

Summary
The identification of familial Parkinson Disease (PD) genes is yielding important molecular pathogenetic insights. In an effort to identify additional PD genes, we studied an eight generation Amish pedigree with apparent autosomal dominant parkinsonism with incomplete penetrance. Phenotypic variability ranged from idiopathic PD to progressive supranuclear palsy (PSP), with the average age at onset 53 years (range of 39 to 74 years). We identified markers on chromosome 3 and 7 that were significant at a genome-wide level by parametric and nonparametric criteria, lod > 3 and non-parametric P-value <0.10, respectively. We also identified markers on chromosomes 10 and 22 with lod > 3. These data suggest that parkinsonism in this pedigree is genetically complex, with contributions from several loci.
Keywords: Parkinson’s disease, progressive supranuclear palsy, parkinsonism, genetic linkage, Amish, population isolate
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease, affecting approximately 120 per 100,000 individuals in the United States, with similar prevalence estimates worldwide (de Lau & Breteler 2006). The cause of PD is unknown, although older age and smoking habit appear to be established risk factors. Pathologically, PD is characterized by the loss of dopaminergic neurons and the presence of ubiquitin positive, intraneuronal cytoplasmic Lewy bodies in the substantia nigra pars compacta, and is distinct from other parkinsonian syndromes such as progressive supranuclear palsy (PSP), multiple system atrophy (MSA), and frontotemporal dementia with parkinsonism (FTDP). Clinically, PD is characterized by the asymmetric onset of bradykinesia, resting tremor, muscular rigidity and later, impaired postural reflexes. While some clinical features help distinguish one form of parkinsonism from another, e.g., symmetric impairment, lack of resting tremor, early cognitive or behavioral changes, dysautonomia, and supranuclear palsy, there is substantial overlap between each syndrome (Jankovic & Tolosa 2007), (Gelb, Oliver, & Gilman 1999).
A strategy for understanding the molecular pathogenesis of PD has been the identification and study of causative genes in rare families (Eriksen, Wszolek, & Petrucelli 2005). Thus far, seven genes have been identified in familial PD cases: α—synuclein (PARK1, PARK4), Parkin (PARK2), UCH-L1 (PARK5), PINK1 (PARK6), DJ-1 (PARK7), LRRK2 (PARK8), and ATP13A2 (PARK9) (Hardy et al. 2006), (Ramirez et al. 2006). Genetic studies conducted on multiplex families in Iceland, as well as smaller multiplex families from North America, have identified contributions from the MAPT gene that encodes tau, as well as additional loci on 1p32 (PARK10) and 2p36 (PARK11) (Scott et al. 2001), (Hicks et al. 2002), (Pankratz et al. 2002).
Advances in genetic approaches has generated increasing interest in the study of isolated populations derived from relatively few founders (Heutink & Oostra 2002). Genetic studies of population isolates have theoretical advantages, including the existence of well-ascertained multi-generational pedigrees, descended from fewer founders and spanning fewer generations (Arcos-Burgos & Muenke 2002). Moreover, the restricted geographical distribution tends to endow population isolates with greater environmental and phenotypic homogeneity. The Amish communities founded in Pennsylvania, Ohio, Indiana and Illinois represent such a population. By virtue of their strong cultural and religious beliefs, they represent a distinct and biologically isolated community, and are well recognized for their contributions to the genetics of Mendelian disorders (Patton 2005). Recent interest has focused on characterizing complex neurodegenerative disorders, including dementia and Parkinson’s disease, in these Amish communities (Ashley-Koch et al. 2005), (Racette et al. 2002). In this report, we present the clinical characteristics of an eight generation pedigree of Amish descent and the results from a genomic screen.
MATERIALS AND METHODS
Recruitment and Genealogy
The study protocol was approved by the Institutional Review Board (IRB) of the Vanderbilt University Medical Center. Written, informed consent was obtained from all participants or their legal guardians. Individuals with parkinsonism were initially identified during ascertainment of an ongoing study of Alzheimer dementia in the index Amish community. With permission of individuals and the participating families, genealogical research was conducted using local Amish directories and family genealogy books. Recruitment of additional subjects was conducted by door-to-door interviews and by advertisement in the Amish community newspaper The Budget. The pedigree was constructed using Cyrillic 2.1 (http://www.cyrillicsoftware.com). The recruitment and ascertainment methods were previously applied successfully in an Alzheimer disease study in the Amish (Pericak-Vance et al. 1996), (Ashley-Koch et al. 2006).
