

Abstract
A major obstacle in the treatment of chronic hepatitis C virus (HCV) infection has been the lack of effective, well-tolerated therapeutics. Notably, the recent development of the HCV cell culture infection system now allows not only for the study of the entire viral life cycle, but also for the screening of inhibitors against all aspects of HCV infection. However, in order to screen libraries of potential antiviral compounds, it is necessary to develop a highly reproducible, accurate assay for HCV infection adaptable for high-throughput screening (HTS) automation. Using an internally quenched 5-FAM/QXL 520 fluorescence resonance energy transfer (FRET) substrate containing the HCV NS3 peptide cleavage sequence, we report the development of a simple, mix-and-measure, homogenous, cell-based HCV infection assay amendable for HTS. This assay makes use of synchronized, nondividing human hepatoma-derived Huh7 cells, which support more-reproducible long-term HCV infection and can be readily scaled down to a 96-well-plate format. We demonstrate that this stable cell culture method eliminates common problems associated with standard cell-based HTS, such as cell culture variability, poor reproducibility, and low signal intensity. Importantly, this HCV FRET assay not only can identify inhibitors that act throughout the viral life cycle as effectively as more-standard HCV assays, such as real-time quantitative PCR and Western blot analysis, but also exhibits a high degree of accuracy with limited signal variation (i.e., Z′ ≥ 0.6), providing the basis for a robust HTS campaign for screening compound libraries and identifying novel HCV antivirals.
Hepatitis C virus (HCV) is an enveloped positive-strand RNA virus that infects and replicates in the liver of ∼170 million individuals worldwide. Although acute infection is typically asymptomatic, ∼80% of patients fail to clear the virus, resulting in a chronic infection associated with significant liver disease, including cirrhosis and hepatocellular carcinoma (HCC) (2). As such, in the United States, HCV-related HCC accounts for over 50% of HCC cases and over 30% of the liver transplants that are performed. With no vaccine available to protect against HCV infection and only a subset of chronically infected patients responding to current treatment options (1), there is an obvious and immediate need for new effective HCV antivirals.
HCV is classified in the family Flaviviridae based on conservation of the viral RNA-dependent RNA polymerase and genome organization (32). The ∼9.6-kb RNA genome encodes a single open reading frame flanked by highly structured 5′ and 3′ untranslated regions. The 5′ untranslated region contains an internal ribosome entry site that is required for translation of an ∼3,010-amino acid viral polyprotein, which is proteolytically cleaved into structural and nonstructural proteins. The nonstructural viral proteins assemble on cytoplasmic cellular membranes to form the viral RNA replication complex in which negative-strand RNA synthesis is believed to occur (15). The negative strand then provides the template for ∼10-fold amplification of positive-strand genomic RNA, which can subsequently be used for additional translation, negative-strand synthesis, or packaging into progeny virus (32).
Since its discovery in 1989 as the causative agent of non-A non-B hepatitis (10), the viral life cycle and host-virus interactions that determine infection outcome have been difficult to study because experimental HCV cell culture infection systems and small animal models have not been available. Nonetheless, significant advancements in the study of HCV have been made over the past 19 years using surrogate systems (4), subgenomic and full-length HCV replicons (6, 7, 20, 33), and pseudotyped particles (HCVpp) (3). While the HCV replicon and HCVpp systems were breakthroughs that overcame key experimental limitations, these systems afforded the study of only viral replication and entry, respectively, and did not recapitulate the entire viral life cycle. It was not until 2005 that the genotype 2a HCV consensus clone (JFH-1) was shown to replicate in the Huh7 human hepatoma-derived cell line and produce infectious HCV in cell culture (HCVcc) (31, 50, 56), recapitulating all aspects of the viral life cycle and providing a promising multifaceted opportunity for drug discovery and high-throughput screening (HTS).
Although numerous types of HCV replicon-based HTS have been developed (8, 11, 16, 19, 24, 29, 30, 34, 38, 43, 58), the need to screen compounds that target all steps of the HCV life cycle is warranted. As such, in this report we describe the development of a cell-based HCVcc HTS assay for identification of HCV antivirals that target any aspect of the viral life cycle, including steps yet to be defined. Rather than using exogenous or foreign enzymatic reporters to measure HCV infection, the assay described herein uses NS3 protease activity as a virally encoded “enzymatic reporter” of HCV infection. This strategy is based on (i) the observation that NS3 protease activity parallels HCV infection kinetics and (ii) the ability of the viral NS3 protein to cleave internally quenched peptide substrates (5, 21), allowing for quantitative measurement of NS3 protease activity by fluorescence resonance energy transfer (FRET), which can be easily adapted to an HTS format. Moreover, to optimize reproducibility and signal intensity while reducing nonspecific effects that screened compounds might have on host cell growth, this assay uniquely incorporates synchronized, nondividing Huh7 cells. Here we show that the resulting mix-and-measure, homogenous, cell-based HCV infection assay can identify inhibitors targeting all aspects of the viral life cycle and exhibits an average assay window with a Z′ factor of greater than 0.6, making it suitable for compound library screening.
