (Gründling and Schneewind, 2007a). An LtaS deletion strain is viable at 30°C when grown in the present of at least 1% NaCl and at 37°C when bacteria are cultured in the presence of 7.5% NaCl or 40% sucrose (Oku et al., 2009). However, S. aureus cells have severe morphological defects in the absence of LTA (Gründling and Schneewind, 2007a; Oku et al., 2009). L. monocytogenes is apparently able to grow in the absence of LTA since we obtained a strain with a clean deletion in lmo0927 (see Fig. 2C). However, during the strain construction, we observed that an lmo0927 deletion strain could only be obtained when bacteria were plated and maintained at 30°C (and not at 37°C) during the final step of the allelic exchange procedure. A more detail growth analysis revealed that strain 10403SΔlmo0927 had already at 30°C a reduced colony size (data not shown) and reduced growth rate as compared with a wild-type strain (Fig. 6A) and growth ceased at 37°C (Fig. 6A). This severe growth defect at 37°C was corroborated by a more than 5-log reduction in the plating efficiency at 37°C [8.7 ± 0.6 × 103 colony-forming units (cfu) ml−1] as compared with 30°C (2.8 ± 0.5 × 109 cfu ml−1). In addition, strain 10403SΔlmo0927 formed short chains at 30°C and chain length increased upon incubation at 37°C (Fig. 6B and D). Transmission electron microscopy (TEM) analysis showed that strain 10403SΔlmo0927 had a pronounced defect in division septum formation (Fig. 7). As already seen previously in electron microscopy images of L. monocytogenes (Edwards and Stevens, 1963), ample membrane material in the form of ‘bubbles’ was observed at the division sites of wild-type cells (Fig. 7A and B). Such membrane bubbles were never observed in 10403SΔlmo0927, regardless of the growth temperature (Fig. 7C–H). The complete absence of membranous material at division sites in the lmo0927 deletion strain was more frequent at the non-permissive growth temperature and 8.5 h after the temperature shift actual cell lysis was observed in several cells (Fig. 7E and F). In the few instances where a clear division site was observed in strain 10403SΔlmo0927, these septa looked abnormal (Fig. 7H). In summary, these data show that LTA plays a crucial role in the cell division process in another Gram-positive pathogen, the rod-shaped bacterium L. monocytogenes.
Fig. 6.

Growth and morphology of wild-type 10403S and 10403SΔlmo0927 L. monocytogenes strains. A. Bacterial growth curves. Overnight cultures of wild-type 10403S (WT) and 10403SΔlmo927 (Δ0927) strains were diluted into fresh BHI medium and cultures incubated at 30°C or 37°C. OD600 values were determined at timed intervals and plotted. B–D. Phase-contrast microscopy images of (B) 10403S (WT) and (C and D) 10403SΔlmo0927 (Δ0927) strains grown for 8.5 h at the indicated temperature.
Fig. 7.

Transmission electron microscopy (TEM) images of wild-type 10403S and 10403SΔlmo0927 L. monocytogenes strains. Overnight cultures of wild-type 10403S (WT) and 10403SΔlmo927 (Δ0927) strains were back-diluted and grown for the indicated time at 30°C or 37°C. Bacteria were fixed and prepared for TEM as described under Experimental procedures and representative images are shown: WT grown for 3.5 h at (A) 30°C and (B) 37°C; Δ0927 grown for 3.5 h at (C) 30°C and (D) 37°C; (E–H) Δ0927 grown for 8.5 h at 37°C. Images were taken at (A–D) 49 000×; (E) 30 000×; (F) 68 000×; (G and H) 98 000× magnification and scale bars are shown.
Discussion
In this study, we have identified and characterized L. monocytogenes proteins required for glycolipid anchor and LTA backbone synthesis and a summary model for their function is shown in Fig. 8.
Fig. 8.

Model for glycolipid and LTA synthesis in L. monocytogenes. The cytoplasmic glycosyltransferases Lmo2555 (LafA, LTA anchor formation protein A; shown in blue) and Lmo2554 (LafB; shown in red) synthesize Glc-DAG and Gal-Glc-DAG, respectively, presumably using nucleotide-activated sugars UDP-Glc and UDP-Gal as substrates. Lmo2553 (LafC, shown in grey) is a membrane protein of unknown function and likely acts downstream of LafA and LafB in the glycolipid synthesis pathway. L. monocytogenes uses a two-enzyme system for the subsequent polyglycerolphosphate LTA chain formation. The LTA primase Lmo0644 (LtaP, shown in light orange) transfers the initial glycerolphosphate (black circle) derived from phosphatidylglycerol (PG) onto Gal-Glc-DAG, resulting in the production of GroP-Gal-Glc-DAG. The LTA synthase Lmo0927 (LtaS, shown in orange) then transfers additional glycerolphosphate residues onto GroP-Gal-Glc-DAG, thereby forming the polyglycerolphosphate backbone chain of LTA.
Based on the structural relation of glycolipids with a single glycerolphosphate subunit (GroP-glycolipids) and their parallels in occurrence with LTA, it has been suggested that GroP-glycolipids are intermediates in the LTA synthesis pathway (Fischer et al., 1978; 1990; Fischer, 1981). This led to the proposal that LTA is synthesized by two enzymes: an LTA primase, which produces the GroP-glycolipid intermediate and an LTA synthase, which extends the polyglycerolphosphate backbone chain on this intermediate (Fischer et al., 1978; 1990; Fischer, 1981). This and alternative models for polyglycerolphosphate chain synthesis and extension have been proposed and recently reviewed by Rahman et al. (2009). Here, we provide evidence that L. monocytogenes uses a two-enzyme system for LTA synthesis and show for the first time distinct enzymatic functions for two LtaS paralogues within the cell. Based on the results presented in Figs 2–4, we propose that LtaP (Lmo0644) acts as an LTA primase and produces GroP-glycolipids and LtaS (Lmo0927) functions as LTA synthase and generates the polyglycerolphophate backbone. However, even in the absence of LtaP (lmo0644 mutant), the L. monocytogenes LtaS enzyme can produce a polyglycerolphosphate polymer (Fig. 2C) and we speculate that LtaS can initiate LTA synthesis on both glycolipids and in the absence of this class of lipids directly on DAG or PG. This notion is supported by the observed LTA profile of an ltaP/lafA (lmo0644/lmo2555) double mutant (Fig. S1A). In the absence of both, glycolipids (lmo2555 mutant) and the LTA primase (lmo0644 mutant), LTA is produced albeit at reduced levels compared with a strain, which only lacks the LTA primase. This indicates that in the absence of LtaP, LtaS can use glycolipids as an anchor and in the absence of both, LtaP and glycolipids, DAG or PG is used. (It should be noted that the observed LTA alterations in the double mutant could be complemented to the levels of the individual single mutants by introduction of the respective complementation vector pPL3-lmo0644 or pPL3-lmo2555; Fig. S1A).