Clinical Data
A standardized interview for PD was conducted by a board-certified genetic counselor on participating individuals or a knowledgeable family informant. Individuals were screened for a history of encephalitis, dopamine-blocking medication exposure within one year before diagnosis, symptoms of normal pressure hydrocephalus (dementia, gait difficulty, and urinary incontinence), or a clinical course with unusual features suggestive of atypical or secondary parkinsonism. Participants were also evaluated for a history of exposure to substances known or suspected to cause parkinsonism, including heavy metals or pesticides. Individuals with a positive symptom history of PD as well as unaffected individuals were personally examined by a board-certified neurologist with subspecialty training in movement disorders. Participants were classified as affected, unaffected or unclear, using published diagnostic criteria based on clinical history and neurologic examination (Gelb, Oliver, & Gilman 1999). Affected individuals had at least two cardinal signs of PD (resting tremor, bradykinesia, or rigidity) and no atypical features of parkinsonism. Individuals with unclear status had only 1 sign of PD, a history of atypical clinical features, or both. Unaffected individuals had no signs of PD. Age at onset was self-reported and defined as the age at which onset of the first symptom suggestive of PD was noted by the affected individual. Levodopa responsiveness was determined based on physician and patient observations. Individuals with uncertain symptom benefit or who never received levodopa therapy were classified as having an unknown response.
The severity of extrapyramidal signs and symptoms was evaluated by Hoehn-Yahr staging (Hoehn & Yahr 1967) and the Unified Parkinson’s Disease Rating Scale (UPDRS-motor subscale) UPDRS-III (Fahn, Elton, & members of the UPDRS Development Committee 1987). Brain-imaging studies were obtained from medical records. Dementia was assessed by the memory-orientation-concentration test (Short-Blessed Test (SBT)) (Katzman et al. 1983). Diagnosis of progressive supranuclear palsy was determined from the NINDS-PSP International Workgroup clinical criteria (Litvan et al. 1996).
Genotyping
DNA samples were prepared from whole blood using standard methods (Puregene, Gentra Systems) and stored using a bar-coded system. The genomic screen was conducted using a preselected set of 364 markers (almost all high quality tri- and tetra-nucleotide repeats) conducted in multiplex sets of two to three markers per sample. PCR was performed using standard thermal cycler protocols on 20 ng of DNA. PCR product was labeled using either prelabeled fluorescent primers or post-PCR fluorescent staining (SybrGold, Molecular Probes) and detected employing an Hitachi FMBIO II.
Error Checking and Statistical Analysis
Markers were read in blinded fashion. Consistency checks were performed using PEDCHECK (O’Connell & Weeks 1998) and either rejected based on quality control, or accepted after re-reading and corrections. The complexity of the pedigree prohibited the calculation of exact lod scores. We instead calculated the parametric lod scores and nonparametric p-values, using approximation methods available in SIMWALK2 (Sobel & Lange 1996), (O’Connell & Weeks 1999) running on the VAnderbilt MultiProcessor Integrated Research Engine (VAMPIRE). Even with this approach and reducing the number of marriage loops from a total of 16 to 8, calculations for a single marker took between 3 and 6 hours of computer time. Marker allele frequencies were calculated from the Amish population under study (Table S1). Haplotypes were constructed manually using pedigree information and assigning phase on the basis of minimizing the number of recombinants.
Sequencing
Genomic sequence data was obtained from the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/). GIGYF2 (TNRC15) mutation screening of exon and exon-intron boundaries containing at least 50 bp of flanking intronic sequences was performed by direct sequencing of PCR products (BigDye Terminator, Applied Biosystems, Inc.) generated from intronic primers designed using Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Two affected subjects and one unaffected subject were sequenced and compared to GenBank data using Sequencher software (Genecodes, VA). Primer sequences not previously published are listed in Table S2. Sequencing of the Parkin gene was performed by direct sequencing of PCR products generated from previously published primers sequences except for exon 3 and 9 (Kitada et al. 1998). Sequencing of reported putative and segregating mutations of the LRRK2 gene (Table S2) was performed under previously described conditions (Zimprich et al. 2004).
RESULTS
Clinical description
We obtained by word of mouth and community announcements the clinical history of approximately 200 individuals over age 65 in an Amish community in middle Ohio. Thirty-three individuals had a history of tremor or parkinsonism. Twenty-four individuals consented to participate in the study. Two were excluded on the basis of clinical findings consistent with essential tremor without parkinsonism. Another individual was excluded on the basis of advanced dementia and parkinsonism without additional clinical history.