MATERIALS AND METHODS
Cells and viruses.
Huh7 cells (56) (also known as Huh7/scr cells [13, 57], obtained from Francis Chisari at The Scripps Research Institute, La Jolla, CA) were cultured in complete Dulbecco's modified Eagle's medium (cDMEM) (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (HyClone), 100 units/ml penicillin, 100 mg/ml streptomycin, and 2 mM l-glutamine (Gibco Invitrogen, Carlsbad, CA) as previously described (56).
The plasmid containing the full-length JFH-1 genome (pJFH1) was kindly provided by T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) and has been previously described (22, 23, 50). Protocols for JFH-1 in vitro transcription and HCV RNA electroporation have been described elsewhere (49). The JFH-1 HCVcc viral stock was generated by infection of naïve Huh7 cells at a multiplicity of infection (MOI) of 0.01 focus-forming units (FFU)/cell, using medium from Huh7 cells electroporated with in vitro-transcribed JFH-1 RNA as previously described (56).
Reagents.
Recombinant human alpha 2a interferon (IFN-α 2a), IFN-β, and IFN-γ (PBL Biomedical Laboratories, New Brunswick, NJ) were resuspended to a concentration of 50 U/μl in complete DMEM supplemented with 10% FBS, aliquoted into single-use tubes, and stored at −80°C. Cyclosporine (CsA) (41) and naringenin (40) were purchased from Sigma (St. Louis, MO) and resuspended to concentrations of 10 mM and 50 mM, respectively, in dimethyl sulfoxide (DMSO) (Sigma). Mycophenolic acid (MA) (17) (Sigma) was resuspended to a concentration of 50 mM in 95% ethyl alcohol. N-Butyldeoxynojirimycin (NB-DNJ) (47) (Sigma) was resuspended to a concentration of 25 mM in distilled H2O. The nucleoside polymerase inhibitor NM107 (36, 37), a gift from Michael J. Otto at Pharmasset, Inc., Princeton, NJ, was resuspended to a concentration of 10 mM in DMSO (Sigma). Reagents were aliquoted into single-use tubes and stored at −20°C. Anti-HCV E2 glycoprotein monoclonal antibody (C1), a gift from Dennis Burton at The Scripps Research Institute, La Jolla, CA, has previously been described (28, 56). The anti-human CD81 monoclonal antibody was purchased from Serotec (Raleigh, NC). Recombinant HCV NS3/4A protease was purchased from Anaspec (San Jose, CA). When added to cells, all reagents were diluted to a specific concentration in cDMEM containing a final DMSO concentration of 1%. Although the inhibitor concentrations chosen were in part determined based on previously published reports, it is relevant to note that these past studies were usually conducted using HCV subgenomic replicons of various genotypes in actively dividing cells, and thus, the reported 50% inhibitory concentrations cannot be directly compared.
The 5-FAM/QXL 520 NS3 FRET substrate (Anaspec) is an internally quenched peptide with a fluorescent donor (5-carboxyfluorescein [5-FAM]) and acceptor (QXL) on opposing sides of the NS3 protease cleavage site. This peptide, like others (12, 21, 25, 35, 43), is modeled upon the NS4A/NS4B site-derived (DEMEECASHL) depsipeptide substrate (5) and has been used to identify inhibitors of HCV NS3 protease activity (44). Specific for the 5-FAM/QXL 520 NS3 FRET substrate, the donor (5-FAM) absorbs energy at 490 nm and emits energy (i.e., fluorescence) at 520 nm. However, when in close contact on an intact peptide, the acceptor (QXL) absorbs the 520 nm energy emitted by the donor, preventing fluorescence. Cleavage of the peptide increases the distance between the fluorophores, resulting in proportional 5-FAM fluorescence. This NS3 FRET substrate allows for enzymatic assays to be performed at high wavelengths, providing increased fluorescence quantum yield, diminished autofluorescence (commonly detected with other fluorophores, such as EDANS), and more sensitivity than other NS3 FRET substrates (12, 21, 25, 35, 43), allowing for the detection of as little as 0.1 pmol of HCV NS3 protease.
Non-HTS HCV infection kinetics assay.
Huh7 cells were seeded at 7 × 104 cells in each well of a 12-well plate (BD Biosciences, San Jose, CA). At 24 h postseeding, cells were infected with JFH-1 HCVcc at an MOI of 0.01 FFU/cell in a total volume of 1 ml cDMEM. Throughout the course of the experiment, infected cells were trypsinized just before reaching confluence and replated at a dilution of 1:3 to maintain active cell growth. At indicated times postinfection (p.i.), medium was harvested from wells for infectivity titration analysis, RNA was isolated from triplicate wells for reverse transcription followed by real-time quantitative PCR (RTqPCR) analysis, and protein was isolated for Western blot (WB) analysis.