In contrast to L. monocytogenes, S. aureus apparently synthesizes LTA with a single enzyme and depletion of LtaSSA does not lead to an accumulation of GroP-glycolipids (M. Wörmann, unpubl. results). The biological significance why some bacteria use a one-enzyme and other bacteria use a multienzyme system for LTA synthesis is not clear at this point, especially considering that an L. monocytogenes ltaP mutant still synthesizes LTA and does not show a growth defect in broth culture. While it is not clear why different Gram-positive bacteria use one or multiple enzymes for LTA synthesis, in general there seems to be a correlation between the number of genome-encoded LtaS-like proteins and bacterial shape; coccoid Staphylococcus spp., Streptococcus spp. (with exception of S. pneumoniae, which does not produce a polyglycerolphosphate-type LTA and does not encode an LtaS-like protein) and Lactococcus lactis strains encode one LtaS protein; ellipsoid-shaped E. faecalis strains, rod-shaped Listeria spp. and with a few exceptions rod-shaped Lactobacillus spp. encode two proteins; and the majority of Bacillus spp., rod-shaped bacteria with a more complex developmental cycle, encode multiple LtaS-like proteins. B. subtilis contains four LtaS paralogues (Gründling and Schneewind, 2007a) and based on published results, it was suggested that the four B. subtilis LtaS paralogues have partially different functions and individual or a combination of these proteins is particular important for proper septum formation during cell division (YlfE) or the sporulation process (YflE and YqgS) (Schirner et al., 2009).
Of the five deletion strains constructed in this study, only deletion of ltaS, which caused a complete absence of LTA (Fig. 2C), had a marked effect on bacterial growth (Figs 6 and 7). The morphological changes observed in L. monocytogenes in the absence of LTA were very similar to those observed in a B. subtilis yflE mutant and seemed to be primarily due to cell division defects (Figs 6 and 7). During the cell division process, a large amount of new membrane and other cell wall material needs to be synthesized at the division site. In wild-type 10403S L. monocytogenes cells, ample membrane material in the form of ‘bubbles’ is observed at the division site (Fig. 7). A striking difference between wild-type and lmo0927 mutant cells is the lack of such large amounts of membranous material at the division site, even at temperatures that are permissive for bacterial growth (Fig. 7). LTA and membrane lipid synthesis are intimately connected, as glycolipids are used as the LTA anchor and the glycerolphosphate subunits of the LTA backbone are likely derived from the headgroup of the membrane lipid phosphatidylglycerol (PG) (Fig. 8). Based on results obtained in this and previous studies (Gründling and Schneewind, 2007a; Oku et al., 2009; Schirner et al., 2009) a unifying scheme has emerged, in which LTA plays an especially important function in the cell division process of Gram-positive bacteria, although the mechanism remains unclear. It has been suggested that LTA ensures that sufficient Mg2+ is available for enzymes acting at the leading edge of the invaginating division septum to function (Schirner et al., 2009). Alternatively, LTA could directly or indirectly contribute to specific membrane properties, which are important for the formation of division septa and the cell division process.
In this study, we further show that LafA (Lmo2555) and LafB (Lmo2554) are necessary for the production of Glc-DAG and Gal-Glc-DAG respectively (Fig. 3A). These enzymes likely use the nucleotide-activated sugars UDP-glucose and UDP-galactose as substrates (see model Fig. 8). A conserved catalytic EX7E signature sequence is present in both LafA (amino acids 293–301) and LafB (amino acids 252–260). This sequence is also found in the characterized glycosyltransferases alMGS and alDGS from A. laidlawii, which belong to the CAZy family 4 of glycosyltransferases (Campbell et al., 1997; Berg et al., 2001; Edman et al., 2003). In the absence of Gal-Glc-DAG, a profound reduction in the overall amount of LTA was observed (Fig. 2B). However, lafA or lafB mutants did not show a reduced growth rate in broth culture suggesting that small amounts of LTA are sufficient for bacterial growth under the conditions tested.
Inactivation of glycolipid-synthesizing enzymes in other Gram-positive bacteria can have different effects on the overall production of LTA. While inactivation of YpfP in S. aureus strains Newman and RN4220 leads to an overall increase in the amount of LTA synthesized and released into the culture supernatant (Kiriukhin et al., 2001; Gründling and Schneewind, 2007b), a drastic reduction in LTA has been reported for a ypfP mutant in the SA113 strain background (Fedtke et al., 2007), which is what we observed for lafA or lafB mutant L. monocytogenes strains. The simplest explanation for this reduction in LTA production is that enzyme(s), which are subsequently needed for the formation of the polyglycerolphosphate polymer, cannot efficiently initiate LTA synthesis in the absence of glycolipids. We suggest that in L. monocytogenes in the absence of glycolipids, neither LtaP nor LtaS can efficiently initiate polyglycerolphosphate LTA backbone synthesis. Therefore, LtaP and LtaS could either have a specific recognition site for glycolipids or alternatively special constraints (the OH-group used for backbone extension will be further removed from the membrane surface in glycolipids as compared with other lipids such as DAG or PG) could dictate which lipid can be used to initiate LTA synthesis. Structural information on the extracellular S. aureus LtaS or B. subtilis YflE domains did not provided any information on potential glycolipid binding sites (Lu et al., 2009; Schirner et al., 2009) and these sites might be embedded within the membrane portion of the protein, for which no structural information is available. For S. aureus strains, differences in LtaS protein sequence could explain observed phenotypic differences in ypfP mutants. However, the LtaS sequence does not vary between sequenced strains, with exception of strain RF122, which has one amino acid change. If not differences in LtaS protein sequence, differences in expression, post-translational modifications or differences in yet to be identified proteins involved in LTA synthesis could explain observed strain differences.