Individuals who met the diagnostic criteria for probable PD were designated “PD” in Table 1 whereas individuals who met the NINDS-SPSP diagnostic criteria for progressive supranuclear palsy were designated “PSP” in Table 1 (Litvan, Agid, Calne, Campbell, Dubois, Duvoisin, Goetz, Golbe, Grafman, Growdon, Hallett, Jankovic, Quinn, Tolosa, & Zee 1996). Those with Probable PD had asymmetric onset of presenting symptoms that consisted rest tremor or stiffness. Focal dystonia, in the form of blepharospasm, was observed in one individual who was treated with levodopa. Individuals in this group who had been prescribed dopaminergic medications were responsive to medications. In those with prolonged disease duration, dyskinesias and other motor complications often developed. An environmental exposure history was obtained in all participants and most subjects in the community lived according to rural agrarian Amish traditions. One individual reported considerable exposure to pesticides although the specific type was unknown. Parkinsonian symptoms did not develop until many years after this exposure and its progressive nature and levodopa responsiveness suggested that this individual was also affected.
Table 1. Clinical Features of subjects included in the genomic screen.
Individuals with essential tremor or with only one clinical feature of parkinsonism were excluded.
| ID | Clinical Diagnosis1 |
Age at Exam |
Age at Onset |
Disease duration |
Clinical features2 | Hoehn-Yahr3 | Notes |
|---|---|---|---|---|---|---|---|
| 1 | PD | 79 | 55 | 24 | RT, BK, Rg, As, LDR | III | |
| 2 | PD | 66 | 47 | 19 | RT, BK, Rg, As, LDR | IV | Significant Pesticide/herbicide exposure |
| 3 | PD | 71 | 53 | 18 | RT, BK, As, LDR | III | Proband. CT head showed mild diffuse cerebral atrophy |
| 4 | PD | 71 | 53 | 18 | BK, Rg, As, LDR | IV | mildly impaired upgaze, blepharospasm noted |
| 5 | PD | 74 | 57 | 17 | RT, BK, Rg, As | IV | |
| 6 | PD | 73 | 58 | 15 | RT, BK, Rg, As, LDR | III | Onset preceded recent stroke by many years |
| 7 | PD | 64 | 53 | 11 | RT, Rg, As, LDR | III | |
| 8 | PD | 57 | 50 | 8 | RT, Rg, As, LDR | I | |
| 9 | PD | 79 | 73 | 6 | As, BK, LDR, Rg | II | |
| 10 | PD | 80 | 75 | 5 | RT, BK, Rg, As, LDR | II | |
| 11 | PD | 70 | 65 | 5 | RT, BK, Rg, As | II | |
| 12 | PD | 71 | 68 | 3 | BK, Rg, LDR | II | |
| 13 | PSP | 79 | 68 | 11 | RT, BK, Rg, As | n/a | Pseudobulbar affect |
| 14 | PSP | 68 | 63 | 5 | BK, Rg, PI | n/a | Downgaze paresis |
| 15 | PSP | 63 | 61 | 2 | BK, Rg, PI | n/a | Initially included as Probable PD but developed downgaze paresis |
PD = Parkinson’s disease, PSP = progressive supranuclear palsy
RT = resting tremor, BK = bradykinesia, Rg = rigidity, LDR = levodopa responsive, As = asymmetry of onset
Hoehn and Yahr scores were obtained in the off status when possible.
Disease severity as judged by the UPDRS motor subscale, showed a trend toward increasing severity with duration of illness. This trend was observed in the PD group even though the severity was partially masked by the symptomatic response to dopaminergic medications. In the PSP group, disease severity appeared much earlier in the course of illness. The correlation observed between disease severity and duration of illness suggests that parkinsonism is a progressive feature in this pedigree. Dementia was a rare feature in the PD group with only one subject with significant dementia after having the disease for 18 years. Two of the three subjects with probable PSP had Short-Blessed test scores compatible with dementia. None of the affected individuals had prominent visual hallucinations or cognitive fluctuations characteristic of diffuse Lewy body disease (DLB).
Brain imaging was obtained when available. One individual had a motor subcortical stroke that occurred several years after the onset of parkinsonism. A brain MRI of this individual showed no vascular or structural lesions that correlated with parkinsonian symptoms. A CT scan of one affected individual showed mild diffuse atrophy. In none of these cases was there observed selected frontal or temporal lobe atrophy suggestive of fronto-temporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), nor was there selective parietal lobe atrophy suggestive of corticobasal ganglionic degeneration (CBD).