RNA isolation and RTqPCR analysis.
Total cellular RNA was isolated using a 1× nucleic acid purification lysis solution (Applied Biosystems, Foster City, CA) and purified using an ABI Prism 6100 nucleic acid PrepStation (Applied Biosystems), per the manufacturer's instructions. One microgram of purified RNA was used for cDNA synthesis using the TaqMan reverse transcription reagents (Applied Biosystems), and a SYBR green RTqPCR was performed using an Applied Biosystems 7300 real-time thermocycler (Applied Biosystems). Thermal cycling consisted of an initial 10-min denaturation step at 95°C followed by 40 cycles of denaturation (15 s at 95°C) and annealing/extension (1 min at 60°C). HCV JFH-1 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) transcript levels were determined relative to a standard curve comprised of serial dilutions of plasmid containing the JFH-1 HCV cDNA or the human GAPDH gene, respectively. The PCR primers used to detect GAPDH and HCV were human GAPDH (NMX002046) (5′-GAAGGTGAAGGTCGGAGTC-3′ [sense] and 5′-GAAGATGGTGATGGGATTTC-3′ [antisense]) and JFH-1 HCV (AB047639) (5′-TCTGCGGAACCGGTGAGTA-3′ [sense] and 5′-TCAGGCAGTACCACAAGGC-3′ [antisense]).
Extracellular infectivity titration assay.
Cell supernatants were serially diluted 10-fold in cDMEM, and 100 μl was used to infect, in triplicate, 4 × 103 naïve Huh7 cells per well in 96-well plates (BD Biosciences). The inoculum was incubated with cells for 24 h at 37°C and then laid over with 150 μl cDMEM containing 0.4% (wt/vol) methylcellulose (Fluka BioChemika, Switzerland) to give a final concentration of 0.25% methylcellulose. Seventy-two h p.i., medium was removed, cells were fixed with 4% paraformaldehyde (Sigma), and immunohistochemical staining for HCV E2 was performed. Briefly, cells were first incubated with 1× PBS containing 0.3% (vol/vol) hydrogen peroxide (Fisher, Fairlawn, NJ) to block endogenous peroxidase. After the cells were rinsed three times with 1× PBS, cells were blocked for 1 h with 1× PBS containing 0.5% (vol/vol) Triton X-100 (Fisher), 3% (wt/vol) bovine serum albumin (Sigma), and 10% (vol/vol) FBS. The HCV E2 glycoprotein was detected by incubation at room temperature with 1× PBS containing 0.5% (vol/vol) Triton X-100 and 3% (wt/vol) bovine serum albumin and a 1:500 dilution of the human monoclonal anti-HCV E2 antibody C1. Bound C1 was subsequently detected by a 1-h incubation with a 1:1,000 dilution of a horseradish peroxidase-conjugated anti-human antibody (Pierce, Rockford, IL) followed by a 30-min incubation with an AEC detection substrate (BD Biosciences). Cells were washed with distilled H2O and visualized using a Zeiss Axiovert microscope (Carl Zeiss, Germany). Viral infectivity titers are expressed as numbers of FFU per milliliter of supernatant (FFU/ml), determined by the average number of E2-positive foci detected in triplicate samples at the highest HCV-positive dilution.
WB analysis.
Cells were harvested in 1.25% Triton X-100 lysis buffer (Triton X-100; 50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 2 mM EDTA) supplemented with a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Fifty micrograms of protein was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Hybond nitrocellulose membranes (Amersham Pharmacia, Piscataway, NJ). Membranes were sequentially blocked with 5% nonfat milk, incubated with a 1:1,000 dilution of either a monoclonal mouse anti-HCV NS3 antibody (clone 9-G2; ViroGen, Watertown, MA) or a monoclonal mouse anti-HCV core antibody (clone C7-50; Affinity BioReagents, Rockford, IL), washed three times with PBS-0.05% Tween 20, incubated with horseradish peroxidase-conjugated goat anti-mouse antibody (Pierce, Rockford, IL), and washed again. Bound antibody complexes were detected with SuperSignal chemiluminescent substrate (Pierce).
High-throughput HCVcc FRET assay.