A third gene, lafC (lmo2553), predicted to code for an integral membrane protein with eight transmembrane helices is part of the laf operon. We show that LafC plays an accessory function in glycolipid and LTA synthesis as inactivation of LafC results in minor changes in the glycolipid profile (Fig. 3A) and production of LTA with a retarded mobility similar to that observed in an ltaP mutant (Fig. 2B and C). Since glycolipids Glc-DAG and Gal-Glc-DAG are produced in a lafC mutant, this protein probably acts downstream of LafA and LafB (see model in Fig. 8). During this study, we also created an ltaP/lafC (lmo0644/lmo2553) double mutant and respective control strains for complementation analysis. LTA analysis revealed that the strain lacking both, the LTA primase LtaP and LtaC, produced only small amounts of LTA, comparable to that of the lafA (lmo2555) mutant, which is unable to synthesize glycolipids (Fig. S1B). This result indicates that either LafC or LtaP is required presumably to modify the glycolipid in such a manner that it can be used by LtaS as the LTA anchor. In several Listeria spp., LTA is anchored to the membrane not only by Gal-Glc-DAG but also by a derivative in which the glucose moiety is lipidated at position 6 with a Ptd group (Fig. 1) (Uchikawa et al., 1986) and LafC could be involved in its synthesis. Such a modification may physically alter the presentation of the anchor thus affecting the ability of LtaS to synthesize LTA. However, additional work is needed to determine the exact molecular function of LafC and to establish the requirement of glycolipids and LTA during infection. Also, determining the precise function of LTA during the cell division process warrants further investigation.
Experimental procedures
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 2. L. monocytogenes mutants were constructed in strain 10403S, which is a streptomycin resistant isolate of the serotype 1/2a strain 10403 (Bishop and Hinrichs, 1987). L. monocytogenes strains were grown in brain heart infusion (BHI) medium at 30°C or 37°C as indicated and Escherichia coli strains were grown in Luria–Bertani (LB) medium at 37°C. When appropriate, the growth medium was supplemented with antibiotics as listed in Table 2.
Table 2.
Bacterial strains used in this study.
| Strain | Relevant features | Reference |
|---|---|---|
| Escherichia coli strains | ||
| XL1 Blue | Cloning strain, TetR – ANG127 | Stratagene |
| CLG190 | Cloning strain, TetR – ANG1141 | D. Boyd |
| SM10 | E. coli strain used for conjugations; KanR – ANG618 | Simon et al. (1983) |
| ANG124 | JM109 pKSV7; allelic exchange vector; AmpR | Smith and Youngman (1992) |
| ANG243 | XL1-Blue with S. aureus integration vector pCL55 | Lee et al. (1991) |
| ANG583 | XL1-Blue pCL55-ltaSSAHis6; S. aureus LtaS (SAV0719) with C-terminal His6 tag; AmpR | This study |
| ANG1378 | CLG190 pKSV7Δlmo0644; AmpR | This study |
| ANG1379 | XL1 Blue pKSV7Δlmo0927; AmpR | This study |
| ANG1382 | XL1 Blue pKSV7Δlmo2553; AmpR | This study |
| ANG1384 | XL1 Blue pKSV7Δlmo2554; AmpR | This study |
| ANG1385 | XL1 Blue pKSV7Δlmo2555; AmpR | This study |
| DH-E898 | XL1 Blue pPL3; L. monocytogenes integration vector; CamR – ANG1276 | Gründling et al. (2004) |
| DH-E899 | XL1 Blue pHPL3; L. monocytogenes integration vector with hyper-spac promoter; CamR – ANG1277 | Gründling et al. (2004) |
| AJW1392 | XL1 Blue pPL3-lmo0644; lmo0644 under native promoter control; CamR | This study |
| AJW1393 | XL1 Blue pPL3-lmo0927; lmo0927 under native promoter control; CamR | This study |
| AJW1396 | XL1 Blue pHPL3-lmo2553; lmo2553 under hyper-spac promoter control; CamR | This study |
| AJW1397 | XL1 Blue pHPL3-lmo2554; lmo2554 under hyper-spac promoter control; CamR | This study |
| AJW1398 | XL1 Blue pPL3-lmo2555; lmo2555 under native promoter control; CamR | This study |
| ANG1399 | XL1 Blue pPL3-lmo0644His6; Lmo0644 with C-terminal His-tag under native promoter control; CamR | This study |
| ANG1401 | XL1 Blue pPL3-lmo0927His6; Lmo0927 with C-terminal His-tag under native promoter control; CamR | This study |
| ANG1406 | XL1 Blue pPL3-ltaSSAHis6; LtaSSA with C-terminal His-tag under native promoter control; CamR | This study |
| ANG1456 | SM10 pPL3; E. coli conjugation strain donor for plasmid pPL3, KanR, CamR | This study |
| ANG1459 | SM10 pPL3-lmo0927; E. coli conjugation strain donor for plasmid pPL3-lmo0927, KanR, CamR | This study |
| Listeria monocytogenes strains | ||
| 10403S | StrepR – ANG1263 | Bishop and Hinrichs (1987) |
| AJW1385 | 10403SΔlmo0644; StrepR | This study |
| ANG1386 | 10403SΔlmo0927; StrepR | This study |
| AJW1389 | 10403SΔlmo2553; StrepR | This study |
| AJW1390 | 10403SΔlmo2554; StrepR | This study |
| AJW1391 | 10403SΔlmo2555; StrepR | This study |
| ANG1411 | 10403SΔlmo0927 pPL3; StrepR, CamR | This study |
| ANG1412 | 10403SΔlmo0927 pPL3-lmo0927; lmo0927 complementation strain; StrepR, CamR | This study |
| AJW1413 | 10403S pPL3; StrepR, CamR | This study |
| AJW1414 | 10403S pHPL3; StrepR, CamR | This study |
| AJW1415 | 10403SΔlmo0644 pPL3-lmo0644; lmo0644 complementation strain; StrepR, CamR | This study |
| AJW1416 | 10403SΔlmo0644 pPL3; StrepR, CamR | This study |
| AJW1417 | 10403SΔlmo2553 pHPL3-lmo2553; lmo2553 complementation strain; StrepR, CamR | This study |
| AJW1418 | 10403SΔlmo2553 pHPL3; StrepR, CamR | This study |
| AJW1419 | 10403SΔlmo2554 pHPL3-lmo2554; lmo2554 complementation strain; StrepR, CamR | This study |
| AJW1420 | 10403SΔlmo2554 pHPL3; StrepR, CamR | This study |
| AJW1421 | 10403SΔlmo2555 pPL3-lmo2555; lmo2555 complementation strain; StrepR, CamR | This study |
| AJW1422 | 10403SΔlmo2555 pPL3; StrepR, CamR | This study |
| AJW1423 | 10403S pPL3-lmo0644His6; StrepR, CamR | This study |
| AJW1424 | 10403S pPL3-lmo0927His6; StrepR, CamR | This study |
| AJW1425 | 10403S pPL3-ltaSSAHis6; StrepR, CamR | This study |
| AJW1496 | 10403S Δlmo0644Δlmo2555; StrepR | This study |
| AJW1497 | 10403S Δlmo0644Δlmo2555 pPL3; StrepR, CamR | This study |
| AJW1498 | 10403S Δlmo0644Δlmo2555 pPL3-lmo0644; lmo0644 complementation strain; StrepR, CamR | This study |
| AJW1499 | 10403S Δlmo0644Δlmo2555 pPL3-lmo2555; lmo2555 complementation strain; StrepR, CamR | This study |
| AJW1501 | 10403S Δlmo0644Δlmo2553; StrepR | This study |
| AJW1502 | 10403S Δlmo0644Δlmo2553 pPL3; StrepR, CamR | This study |
| AJW1503 | 10403S Δlmo0644Δlmo2553 pPL3-lmo0644; lmo0644 complementation strain; StrepR, CamR | This study |
| AJW1504 | 10403S Δlmo0644Δlmo2553 pHPL3-lmo2553; lmo2553 complementation strain; StrepR, CamR | This study |
| Other strains | ||
| RN4220 | Transformable S. aureus laboratory strain – ANG113 | Kreiswirth et al. (1983) |
Antibiotics were used at the following concentrations: for E. coli cultures: ampicillin (AmpR) 100 μg ml−1; kanamycin (KanR) 30 μg ml−1; tetracycline (TetR) 10 μg ml−1; for L. monocytogenes cultures: chloramphenicol (CamR) 7.5 or 10 μg ml−1; streptomycin 200 μg ml−1 (StrepR) for the conjugation experiment.