Genealogical information was used to link the individuals into one large complex pedigree spanning eight generations (Figure 1). Where clinical histories were available for deceased individuals, parkinsonism was evident in some. No clear pattern of inheritance was evident. A sub-pedigree could be constructed from the individuals with PSP, tracing to a common ancestor across six generations. Age at onset information was limited for many historical cases and therefore anticipation could not be assessed (Table 1).
Figure 1.
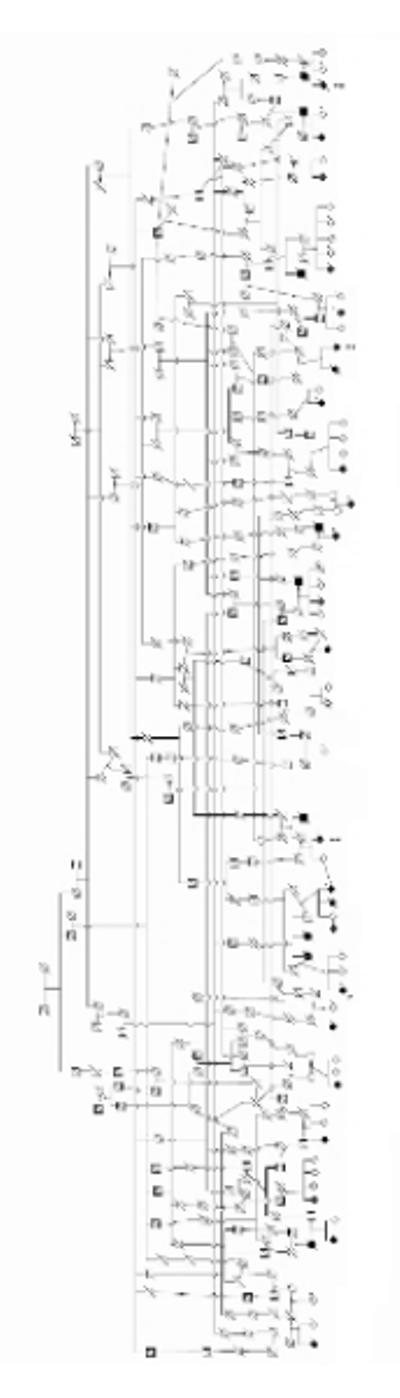
Pedigree of extended family with parkinsonism. Arrowhead denotes the proband. Single asterisks (*) denote individuals included in the genomic screen. Double asterisks (**) denote PSP. Shaded figures represent affected individuals confirmed by examination and/or medical records. Not all unaffected individuals are displayed. Diamonds denote de-identified gender status. (Lange & Lange 2004)
Simulation Study
Although simulation studies at best are a rough approximation, we sought to determine if this pedigree contained sufficient power for linkage analysis. The complexity of the pedigree required an approximation algorithm approach (Simwalk2) (Sobel & Lange 1996). It also required using a simplified diallelic marker to allow computation; such a simplification will underestimate power since the overall heterozygosity will be low. Initial lod scores were consistently lower using a recessive rather than a dominant model and was therefore the more conservative model to estimate power. Using 100 data replicates with a disease allele frequency of 0.05, an autosomal recessive inheritance with low penetrance, a marker allele frequency of 0.10 and a 5% recombination, we obtained a mean lod score of 3.65 (max 5.39) with excellent power (>95) to detect a lod score >3.0. The false positive rate was 0. In fact the highest lod score for an unlinked marker was 0.95 among any of the 100 runs.
Analysis of known PARK loci and Parkinson Genes
To evaluate the possibility that PD in this pedigree was linked to known PARK loci, we genotyped microsatellite markers surrounding PARK1 through PARK8 and found that none of the markers returned significant lod scores (not shown). We also sequenced the Parkin gene, exons containing putative and segregating mutations in LRRK2, GIGYF2 (TNRC15), the PARK11 candidate gene GIGYF2 (TNRC15) on chromosome 1 and the MAPT gene, frequently associated with progressive supranuclear palsy, did not reveal any mutations that were associated with affection status (Table S2).