Huh7 cells were seeded in 96-well BioCoat culture black plates with clear bottoms (BD Biosciences) at a density of 8 × 103 cells/well in cDMEM. Upon reaching 90% confluence, medium was replaced with 200 μl cDMEM supplemented with 1% DMSO (Sigma), and cells were cultured for an additional 20 days, with the medium replaced every 2 days as previously described (9, 46). For inhibition experiments, cultures were inoculated with HCVcc JFH-1 at an MOI of 0.05 FFU/cell. Unless otherwise indicated, uninfected and infected cultures were mock treated or treated with specified compounds 0, 2, and 4 days p.i. On day 6 p.i., medium was removed from culture plates and cells were lysed in 50 μl 1.25% Triton X-100 lysis buffer (Triton X-100; 50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 2 mM EDTA) and immediately frozen (−80°C). Initially, a panel of lysis buffers and conditions was rigorously tested to determine the optimal parameters to ensure maximum lysis with a minimum nonspecific FRET background. Based on these analyses, we concluded that cultures could be immediately lysed using 1.25% Triton X-100 lysis buffer, without washing them, after removal of phenol-red-containing cDMEM (see Fig. S1 in the supplemental material).
For FRET analysis, plates were brought to room temperature and then placed in a Fluostar Optima microplate reader (BMG Labtech, Durham, NC), which automatically injects into each well 50 μl of the 5-FAM/QXL 520 NS3 FRET substrate (Anaspec), diluted to a final concentration of 5 μM. Then 5-FAM dequenching was measured at 490 nm (excitation) and 520 nm (emission) for 20 cycles in kinetic mode. Reported relative fluorescence units (RFU) were determined by end-point analysis of RFU at approximately cycle 20, at which the average signal-to-noise ratio was 86.
Cytotoxicity assay.
As an important secondary screen for cytotoxicity, the Toxilight bioassay kit (Lonza, Walkersville, MD), a bioluminescence-based assay which measures adenylate kinase (AK) released from damaged cells, was used to assess drug-induced cellular toxicity, per the manufacturer's instructions. Briefly, 20 μl of supernatant was collected on day 6 p.i. and transferred to white 96-well plates (BD Biosciences). One hundred μl of AK detection reagent was then added to each well, and luminescence, expressed as relative light units (RLU), was measured (Fluostar Optima).
Calculations.
All RFU and RLU values were background subtracted (1.25% Triton X-100 lysis buffer alone and medium alone, respectively). RFU values from nontreated HCV-infected wells and RLU values from Triton X-100-treated wells were considered 100% maximum activity. Signals from other wells are expressed as a percentage of the appropriate maximum. The Z′ was calculated using the equation 1 − [(3σc+ + 3σc−)/(μc+ − μc−)], where 3σc+ is the standard deviation of the signal (nontreated), 3σc− is the standard deviation of the background (treated), μc+ is the average RFU of the signal (nontreated), and μc− is the average RFU of the background (treated).
RESULTS
HCV NS3 protein levels parallel HCVcc infection kinetics.
NS3 protease activity has previously been shown to be an accurate readout for HCV replication in replicon-based cell culture systems, providing the same calculation of the 50% effective concentration for IFN-β inhibition as that obtained by RTqPCR analysis of replicon RNA (43). To determine if viral protein levels can also be used to monitor HCVcc infection, the kinetics of HCV protein accumulation in Huh7 cells infected with HCVcc at an MOI of 0.01 FFU/cell was assessed by WB analysis and compared to HCV RNA expansion and de novo production of infectious HCVcc. Fig. 1A illustrates that HCV RNA levels and infectious virus production correlate well with HCV protein levels (Fig. 1B), in particular those of NS3, suggesting that it is possible to use NS3 protease activity as a virally encoded “enzymatic reporter” of HCVcc infection, rather than using a genetically engineered HCVcc encoding an exogenous reporter, such as luciferase (26, 48, 55).
FIG. 1.
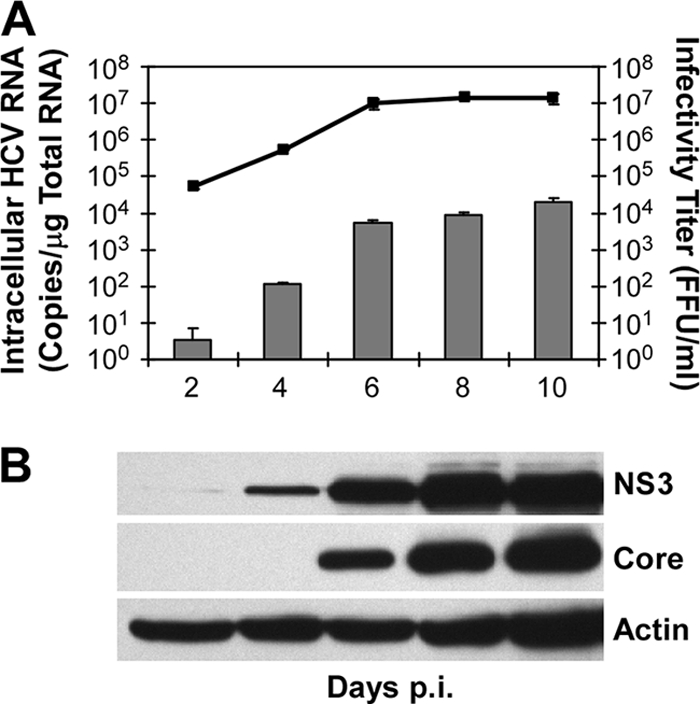
HCV NS3 protein accumulation parallels infection kinetics. (A) Huh7 cells were infected with JFH-1 HCVcc at an MOI of 0.01 FFU/cell. Culture supernatant, intracellular RNA, and cellular protein were collected at the indicated times p.i. Intracellular HCV RNA was analyzed by RTqPCR and is displayed as HCV RNA copies/μg total cellular RNA (means ± standard errors of the means; n = 3). Infectivity titers, expressed as FFU/ml, were determined by immunohistochemical analysis of 10-fold serially diluted culture supernatants on naïve Huh7 cells. The lower limit of detection of the assay was 10 FFU/ml. (B) HCV NS3, core, and cellular actin protein levels were determined by WB analysis.