Plasmid and strain construction
Pfu polymerase (Stratagene) was used for PCR amplification of DNA fragments subsequently used for cloning. Restriction enzymes were purchased from New England Biolabs and used according to the manufacturer's instructions. All plasmids were constructed initially in E. coli strains XL1-Blue or CLG190. Allelic exchange plasmids for the construction of strains 10403SΔlmo0644, 10403SΔlmo0927, 10403SΔlmo2553, 10403SΔlmo2554 and 10403SΔlmo2555 containing an in-frame deletion in the respective gene were constructed by the two-step PCR SOE method (Horton et al., 1989) using primers listed in Table 3. Briefly, primers 5-KpnI-LMO0644, 5-int-LMO0644, 3-BamHI-LMO0644 and 3-int-LMO0644-10403S (for deletion of lmo0644); 5-KpnI-LMO0927, 5-int-LMO0927, 3-BamHI-LMO0927 and 3-int-LMO0927 (for deletion of lmo0927); 5-KpnI-LMO2553, 5-int-LMO2553, 3-BamHI-LMO2553 and 3-int-LMO2553 (for deletion of lmo2553); 5-KpnI-LMO2554, 5-int-LMO2554, 3-BamHI-LMO2554 and 3-int-LMO2554 (for deletion of lmo2554); 5-KpnI-LMO2555, 5-int-LMO2555, 3-BamHI-LMO2555 and 3-int-LMO2555 (for deletion of lmo2555) were used to amplify and fuse approximately 950 bp upstream and downstream regions of relevant genes using 10403S chromosomal DNA in PCR reactions. The resulting PCR products were digested with the restriction enzymes KpnI and BamHI and ligated with the allelic exchange vector pKSV7 (Smith and Youngman, 1992) that had been digested with the same restriction enzymes, resulting in plasmids pKSV7Δlmo0644, pKSV7Δlmo0927, pKSV7Δlmo2553, pKSV7Δlmo2554 and pKSV7Δlmo2555. These plasmids were introduced by electroporation (Park and Stewart, 1990) into strain 10403S and a previously described allelic exchange method (Camilli et al., 1993) was used to create strains 10403SΔlmo0644, 10403SΔlmo0927, 10403SΔlmo2553, 10403SΔlmo2554 and 10403SΔlmo2555. Plasmids pKSV7Δlmo2553 and pKSV7Δlmo2555 and strain 10403SΔlmo0644 were used to create double mutants 10403SΔlmo0644/2553 and 10403SΔlmo0644/2555 respectively. Deletions were confirmed by PCR using primer pairs listed in Table 3, which bind outside the region of homology used for allelic exchange. For complementation analysis, genes lmo0644, lmo0927 and lmo2555 were cloned with their native promoter into the L. monocytogenes single-site integration vector pPL3 (Gründling et al., 2004), while genes lmo2553 and lmo2554 were cloned under the control of the hyper-spac promoter into the integration vector pHPL3 (Gründling et al., 2004). Briefly, primers 5-BamHI-LMO0644_pPL3 and 3-KpnI-LMO0644_pPL3 (for complementation of lmo0644), 5-SalI-LMO0927_pPL3 and 3-KpnI-LMO0927_pPL3 (for complementation of lmo0927), 5-BamHI-LMO2553_pPL3HSPAC and 3-KpnI-LMO2553_pPL3HSPAC (for complementation of lmo2553), 5-BamHI-LMO2554_pPL3HSPAC and 3-KpnI-LMO2554_pPL3HSPAC (for complementation of lmo2554) and 5-BamHI-LMO2555_pPL3 and 3-KpnI-LMO2555_pPL3 (for complementation of lmo2555) were used to amplify relevant fragments from chromosomal DNA of strain 10403S. The resulting PCR products were digested with the restriction enzymes KpnI and BamHI or KpnI and SalI for the lmo0927 PCR product and ligated either with pPL3 or with pHPL3, which were digested with the same restriction enzymes, resulting in plasmids pPL3-lmo0644, pPL3-lmo0927, pHPL3-lmo2553, pHPL3-lmo2554 and pPL3-lmo2555. Plasmids pPL3-lmo0644, pHPL3-lmo2553, pHPL3-lmo2554 and pPL3-lmo2555 were electroporated into the relevant deletion strains resulting in complementation strains listed in Table 2. Plasmid pPL3-lmo0927 was introduced into strain 10403SΔlmo0927 by conjugation (Lauer et al., 2002) resulting in strain 10403SΔlmo0927 pPL3-lmo0927. As controls, empty pPL3 and pHPL3 vectors were introduced into wild-type 10403S and relevant deletion strains by electroporation or conjugation for strain 10403SΔlmo0927. For expression of C-terminally His-tagged Lmo0644 and Lmo0927 proteins under their native promoter control, plasmids pPL3-lmo0644His6 and pPL3-lmo0927His6 were constructed. The C-terminal His6 tag was introduced by PCR using primer pair 5-BamHI-LMO644_pPL3/3-SalI-LMO0644-C-His and 5-PstI-LMO0927-withP/3-SalI-LMO0927-C-His and 10403S chromosomal DNA. The resulting PCR products were cut with BamHI and SalI (for lmo0644His6) or PstI and SalI (for lmo0927His6) and inserted into vector pPL3 cut with the same enzymes. As control, the S. aureus ltaS gene, annotated as SAV0719 in the MU50 genome, was also cloned as C-terminal His6 fusion under its native promoter into the L. monocytogenes integration vector pPL3. Plasmid pPL3-ltaSSAHis6 was constructed by amplifying ltaS from S. aureus RN4220 chromosomal DNA using primer pair 5-BamHI + P SAV0719 and 3-KpnI-His6-719 and the PCR product was cut and cloned as BamHI and KpnI fragment into vector pCL55 (Lee et al., 1991), resulting in plasmid pCL55-ltaSSAHis6. The BamHI/KpnI fragment was subsequently excised and cloned into pPL3, resulting in plasmid pPL3-ltaSSAHis6. Plasmids for expression of His-tag protein fusions were initially recovered in E. coli strain XL1-Blue and subsequently introduced by electroporation into the L. monocytogenes strain 10403S, resulting in strains 10403S pPL3-lmo0644His6 and 10403S pPL3-lmo0927His6, 10403S pPL3-ltaSSAHis6. The DNA sequences of all inserts were verified by automated fluorescence sequencing at the MRC Clinical Sciences Centre Genomics Core Laboratory, Imperial College London.