Genomic Screen
To test the possibility that genetic loci were linked to parkinsonism in this pedigree, we performed a genome-wide scan at a density of ~10 cM. While the genome scan was being performed, one of the subjects originally identified as PD developed clinical features consistent with PSP. We also ascertained two additional subjects who met diagnostic criteria for possible PSP. It was not clear whether these subjects should be classified as unaffected individuals representing phenocopies of the phenotype, or if they were affected individuals representing a broader phenotype with phenotypic variability. We therefore analyzed the data under two models, classifying PSP as either unaffected or affected (Table 2). Two markers, D7S2195 and D10S2325, showed lod > 3 irrespective of affection status of the PSP individuals. Two additional markers, D3S1764 and D22S425 showed lod > 3 under the model classifying PSP as affected. Using additional nonparametric test statistics optimized for additive models (Entropy and NPL-PAIR), only D3S1764 and D7S2195 were significant (p < 0.10) under the model classifying PSP as affected. These results suggest that the markers on chromosome 3 and 7 were the most promising but that the status of the other markers could not be excluded. Multipoint analysis was not possible because of the extreme complexity of the pedigree.
Table 2.
Genomic screen results
| PSP as unaffected | |||||||
|---|---|---|---|---|---|---|---|
| Parametric LOD scores |
Empirical P-values of nonparametric marker allele sharing statistics |
||||||
| Marker | Map (cM) | AD1 | AR2 | BLOCKS3 | MAX-TREE4 | ENTROPY5 | NPL-PAIR6 |
| D3S1764 | 153 | 2.81 | 2.13 | 0.228 | 0.315 | 0.178 | 0.177 |
| D7S2195 | 155 | 3.50 | 2.22 | 0.154 | 0.247 | 0.116 | 0.117 |
| D10S2325 | 33 | 4.09 | 1.99 | 0.256 | 0.365 | 0.209 | 0.208 |
| D22S425 | 14 | 1.95 | 1.06 | 0.370 | 0.531 | 0.320 | 0.323 |
| PSP as affected | |||||||
|---|---|---|---|---|---|---|---|
| Parametric LOD scores |
Empirical P-values of nonparametric marker allele sharing statistics |
||||||
| Marker | Map (cM) | AD1 | AR2 | BLOCKS3 | MAX-TREE4 | ENTROPY5 | NPL-PAIR6 |
| D3S1764 | 153 | 3.60 | 2.42 | 0.123 | 0.083 | 0.065 | 0.057 |
| D7S2195 | 155 | 3.88 | 2.63 | 0.110 | 0.153 | 0.073 | 0.074 |
| D10S2325 | 33 | 3.09 | 1.84 | 0.246 | 0.220 | 0.172 | 0.163 |
| D22S425 | 14 | 3.06 | 1.32 | 0.328 | 0.390 | 0.266 | 0.268 |
Parametric and nonparametric linkage analysis results from SIMWALK2 (Lange and Lange, 2004)
Parametric and nonparametric linkage analysis results from SIMWALK2 (Lange and Lange, 2004)
Parametric and nonparametric linkage analysis results from SIMWALK2 (Lange and Lange, 2004)
Parametric and nonparametric linkage analysis results from SIMWALK2 (Lange and Lange, 2004)
Parametric and nonparametric linkage analysis results from SIMWALK2 (Lange and Lange, 2004)
Parametric and nonparametric linkage analysis results from SIMWALK2 (Lange and Lange, 2004)
Parametric linkage analysis under an autosomal dominant model
Parametric linkage analysis under an autosomal recessive model
Nonparametric linkage analysis test statistic optimized for recessive models.
Nonparametric linkage analysis test statistic optimized for dominant models.
Nonparametric linkage analysis test statistic optimized for additive models.
Nonparametric linkage analysis test statistic optimized for additive models.
Haplotype analysis was conducted to further characterize the identified regions of interest (Table 3). Interestingly, there was a definite trend where the number of unaffected became much less common as the haplotype moved across the peak marker for chromosomes 3, 10, and 22. The most interesting haplotypes were D3S1764-D3S1744, D10S189-D10S2325-D10S1423, and D22S425-D22S1685 where there were substantial differences in the ratio of affected vs. unaffected. These results suggest that genes contributing to the affected status lie between these respective markers.
Table 3. Haplotype analysis.