NS3 FRET signal increases linearly with intracellular NS3 protein levels.
To verify the feasibility of using FRET to quantitatively measure HCV NS3 protein levels, we first confirmed that NS3-dependent cleavage of an internally quenched peptide substrate would produce a FRET signal linear with the amount of NS3 present. Purified recombinant NS3/4A protease was serially diluted and incubated with the 5-FAM/QXL 520 NS3 FRET peptide substrate, and NS3 FRET activity was measured. As shown in Fig. 2A, the FRET signal increased with increasing amounts of purified NS3, revealing a linear correlation (Fig. 2A, inset; R2 = 0.999) between NS3 protein levels and FRET activity. To determine if such a linear correlation could be achieved with intracellular NS3 protein, NS3 FRET activity was determined using lysates from serially diluted sgJFH-1 replicon cells or Huh7 cells infected with HCVcc JFH-1 at increasing MOIs (0.05, 0.10, 0.50, and 1.0 FFU/cell). Similar to the results obtained using purified NS3/4A protease, the FRET assay exhibited a linear signal (Fig. 2B and C; R2 = 0.996 and 0.999, respectively) relative to intracellular NS3 protease concentrations.
FIG. 2.

FRET signal increases linearly with NS3 protein levels. (A) Recombinant NS3 standard curve showing NS3-dependent cleavage of quenched peptide substrate produces FRET signal proportional to NS3 levels. One unit of NS3 equals 3.036 ng of recombinant NS3/4A. (B) Serial dilutions of sgJFH replicon cell lysate (equal to 8 × 103 to 1.5 × 105 cells) exhibit FRET signals linear to cell numbers. (C) Huh7 cells infected with increasing MOIs (0.05, 0.1, 0.5, and 1.0 FFU/cell) of HCVcc JFH-1 and lysed at day 6 p.i. exhibit FRET signals proportional to the infection MOI. Shown are the relative fluorescent units (RFU) minus the HCV-negative background detected at cycle 20. Insets represent linear representation of data and R2 values.
Cell-based HCV infection assay.
Having identified a suitable assay readout, we next sought to define the optimal cell culture conditions necessary for a cell-based HCV infection HTS assay. Since cell culture variability and nonspecific effects of compounds on cell growth can be a problem for cell-based HTS, particularly for HCV-based assays where confluence and changes in the state of the host cell can have a negative effect on viral replication (42, 45, 46, 51), we chose to utilize nondividing Huh7 cells for our cell-based HCV infection assay. As previously described (46), treatment of Huh7 cells with 1% DMSO for 20 days induces cell growth arrest, allowing nondividing, HCV-permissive Huh7 cells to be maintained at a stable cell number for extended periods of time (>100 days) (Fig. 3). Importantly, we show here that when replicate 96-well cultures of nondividing, G0-synchronized Huh7 cells were infected with HCVcc at an MOI of 0.05 FFU/cell, high reproducibility between wells was observed with HCV NS3 protein (FRET) and RNA (RTqPCR) at day 6 p.i. (Fig. 4A) and de novo infectious virus titers at days 3, 5, 7, and 25 p.i. (Fig. 4B), thus minimizing the well-to-well variability commonly associated with cell-based HTS assays, which typically utilize rapidly dividing unsynchronized cell cultures.
FIG. 3.

Diagram of the nondividing Huh7 cell culture system. To establish nondividing cultures, Huh7 cells are seeded at 80% confluence on collagen-coated plasticware. Upon reaching 90% confluence, media are replaced with complete DMEM supplemented with 1% DMSO, and cultures are incubated for an additional 20 days. Six days after the initiation of DMSO treatment, cultures reach steady-state cell numbers.
FIG. 4.