Table 3.
Primers used in this study.
| Number | Name | Sequence |
|---|---|---|
| ANG383 | 5-KpnI-LMO0644 | GGGGTACCGGAGGAAACGGCATCAAAACCTAAATAAGCAAAG |
| ANG384 | 5-int-LMO0644 | CGTAATGGTAAATTAATAATTAGTAAAAAATAAAATAAAATCAA |
| ANG637 | 3-int-LMO0644–10403S | TTACTAATTATTAATTTACCATTACGAGACGAAGATAAATAA |
| ANG386 | 3-BamHI-LMO0644 | CGGGATCCGGCGCACTGTTTATCGTTATCGTTGGCTAC |
| ANG376 | 5-KpnI-LMO0927 | GGGGTACCGTAGCTCTTCTTATGAAGCAAAGAAAATCAGTG |
| ANG377 | 5-int-LMO0927 | AGTTGATTTTTTCGTTTGGATTTTTATTTTCCAATCCTTCAT |
| ANG378 | 3-int-LMO0927 | AAAATCCAAACGAAAAAATCAACTGATTCATCCGATAAATAA |
| ANG379 | 3-BamHI-LMO0927 | CGGGATCCCGTTATCGTGCCACAAGTGTTATTTTGTGG |
| ANG544 | 5-KpnI-LMO2553 | GGGGTACCGTTTTGAGAAATCGGATATCACGCATTACC |
| ANG545 | 5-int-LMO2553 | AGGTGTTTTTGCAAATAAGTTTTTCTTTGCGCCTCCACTCAT |
| ANG546 | 3-int-LMO2553 | AAAAACTTATTTGCAAAAACACCTGCAAAAAATTTACCATAG |
| ANG547 | 3-BamHI-LMO2553 | CGGGATCCCGCAATGATTCCCTCTAAGTGAGTTGGGAT |
| ANG551 | 5-KpnI-LMO2554 | GGGGTACCTTACTTGCATTATATTGCGAAAGGTAAAATTTTG |
| ANG552 | 5-int-LMO2554 | TCGATCCTCTGATGCCGAAGATAGCATTGTCAACTTAATCAC |
| ANG553 | 3-int-LMO2554 | CTATCTTCGGCATCAGAGGATCGACTAGCTGAAATATGGTT |
| ANG554 | 3-BamHI-LMO2554 | CGGGATCCAGTGTACTCAGCTCCACCGGCCCCGCCTGG |
| ANG558 | 5-KpnI-LMO2555 | GGGGTACCGCTTTGATTCTTGTAAAGCGGCTATCGATG |
| ANG559 | 5-int-LMO2555 | AACGTGTGTAGAGTAGGTATCCGTAAAAATCCCTATATTCAT |
| ANG560 | 3-int-LMO2555 | ACGGATACCTACTCTACACACGTTCAAAGGAAAGAGAGGTCA |
| ANG561 | 3-BamHI-LMO2555 | CGGGATCCCCCTAATAAATCCAGGGTTATCTACTTCTTTCAC |
| ANG651 | 5-BamHI-LMO0644_pPL3 | CGGGATCCTTTGTCTCCTACCTTTTTACATTCTTC |
| ANG652 | 3-KpnI-LMO0644_pPL3 | GGGGTACCTTATTTATCTTCGTCTCGTAATGGTAAATTG |
| ANG653 | 5-SalI-LMO0927_pPL3 | ACGCGTCGACCTAGCAGACTTCCATTCCAAATGGTTC |
| ANG654 | 3-KpnI-LMO0927_pPL3 | GGGGTACCTTATTTATCGGATGAATCAGTTGATTTTTTC |
| ANG659 | 5-BamHI-LMO2553_pPL3HSPAC | CGGGATCCCAAGGATTATTAACGAAGGAGTGAAAG |
| ANG660 | 3-KpnI-LMO2553_pPL3HSPAC | GGGGTACCCTATGGTAAATTTTTTGCAGGTGTTTTTGC |
| ANG661 | 5-BamHI-LMO2554_pPL3HSPAC | CGGGATCCACGTTCAAAGGAAAGAGAGGTCATC |
| ANG662 | 3-KpnI-LMO2554_pPL3HSPAC | GGGGTACCTCACTCCTTCGTTAATAATCCTTGAT |
| ANG663 | 5-BamHI-LMO2555_pPL3 | CGGGATCCGGTTCTAACGGTAAAGCGTAAGACGAAC |
| ANG664 | 3-KpnI-LMO2555_pPL3 | GGGGTACCTTAATCACGCCGCGATGACCTCTCTTTCC |
| ANG673 | 3-SalI-LMO0644-C-His | ACGCGTCGACTTAGTGATGGTGATGGTGATGACCTTTATCTT CGTCTCGTAATGGTAAATTG |
| ANG674 | 5-PstI-LMO0927-withP | AACTGCAGCTAGCAGACTTCCATTCCAAATGGTTC |
| ANG676 | 3-SalI-LMO0927-C-His | ACGCGTCGACTTAGTGATGGTGATGGTGATGACCTTTATCGGAT GAATCAGTTGATTTTTTC |
| ANG086 | 5-BamHI + P SAV0719 | CGGGATCCGGAATAGAATATAGAATGCAATTAGAAATG |
| ANG419 | 3-KpnI-His6-719 | GGGGTACCTTAGTGATGGTGATGGTGATGACCTTTTTTAGAG TTTGCTTTAGGTCCTG |
| Primers for verifying deletion strains | ||
| ANG380 | 5-check-LMO0927 | CTTTAACATATGATTCCTCCTTGTAAC |
| ANG381 | 3-check-LMO0927 | CTTTCTACTTTTGCAAATAATGAATTTCAAATC |
| ANG387 | 5-check-LMO0644 | CGGCATCGTCCGTTGCGGATCTTTCAC |
| ANG388 | 3-check-LMO0644 | GCCGCGCCGCACTGGAAGATACGATGAC |
| ANG548 | 5-check-LMO2553 | GTAAAAGGTCAGGGTGTGGCATCAG |
| ANG549 | 3-check-LMO2553 | CAACTTTTTTTATATTCTCTACTTCACC |
| ANG555 | 5-check-LMO2554 | TAGGTCTTTTAGGTAAGCGAATTG |
| ANG556 | 3-check-LMO2554 | CTCCTGCACCAAAAACGATACAAC |
| ANG562 | 5-check-LMO2555 | ACTGAAGGACTTGTAGAAGACCTG |
| ANG563 | 3-check-LMO2555 | CTAGTCGATCCTCTGAATAATAAG |
Restriction sites in primer sequences are underlined and shown in bold.