Comparison of the observed haplotype frequency in affected (A) and unaffected (U) individuals and their relative ratio (A/U) where (n) is the maximum number of possible haplotypes being compared.
| Marker | Haplotype | A | U | A/U | n | |||
|---|---|---|---|---|---|---|---|---|
| D3S4523 | 1 | |||||||
| D3S2322 | 4 | 4 | 0.38 | 0.40 | 0.93 | 15 | ||
| D3S1764 | 4 | 4 | 0.25 | 0.17 | 1.50 | 19 | ||
| D3S1744 | 4 | 4 | 0.21 | 0.05 | 4.38 | 26 | ||
| D3S1763 | 5 | 0.08 | 0.10 | 0.88 | 29 | |||
| D7S1804 | 4 | |||||||
| D7S2202 | 4 | 4 | 0.13 | 0.10 | 1.31 | 26 | ||
| D7S2195 | 14 | 14 | 0.21 | 0.17 | 1.25 | 28 | ||
| D7S3070 | 4 | 4 | 0.17 | 0.20 | 0.83 | 30 | ||
| D7S3058 | 2 | 0.04 | 0.10 | 0.44 | 35 | |||
| D10S1435 | 3 | |||||||
| D10S189 | 2 | 2 | 0.25 | 0.31 | 0.81 | 17 | ||
| D10S2325 | 6 | 6 | 0.13 | 0.02 | 5.25 | 25 | ||
| D10S1423 | 3 | 3 | 0.13 | 0.02 | 5.25 | 25 | ||
| D10S1426 | 3 | 0.17 | 0.12 | 1.40 | 19 | |||
| D22S420 | 4 | |||||||
| D22S425 | 6 | 6 | 0.17 | 0.17 | 1.00 | 25 | ||
| D22S1685 | 4 | 4 | 0.13 | 0.02 | 5.25 | 18 | ||
| D22S689 | 5 | 0.08 | 0.07 | 1.17 | 14 | |||
DISCUSSION
In this paper, we describe an extended Amish pedigree with apparent autosomal dominant parkinsonism with variable penetrance. Simulation analysis under conservative estimates (autosomal recessive model) indicated that this pedigree was adequately powered to detect linkage with a low false positive rate. The genome-wide scan identified markers on chromosomes 3, 7, 10 and 22 with parametric lod score > 3. While false positive lod scores due to pedigree errors are possible, we consider this unlikely. In support of this, Pollin et al. found that genealogical information of the Anabaptist Genealogy Database to be reliable, with nearly all deviations from the founder haplotypes (at least in the male lineage) to be consistent with mutation rates rather than errors (Pollin et al 2008). Although less sensitive, nonparametric analysis was significant for markers on chromosome 3 and 7 (p < 0.10). Haplotype analysis supported the existence of genes between markers D3S1764-D3S1744, D10S189-D10S1423, and D22S425-D22S1685, respectively, and suggested the contribution of multiple genes in this pedigree.
None of these markers were reported in previous whole genome linkage studies. Scott et al. reported a mlod of 1.62 for D3S2460 (135 cM) in levodopa-nonresponsive PD (Scott, Nance, Watts, Hubble, Koller, Lyons, Pahwa, Stern, Colcher, Hiner, Jankovic, Ondo, Allen, Jr., Goetz, Small, Masterman, Mastaglia, Laing, Stajich, Slotterbeck, Booze, Ribble, Rampersaud, West, Gibson, Middleton, Roses, Haines, Scott, Vance, & Pericak-Vance 2001) and Pankratz et al. reported a lod 1.3 for D10S1208 (63cM) in a whole genome scan of multiplex PD negative for parkin mutations (Pankratz, Nichols, Uniacke, Halter, Rudolph, Shults, Conneally, & Foroud 2002). Chromosome 22 (D22S685, 37 cM) was previously reported to contain a QTL for age at onset (Li et al. 2002). None of these previously reported markers are located near the markers we have identified in this study.
The parkinsonian phenotype in this pedigree includes three cases of PSP. Although usually considered clinically and pathologically distinct from PD, the inclusion of the PSP cases in our analysis did not adversely affect lod scores, and suggests the presence of phenotypic heterogeneity, which has been observed in other genetic forms of parkinsonism, including α-synuclein, LRRK2, and parkin gene mutations (Nishioka et al. 2006), (Fuchs et al. 2007), (Whaley et al. 2006), (Papapetropoulos et al. 2006), (Kunishige et al. 2004). We did not observe linkage to markers associated with α-synuclein, LRRK2 or Parkin. Sequencing of known segregating LRRK2 mutations and the entire Parkin coding region excludes the role of these genes. Compound heterozygosity for autosomal recessive genes, such as DJ1 and PINK1, could result in an apparent autosomal dominant inheritance pattern on account of cosegregation across several meioses. Although not feasible computationally, a joint linkage analysis across these two regions would address this possibility.