Reproducibility of HCV infection in nondividing Huh7 cell cultures. Huh7 cells were plated in a 96-well format, DMSO-treated for 20 days, and then infected with HCVcc JFH-1 at an MOI of 0.05 FFU/cell. (A) HCV RNA and NS3 protease activity were measured 6 days p.i. in 42 replicates. Intracellular HCV RNA was analyzed by RTqPCR and displayed as HCV RNA copies/μg total cellular RNA. NS3 protease activity, expressed as RFU, was determined by FRET analysis. (B) HCVcc infectivity titers on days 3, 5, 7, and 25 p.i., expressed as FFU/ml, were determined by immunohistochemical analysis of 10-fold serially diluted culture supernatant replicates on naïve Huh7 cells.
To determine assay conditions under which HCV NS3 protease activity can be used to quantitatively assess HCVcc infection progression, the kinetics of NS3 protease activity in DMSO-treated Huh7 cells was assessed after infection with increasing MOIs of HCVcc. Specifically, nondividing cultures of Huh7 cells were infected with HCVcc at an MOI of 0.01, 0.05, or 0.1 FFU/cell, and HCV RNA levels and NS3 protease activity were measured daily for 10 days by RTqPCR and FRET, respectively (Fig. 5A). As expected, HCV RNA levels increased exponentially from day 1 to day 8 p.i. in an MOI-dependent manner, reaching steady-state levels of ∼1 × 107 copies/μg RNA by days 6 to 8 p.i. Likewise, HCV NS3 protease activity, as determined by 5-FAM dequenching, also demonstrated a steady increase through day 10 p.i. (Fig. 5A) and then, like HCV RNA levels, remained at a constant plateau level at later time points that were examined (data not shown). Notably, similar to that of HCV RNA expansion, a linear increase in HCV NS3 protease activity up to day 8 p.i. at MOIs of 0.01 and 0.05 (R2 = 0.999 and 0.989, respectively) was observed. When plotted as a function of one another (Fig. 5B), a linear correlation between HCV RNA expansion and NS3 protease activity was observed (R2 value = 0.983), confirming that HCV NS3 protease activity directly parallels HCV RNA expansion over an extended period in which quantitative end-point HTS analyses of HCV infection can be performed.
FIG. 5.
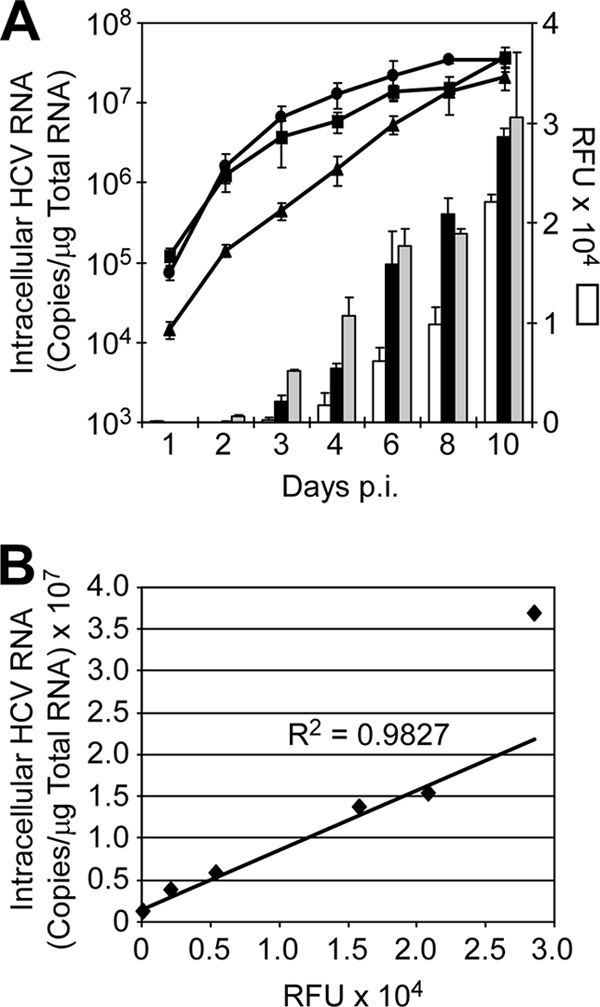
NS3 FRET analysis of HCVcc infection in nondividing Huh7 cells. (A) Huh7 cells were plated in a 96-well format, DMSO treated for 20 days, and then infected with HCVcc at MOIs of 0.01 (diamonds, white bars), 0.05 (squares, black bars), and 0.1 (circles, gray bars) FFU/cell. On indicated days p.i., triplicate wells were harvested. Intracellular HCV RNA was determined by RTqPCR (lines), and NS3 protease activity was monitored by FRET analysis (bars). Results are graphed as mean ± standard error of the mean. (B) Linear representation of HCV copies/μg RNA versus RFU generated from data points corresponding to cultures infected with HCVcc at an MOI of 0.05 FFU/cell. R2 value was determined from data points corresponding to days 1 to 8 p.i.