LTA and protein detection by Western blot
Lipoteichoic acid and protein detection by Western blot was undertaken essentially as previously described (Gründling and Schneewind, 2007b). In brief, for sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Western blot analysis of cell-associated LTA and His-tagged proteins, 1 ml of overnight culture was mixed with 0.5 ml of 0.1 mm glass beads and lysed by vortexing for 45 min in the cold. Glass beads were sedimented by centrifugation at 200 g for 1 min, and 0.5 ml of the resultant supernatant transferred to a fresh tube. Bacterial debris and LTA were sedimented by centrifugation at 17 000 g for 15 min and suspended in protein sample buffer containing 2% SDS normalized for OD600; that is, samples from a culture with an OD600 of 2 were suspended in 50 μl of sample buffer. Samples were boiled for 20 min, centrifuged at 17 000 g for 5 min and 10 μl of samples loaded onto SDS-PAA gels. To determine the amount of LTA shed into the culture medium, 500 μl of culture was first centrifuged at 17 000 g for 5 min to pellet bacteria. Culture supernatant (100 μl) was removed, mixed with 100 μl of 2× protein sample buffer, boiled for 30 min and insoluble material removed by centrifugation at 17 000 g for 5 min. Supernatant samples were normalized based on OD600 of 2, in that 10 μl of a culture of OD600 of 2 was loaded. To determine if the His-tagged proteins were shed into the supernatant, 1.4 ml of culture was centrifuged at 17 000 g for 10 min to pellet the bacteria. One millilitre of the supernatant was transferred to a new tube, mixed with 100 μl of 100% trichloroacetic acid (TCA), vortexed, incubated on ice for 1 h and centrifuged for 10 min at 17 000 g. The supernatant was aspirated and the TCA precipitated pellet was washed twice with 1 ml of ice-cold acetone. Between wash steps, samples were incubated on ice for 1 h and debris collected by centrifugation as described above. After the final centrifugation step, pellets were air dried and suspended in 2× protein sample buffer normalized for OD600; that is, samples from a culture with an OD600 of 2 were suspended in 100 μl of sample buffer. The samples were boiled for 30 min and 10 μl analysed by Western blot. LTA samples were routinely loaded onto 15% SDS-PAA gels and probed with polyglycerolphosphate-specific LTA antibody (Clone 55 from Hycult biotechnology) and HRP-conjugated anti-mouse IgG (Cell Signalling Technologies, USA) used at 1:2000 and 1:10 000 dilutions respectively. His-tagged protein samples were routinely loaded onto 10% SDS-PAA gels and probed with HRP-conjugated His-tag-specific antibody (Sigma) used at a 1:10 000 dilution and Western blots were developed by enhanced chemiluminesce (ECL). Western blots were performed with at least three independently grown cultures in at least two independent experiments and representative images are shown.
Growth curves and determination of colony-forming units
Wild-type and mutant L. monocytogenes cultures were grown overnight at 30°C in 4 ml of BHI medium. Next day, cultures were diluted to a starting OD600 of 0.07 into 25 ml of BHI medium, incubated with shaking at 30°C or 37°C and OD600 values determined at timed intervals. Growth curves were performed in duplicate and representative graphs are shown. To determine the number of colony-forming units (cfu) per ml of culture normalized for OD600 of 2, the optical density of overnight cultures grown at 30°C was determined, cultures normalized based on OD600 readings and a dilution series prepared in phosphate-buffered saline (PBS) pH 7.4. Fifty microlitres of appropriate dilutions were plated in duplicate onto BHI plates and plates incubated at 30°C or 37°C. Colonies were enumerated after 24 or 48 h growth for wild-type and 10403SΔlmo0927 L. monocytogenes strains, respectively, and average values plus standard deviations for three independently grown cultures are given.
Wide-field and transmission electron microscopy
For wide-field microscopy, strains 10403S and 10403SΔlmo927 were grown overnight at 30°C in 4 ml of BHI medium. Next day, cultures were diluted 1:100 or 1:50 (for strain 10403SΔlmo0927 for subsequent growth at 37°C) into 25 ml of fresh medium and grown either at 30°C or 37°C. Eight and a half hours after back dilution, culture aliquots were removed, washed once with 1 ml of PBS pH 7.4 buffer and viewed under a 100× objective on a Nikon Elipse E600 microscope and images taken with a Nikon DXM1200 digital camera. Experiments were performed with at least three independently grown cultures in at least two independent experiments and representative images are shown.