Racette et al. described a large Old Order Amish pedigree with multi-incident PD (Racette, Rundle, Wang, Goate, Saccone, Farrer, Lincoln, Hussey, Smemo, Lin, Suarez, Parsian, & Perlmutter 2002), (Racette et al. 2006). To our knowledge, our pedigree is not related to any members of this pedigree. Initial screening of their pedigree also suggested that genetic factors are likely to be responsible for PD in their population but the results of a genomic screen have not been reported. Interestingly, ascertainment of other Amish communities have revealed considerable variation in the prevalence of PD, suggesting the possibility of familial clustering and genetic risk for PD in this Amish community (Scott, W., personal communications). Due to social and cultural beliefs, autopsy studies are rarely conducted in this population, thus pathological confirmation has been difficult to obtain.
Supplementary Material
Acknowledgments
We dedicate this paper to the Amish communities in Holmes County and surrounding area, without whom none of this work would be possible. S.L.L. is supported by the Vanderbilt Physician Scientist Development Program and K08-NS01441S. J.L.H. is supported by R01 AG19085. S.L.L. and J.L.H. are also supported by a grant from the Michael J. Fox Foundation. The authors thank Thomas L. Davis, M.D. for introducing us to the Amish community and its leaders.
REFERENCES
- Arcos-Burgos M, Muenke M. Genetics of population isolates. Clin.Genet. 2002;vol. 61(no 4):233–247. doi: 10.1034/j.1399-0004.2002.610401.x. [DOI] [PubMed] [Google Scholar]
- Agarwala R, Biesecker LG, Schäffer AA. Anabaptist genealogy database. Am. J. Med. Genet. C Semin. Med. Genet. vol. 121(No 1):23–7. doi: 10.1002/ajmg.c.20004. [DOI] [PubMed] [Google Scholar]
- Ashley-Koch AE, Mei H, Jaworski J, Ma DQ, Ritchie MD, Menold MM, Delong GR, Abramson RK, Wright HH, Hussman JP, Cuccaro ML, Gilbert JR, Martin ER, Pericak-Vance MA. An analysis paradigm for investigating multi-locus effects in complex disease: examination of three GABA receptor subunit genes on 15q11-q13 as risk factors for autistic disorder. Ann.Hum.Genet. 2006;vol. 70(no Pt 3):281–292. doi: 10.1111/j.1469-1809.2006.00253.x. [DOI] [PubMed] [Google Scholar]
- Ashley-Koch AE, Shao Y, Rimmler JB, Gaskell PC, Welsh-Bohmer KA, Jackson CE, Scott WK, Haines JL, Pericak-Vance MA. An autosomal genomic screen for dementia in an extended Amish family. Neurosci.Lett. 2005;vol. 379(no 3):199–204. doi: 10.1016/j.neulet.2004.12.065. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;vol. 5(no 6):525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Wszolek Z, Petrucelli L. Molecular pathogenesis of Parkinson disease. Arch.Neurol. 2005;vol. 62(no 3):353–357. doi: 10.1001/archneur.62.3.353. [DOI] [PubMed] [Google Scholar]
- Fahn S, Elton R, members of the UPDRS Development Committee 1987 . Recent Developments in Parkinson’s Disease. In: Fahn S, Marsden CD, Calne DB, editors. Recent Developments in Parkinson’s Disease. vol. 2. MacMillan Helathcare Information; Florham Park, NJ: pp. 153–163. [Google Scholar]
- Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;vol. 68(no 12):916–922. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch.Neurol. 1999;vol. 56(no 1):33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson’s disease and parkinsonism. Ann.Neurol. 2006;vol. 60(no 4):389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- Heutink P, Oostra BA. Gene finding in genetically isolated populations. Hum.Mol.Genet. 2002;vol. 11(no 20):2507–2515. doi: 10.1093/hmg/11.20.2507. [DOI] [PubMed] [Google Scholar]
- Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J, Frigge ML, Kong A, Gulcher JR, Stefansson K, Sveinbjornsdottir S. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann.Neurol. 2002;vol. 52(no 5):549–555. doi: 10.1002/ana.10324. [DOI] [PubMed] [Google Scholar]
- Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology. 1967;vol. 17(no 5):427–442. doi: 10.1212/wnl.17.5.427. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Tolosa E. Parkinson’s disease and movement disorders. 5th ed Lippincott Williams & Wilkins; Philadelphia: 2007. [Google Scholar]