Low-MOI HCVcc HTS FRET assay can quantitatively identify inhibitors that target any aspect of the HCV life cycle.
Based on the ability to reproducibly perform low-MOI HCV infection over several days in nondividing Huh7 cells, we designed a novel HCV infection HTS assay whereby compounds are added during the exponential phase of HCV spread throughout the culture, with NS3 protease activity being assessed at day 6 p.i. after multiple rounds of infection and reinfection (Fig. 6A). The rationale is that, unlike studies which use a high-MOI approach and are limited to a single cycle of virus replication, inhibitors that target any aspect of the viral life cycle (e.g., entry, replication, assembly, egress, and spread) should be detectable using a low-MOI approach. To validate this HTS experimental design (Fig. 6A), we compared the ability of the cell-based FRET assay to identify inhibitors of HCV to those of standard RTqPCR and WB analyses and also confirmed that the low-MOI 6-day experimental strategy could effectively detect inhibitors that target any aspect of the viral life cycle.
FIG. 6.
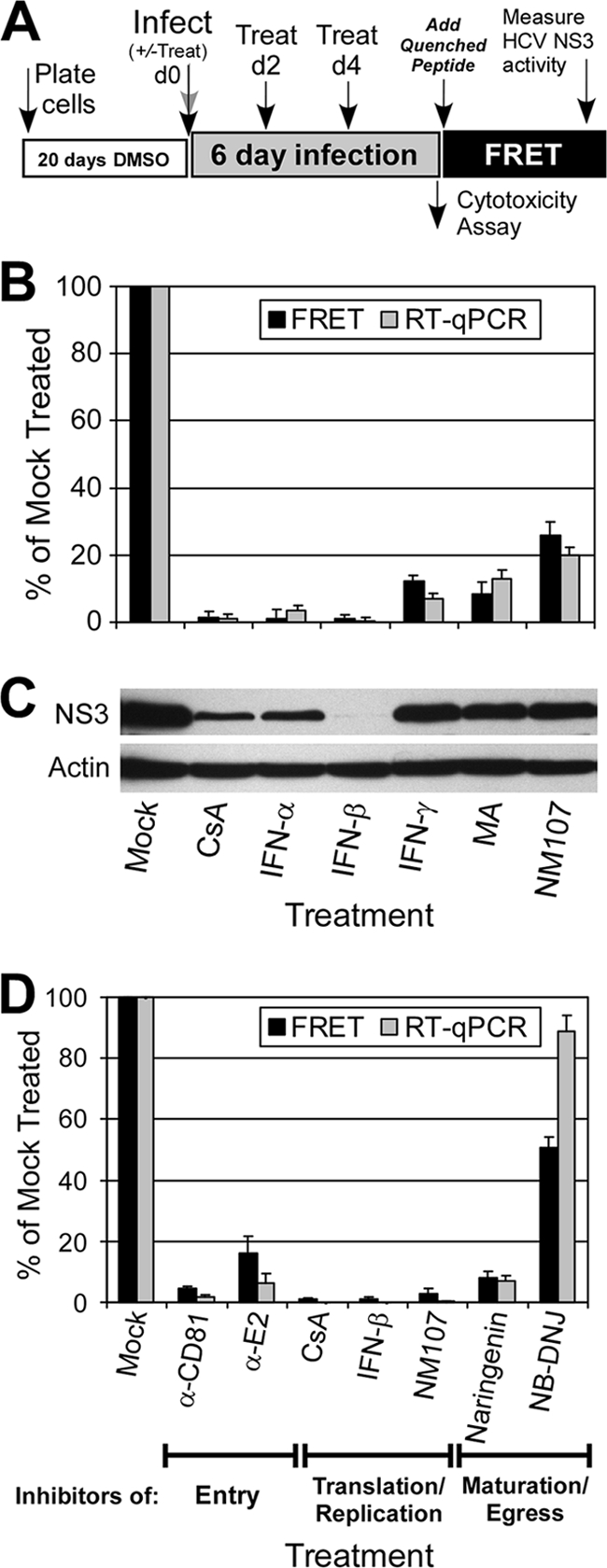
Quantitative identification of inhibitors that act throughout the HCV life cycle. (A) HTS experimental design. (B) DMSO-Huh7 cells were infected with HCVcc at 0.05 FFU/cell and treated with 2.5 μM CsA, 100 U/ml IFN-α, 100 U/ml IFN-β, 100 U/ml IFN-γ, 10 μM MA, or 18.5 μM NM107. Compounds were added at 2 days p.i. and were replenished in fresh medium at day 4 p.i. At day 6 p.i., triplicate cultures were assayed for HCV RNA levels by RTqPCR and for NS3 protein levels by FRET. Data are presented as a percentage of mock-treated cells. (C) HCV NS3 and cellular actin protein levels in parallel cultures were determined by WB analysis. (D) DMSO-Huh7 cells were infected with HCVcc at 0.05 FFU/cell and treated with HCV inhibitors that act at different stages of HCV infection: 50 μg/ml α-CD81, 100 μg/ml α-E2, 2.5 μM CsA, 250 U/ml IFN-β, 18.5 μM NM107, 200 μM naringenin, and 500 μM NB-DNJ. Compounds were added at the time of infection and were replenished every 2 days over the 6-day assay. At day 6 p.i., triplicate cultures were assayed for HCV RNA levels by RTqPCR and for NS3 protein levels by FRET. Data are presented as a percentage of mock-treated cells.