Samples for TEM were prepared similar as described in Thomaides et al. (2001). Briefly, L. monocytogenes strains were grown overnight at 30°C, back-diluted between 1:100 and 1:25 into 200 ml of BHI medium and grown for 3.5 or 8.5 h at 30°C or 37°C as indicated in the text. Bacteria from an equivalent of a 100 ml culture with an OD600 of 0.5 were collected by centrifugation for 10 min at 8000 g, washed twice with 10 ml of 0.2 M sodium cacodylate buffer pH 7.1 and finally suspended in 2 ml of 0.2 M sodium cacodylate buffer pH 7.1 containing 4% glutaraldehyde. Bacteria were fixed for 4 h at 4°C without shaking and subsequently collected by centrifugation for 5 min at 10 000 g. Bacteria were suspended in 1 ml of 0.2 M sodium cacodylate buffer pH 7.1, transferred to 35 mm round tissue culture dishes, overlaid with 1 ml of 0.2 M sodium cacodylate buffer pH 7.1 containing 4% glutaraldehyde and incubated for 1 h at room temperature. After this second fixation step, bacterial layers were washed six times with 2 ml of 0.2 M sodium cacodylate buffer pH 7.1 and processed for electron microscopy as previously described (Thomaides et al., 2001). Images were taken on an FEI Tecnai GZ transmission electron microscope at the Henry Wellcome Trust Imaging Centre, St Mary's Campus, Imperial College London.
Membrane lipid extraction and detection of glycolipids by TLC
For L. monocytogenes membrane lipid extraction and glycolipid analysis, bacteria from 200 ml of cultures grown for 20–24 h at 30°C were collected by centrifugation for 10 min at 8000 g. Bacteria were washed once with 10–20 ml of ice-cold 0.1 M sodium citrate buffer pH 4.7, suspended in 3 ml of 0.1 M sodium citrate buffer pH 4.7 and dispensed into three 2 ml Fast Prep tubes containing 0.1 mm glass beads (∼0.5 ml). Bacteria were lysed and lipids extracted as previously described using a modified Bligh-Dyer method (Kates, 1972; Gründling and Schneewind, 2007b). Dried lipids were suspend either in chloroform or in a 1:1 chloroform : methanol mix at a concentration of 50 mg ml−1. Ten microlitres corresponding to a total of 500 μg of lipids were spotted onto pre-run silica gel Å60 plates (Macherey-Nagel), lipids separated using a chloroform : methanol : H2O (65:25:4) solvent system and glycolipids visualized by spraying plates with 0.5% α-naphthol in 50% methanol and then with 95% H2SO4 (Gründling and Schneewind, 2007b; Kates, 1972). Experiments were performed with at least three independently grown cultures in at least two independent experiments and representative images are shown.
Lipid analysis by MALDI mass spectrometry
For MALDI analysis to determine and confirm the structure of different glycolipids, a total of 4 mg of lipids were spotted (4 × 20 μl) and separated by TLC as described above and different lipids were further purified after scraping the silica gel from appropriate areas. Areas containing glycolipids were determined by developing one lane run in parallel with α-naphthol and H2SO4 and lipids were extracted from the silica gel with chloroform/methanol as previously described (Gründling and Schneewind, 2007b). Dried lipids were suspended in 10 μl of 0.5 M 2,5-dihydroxybenzoic acid (DHB) MALDI matrix dissolved in 1:1 methanol : chloroform and 1 μl was spotted directly onto MALDI plates or diluted 1:10 using 0.5 M DHB matrix and 1 μl spotted. Spotted MALDI plates were run on a MALDI micro MX™ machine (Waters, UK) available at the Proteomics Facility at Imperial College London. Using an automated program, 10 spectra were recorded for each spot in the reflector positive ion mode. As calibration standard, 25–50 pmoles of bradykinin peptide standard (Sigma) with an absolute mass of 757.3997 (M+H+) was spotted in α-cyano-4-hydroxycinnamic acid (CHCA) matrix, which was suspended at 10 mg ml−1 in 70% acetonitrile 0.1% TFA. Mass signals for lipids were manually corrected for observed mass difference of the internal peptide standard. Representative data from two independent experiments are shown.
Acknowledgments
We would like to thank Dr Judit Nagy for her technical assistance with the MALDI mass spectrometry instrument, Michael S. Hollinshead for his excellent assistant with the electron microscopy and Dr Christiaan van Ooij and Dr Rebecca Corrigan for valuable discussions and critical reading of the manuscript. This work was supported by the Medical Research council grant G0701212 and the Wellcome Trust grant WT084483 to A.G.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abachin E, Poyart C, Pellegrini E, Milohanic E, Fiedler F, Berche P, Trieu-Cuot P. Formation of d-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol Microbiol. 2002;43:1–14. doi: 10.1046/j.1365-2958.2002.02723.x. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Edman M, Li L, Wikstrom M, Wieslander A. Sequence properties of the 1,2-diacylglycerol 3-glucosyltransferase from Acholeplasma laidlawii membranes. Recognition of a large group of lipid glycosyltransferases in eubacteria and archaea. J Biol Chem. 2001;276:22056–22063. doi: 10.1074/jbc.M102576200. [DOI] [PubMed] [Google Scholar]
- Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- Braun L, Dramsi S, Dehoux P, Bierne H, Lindahl G, Cossart P. InlB: an invasion protein of Listeria monocytogenes with a novel type of surface association. Mol Microbiol. 1997;25:285–294. doi: 10.1046/j.1365-2958.1997.4621825.x. [DOI] [PubMed] [Google Scholar]
- Braun L, Ohayon H, Cossart P. The InIB protein of Listeria monocytogenes is sufficient to promote entry into mammalian cells. Mol Microbiol. 1998;27:1077–1087. doi: 10.1046/j.1365-2958.1998.00750.x. [DOI] [PubMed] [Google Scholar]
- Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Davies GJ, Bulone V, Henrissat B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J. 1997;326(Part 3):929–939. doi: 10.1042/bj3260929u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran KS, Engelson EJ, Khosravi A, Maisey HC, Fedtke I, Equils O, et al. Blood–brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J Clin Invest. 2005;115:2499–2507. doi: 10.1172/JCI23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman M, Berg S, Storm P, Wikström M, Vikström S, Öhman A, Wieslander A. Structural features of glycosyltransferases synthesizing major bilayer and nonbilayer-prone membrane lipids in Acholeplasma laidlawii and Streptococcus pneumoniae. J Biol Chem. 2003;278:8420–8428. doi: 10.1074/jbc.M211492200. [DOI] [PubMed] [Google Scholar]