- Katzman R, Brown T, Fuld P, Peck A, Schechter R, Schimmel H. Validation of a short Orientation-Memory-Concentration Test of cognitive impairment. Am.J.Psychiatry. 1983;vol. 140(no 6):734–739. doi: 10.1176/ajp.140.6.734. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;vol. 392(no 6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kunishige M, Mitsui T, Kuroda Y, Yoshida S, Kosaka M, Matsumoto T. Expanding phenotype and clinical heterogeneity in patients with identical mutation of the parkin gene. Eur.Neurol. 2004;vol. 51(no 3):183–185. doi: 10.1159/000077671. [DOI] [PubMed] [Google Scholar]
- Lange EM, Lange K. Powerful allele sharing statistics for nonparametric linkage analysis. Hum.Hered. 2004;vol. 57(no 1):49–58. doi: 10.1159/000077389. [DOI] [PubMed] [Google Scholar]
- Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen FA, Jr., Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Age at onset in two common neurodegenerative diseases is genetically controlled. Am.J.Hum.Genet. 2002;vol. 70(no 4):985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E, Zee DS. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;vol. 47(no 1):1–9. doi: 10.1212/wnl.47.1.1. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, Mizoguchi K, Murata M, Toda T, Imoto I, Inazawa J, Mizuno Y, Hattori N. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann.Neurol. 2006;vol. 59(no 2):298–309. doi: 10.1002/ana.20753. [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am.J.Hum.Genet. 1998;vol. 63(no 1):259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. An optimal algorithm for automatic genotype elimination. Am.J.Hum.Genet. 1999;vol. 65(no 6):1733–1740. doi: 10.1086/302663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, Conneally PM, Foroud T. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am.J.Hum.Genet. 2002;vol. 71(no 1):124–135. doi: 10.1086/341282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos S, Singer C, Ross OA, Toft M, Johnson JL, Farrer MJ, Mash DC. Clinical heterogeneity of the LRRK2 G2019S mutation. Arch.Neurol. 2006;vol. 63(no 9):1242–1246. doi: 10.1001/archneur.63.9.1242. [DOI] [PubMed] [Google Scholar]
- Patton MA. Genetic studies in the Amish community. Ann.Hum.Biol. 2005;vol. 32(no 2):163–167. doi: 10.1080/03014460500075274. [DOI] [PubMed] [Google Scholar]
- Pericak-Vance MA, Johnson CC, Rimmler JB, Saunders AM, Robinson LC, D’Hondt EG, Jackson CE, Haines JL. Alzheimer’s disease and apolipoprotein E-4 allele in an Amish population. Ann.Neurol. 1996;vol. 39(no 6):700–704. doi: 10.1002/ana.410390605. [DOI] [PubMed] [Google Scholar]
- Pollin TI, McBride DJ, Agarwala R, Schäffer AA, Shuldiner AR, Mitchell BD, O’Connell JR. Investigations of the Y chromosome, male founder structure and YSTR mutation rates in the Old Order Amish. Hum. Hered. vol. 65(no 2):91–104. doi: 10.1159/000108941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racette BA, Good L, Antenor JA, Gee-Minnich L, Moerlein SM, Videen TO, Perlmutter JS. [18F]FDOPA PET as an endophenotype for Parkinson’s Disease linkage studies. Am.J.Med.Genet.B Neuropsychiatr.Genet. 2006;vol. 141(no 3):245–249. doi: 10.1002/ajmg.b.30293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racette BA, Rundle M, Wang JC, Goate A, Saccone NL, Farrer M, Lincoln S, Hussey J, Smemo S, Lin J, Suarez B, Parsian A, Perlmutter JS. A multi-incident, Old-Order Amish family with PD. Neurology. 2002;vol. 58(no 4):568–574. doi: 10.1212/wnl.58.4.568. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat.Genet. 2006;vol. 38(no 10):1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, Hiner BC, Jankovic J, Ondo WG, Allen FH, Jr., Goetz CG, Small GW, Masterman D, Mastaglia F, Laing NG, Stajich JM, Slotterbeck B, Booze MW, Ribble RC, Rampersaud E, West SG, Gibson RA, Middleton LT, Roses AD, Haines JL, Scott BL, Vance JM, Pericak-Vance MA. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA. 2001;vol. 286(no 18):2239–2244. doi: 10.1001/jama.286.18.2239. [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am.J.Hum.Genet. 1996;vol. 58(no 6):1323–1337. [PMC free article] [PubMed] [Google Scholar]
- Whaley NR, Uitti RJ, Dickson DW, Farrer MJ, Wszolek ZK. Clinical and pathologic features of families with LRRK2-associated Parkinson’s disease. J.Neural Transm.Suppl. 2006;(no 70):221–229. doi: 10.1007/978-3-211-45295-0_34. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;vol. 44(no 4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.