- Edwards MR, Stevens RW. Fine structure of Listeria monocytogenes. J Bacteriol. 1963;86:414–428. doi: 10.1128/jb.86.3.414-428.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedtke I, Mader D, Kohler T, Moll H, Nicholson G, Biswas R, et al. A Staphylococcus aureus ypfP mutant with strongly reduced lipoteichoic acid (LTA) content: LTA governs bacterial surface properties and autolysin activity. Mol Microbiol. 2007;65:1078–1091. doi: 10.1111/j.1365-2958.2007.05854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W. Glycerophosphoglycolipids, presumptive biosynthetic precursors of lipoteichoic acids. In: Shockman GD, Wicken AJ, editors. Chemistry and Biological Activities of Bacterial Surface Amphiphiles. New York: Academic Press; 1981. pp. 209–228. [Google Scholar]
- Fischer W. Physiology of lipoteichoic acids in bacteria. Adv Microb Physiol. 1988;29:233–302. doi: 10.1016/s0065-2911(08)60349-5. [DOI] [PubMed] [Google Scholar]
- Fischer W. Bacterial phosphoglycolipids and lipoteichoic acids. In: Hanahan D, editor. Handbook of Lipid Research. New York: Plenum Press; 1990. pp. 123–234. [Google Scholar]
- Fischer W, Leopold K. Polar lipids of four Listeria species containing l-lysylcardiolipin, a novel lipid structure, and other unique phospholipids. Int J Syst Bacteriol. 1999;49(Part 2):653–662. doi: 10.1099/00207713-49-2-653. [DOI] [PubMed] [Google Scholar]
- Fischer W, Nakano M, Laine RA, Bohrer W. On the relationship between glycerophosphoglycolipids and lipoteichoic acids in Gram-positive bacteria. I. The occurrence of phosphoglycolipids. Biochim Biophys Acta. 1978;528:288–297. doi: 10.1016/0005-2760(78)90018-8. [DOI] [PubMed] [Google Scholar]
- Fischer W, Mannsfeld T, Hagen G. On the basic structure of poly(glycerophosphate) lipoteichoic acids. Biochem Cell Biol. 1990;68:33–43. doi: 10.1139/o90-005. [DOI] [PubMed] [Google Scholar]
- Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, et al. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- Gründling A, Schneewind O. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc Natl Acad Sci USA. 2007a;104:8478–8483. doi: 10.1073/pnas.0701821104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Schneewind O. Genes required for glycolipid synthesis and lipoteichoic acid anchoring in Staphylococcus aureus. J Bacteriol. 2007b;189:2521–2530. doi: 10.1128/JB.01683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Burrack LS, Bouwer HG, Higgins DE. Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. Proc Natl Acad Sci USA. 2004;101:12318–12323. doi: 10.1073/pnas.0404924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hether NW, Jackson LL. Lipoteichoic acid from Listeria monocytogenes. J Bacteriol. 1983;156:809–817. doi: 10.1128/jb.156.2.809-817.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Jonquieres R, Bierne H, Fiedler F, Gounon P, Cossart P. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of gram-positive bacteria. Mol Microbiol. 1999;34:902–914. doi: 10.1046/j.1365-2958.1999.01652.x. [DOI] [PubMed] [Google Scholar]
- Jorasch P, Wolter FP, Zahringer U, Heinz E. A UDP glucosyltransferase from Bacillus subtilis successively transfers up to four glucose residues to 1,2-diacylglycerol: expression of ypfP in Escherichia coli and structural analysis of its reaction products. Mol Microbiol. 1998;29:419–430. doi: 10.1046/j.1365-2958.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- Jorasch P, Warnecke DC, Lindner B, Zahringer U, Heinz E. Novel processive and nonprocessive glycosyltransferases from Staphylococcus aureus and Arabidopsis thaliana synthesize glycoglycerolipids, glycophospholipids, glycosphingolipids and glycosylsterols. Eur J Biochem. 2000;267:3770–3783. doi: 10.1046/j.1432-1327.2000.01414.x. [DOI] [PubMed] [Google Scholar]
- Kates M. Techniques of lipidology. In: Work TS, Work E, editors. Laboratory Techniques in Biochemistry and Molecular Biology. New York: American Elsevier; 1972. pp. 347–469. [Google Scholar]
- Kiriukhin MY, Debabov DV, Shinabarger DL, Neuhaus FC. Biosynthesis of the glycolipid anchor in lipoteichoic acid of Staphylococcus aureus RN4220: role of YpfP, the diglucosyldiacylglycerol synthase. J Bacteriol. 2001;183:3506–3514. doi: 10.1128/JB.183.11.3506-3514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaric N, Carroll KK. Phospholipids of Listeria monocytogenes. Biochim Biophys Acta. 1971;239:428–442. doi: 10.1016/0005-2760(71)90035-x. [DOI] [PubMed] [Google Scholar]
- Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- Lauer P, Chow MYN, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Buranen SL, Ye ZH. Construction of single-copy integration vectors for Staphylococcus aureus. Gene. 1991;103:101–105. doi: 10.1016/0378-1119(91)90399-v. [DOI] [PubMed] [Google Scholar]
- Lu D, Wörmann ME, Zhang X, Schneewind O, Gründling A, Freemont PS. Structure-based mechanism of lipoteichoic acid synthesis by Staphylococcus aureus LtaS. Proc Natl Acad Sci USA. 2009;106:1584–1589. doi: 10.1073/pnas.0809020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, Schneewind O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku Y, Kurokawa K, Matsuo M, Yamada S, Lee BL, Sekimizu K. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J Bacteriol. 2009;191:141–151. doi: 10.1128/JB.01221-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SF, Stewart GS. High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene. 1990;94:129–132. doi: 10.1016/0378-1119(90)90479-b. [DOI] [PubMed] [Google Scholar]
- Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 1999;274:8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- Rahman O, Dover LG, Sutcliffe IC. Lipoteichoic acid biosynthesis: two steps forwards, one step sideways? Trends Microbiol. 2009;17:219–225. doi: 10.1016/j.tim.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Schirner K, Marles-Wright J, Lewis RJ, Errington J. Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 2009;28:830–842. doi: 10.1038/emboj.2009.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vitro genetic engineering: transposon mutagenesis in Gram negative bacteria. Biotechnology. 1983;1:784–791. [Google Scholar]
- Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- Theilacker C, Sanchez-Carballo P, Toma I, Fabretti F, Sava I, Kropec A, et al. Glycolipids are involved in biofilm accumulation and prolonged bacteraemia in Enterococcus faecalis. Mol Microbiol. 2009;71:1055–1069. doi: 10.1111/j.1365-2958.2008.06587.x. [DOI] [PubMed] [Google Scholar]
- Thomaides HB, Freeman M, El Karoui M, Errington J. Division site selection protein DivIVA of Bacillus subtilis has a second distinct function in chromosome segregation during sporulation. Genes Dev. 2001;15:1662–1673. doi: 10.1101/gad.197501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikawa K, Sekikawa I, Azuma I. Structural studies on lipoteichoic acids from four Listeria strains. J Bacteriol. 1986;168:115–122. doi: 10.1128/jb.168.1.115-122.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev. 2008;6:276–287. doi: 10.1038/nrmicro1861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.