First, using known HCV inhibitors (17, 36, 41, 56), we simply tested whether the HCV NS3 FRET assay was able to identify HCV inhibitors analogous to those of non-HTS assays, such as RTqPCR (Fig. 6B) and WB (Fig. 6C). For this, we used IFN-α, -β, and -γ and three HCV replication inhibitors: the immunosuppressive drugs CsA (41) and MA (17) and the HCV-specific nucleoside polymerase inhibitor NM107 (36, 37). Impressively, in the case of IFN-α and -β and CsA, both the NS3 FRET assay and RTqPCR indicated over 98% inhibition. Likewise, both assays measured a comparable inhibition range of 74 to 93% for IFN-γ, MA, and NM107. While less quantitative in nature, WB analysis of NS3 proteins levels also nicely paralleled the NS3 FRET protease activity detected, demonstrating equivalency between the HCV NS3 FRET assay and standard analyses in accurately identifying HCV inhibitors at the level of percentage of inhibition.
To confirm that our low-MOI (e.g., 0.05-FFU/cell) 6-day infection strategy would effectively identify inhibitors that target any aspect of the viral life cycle (e.g., entry, replication, assembly, egress, and spread), we further measured HCV NS3 activity following treatment of cells with a panel of HCV antivirals shown to target different aspects of the viral life cycle (17, 36, 40, 41, 47, 56). As illustrated in Fig. 6A, DMSO-Huh7 cells were infected with HCVcc at 0.05 FFU/cell; compounds were added either at the time of infection (co-) or 2 days p.i. and replenished every 2 days over the 6-day assay; and HCV RNA and NS3 protein levels were measured by RTqPCR and by FRET, respectively, 6 days p.i. Notably, when added either at the time of infection (Fig. 6D) or 2 days p.i. (see Fig. S2 in the supplemental material), all tested inhibitors efficiently reduced both HCV RNA replication and NS3 protease activity to equivalent levels; however, as expected, the effect of inhibitors that targeted HCV entry (i.e., α-CD81 and α-E2) was less pronounced when added p.i. (see Fig. S2 in the supplemental material).
In addition, since secondary toxicity screens are a necessary component of any HTS campaign, we incorporated a luminescence-based cellular toxicity assay (ToxiLight; Lonza) to assess compound-mediated cytotoxicity. Notably, since this assay quantitatively measures AK release into the culture medium from damaged cells, cellular toxicity and FRET can be measured from the same well by simply removing 20 μl of the culture medium prior to cell lysis (Fig. 6A). Using this assay, we confirmed that none of the compounds tested exhibited any nonspecific cytotoxic effect, compared to a positive control culture treated with 10% Triton X-100 (see Fig. S2 in the supplemental material). Thus, these data together demonstrate the utility of this cell-based HCVcc HTS assay for identifying inhibitors that target all aspects of the viral life cycle and the compatibility of the assay design for assessing compound-mediated cytotoxicity.
Statistical validation of HCV FRET assay performance.
The quality of an HTS assay can be determined according to its primary goal, which is to distinguish hits from nonhits. The Z′ statistic is a measure of the distance between the standard deviations for the positive (signal) and negative (noise) controls of the assay. This value not only reflects the size of the window between the positive and negative controls but also assesses the noise/error associated with the control assays. To determine the Z′ value of our cell-based HCVcc FRET assay, full plates containing untreated and treated samples were analyzed. IFN-β was used as a positive control inhibitor of HCV replication, and the Z′ value was calculated using the equation shown in Fig. 7A. The data for three representative plates are graphically plotted (Fig. 7B to D), showing the respective Z′ values of 0.604, 0.643, and 0.654 obtained for each plate and an average signal-to-background ratio of 7. Notably, similar Z′ values (i.e., ≥0.6) were obtained when CsA was used as an alternate inhibitor (data not shown). Taken together, these data indicate an acceptable signal-to-noise window and therefore satisfy the criteria for an HTS assay.
FIG. 7.
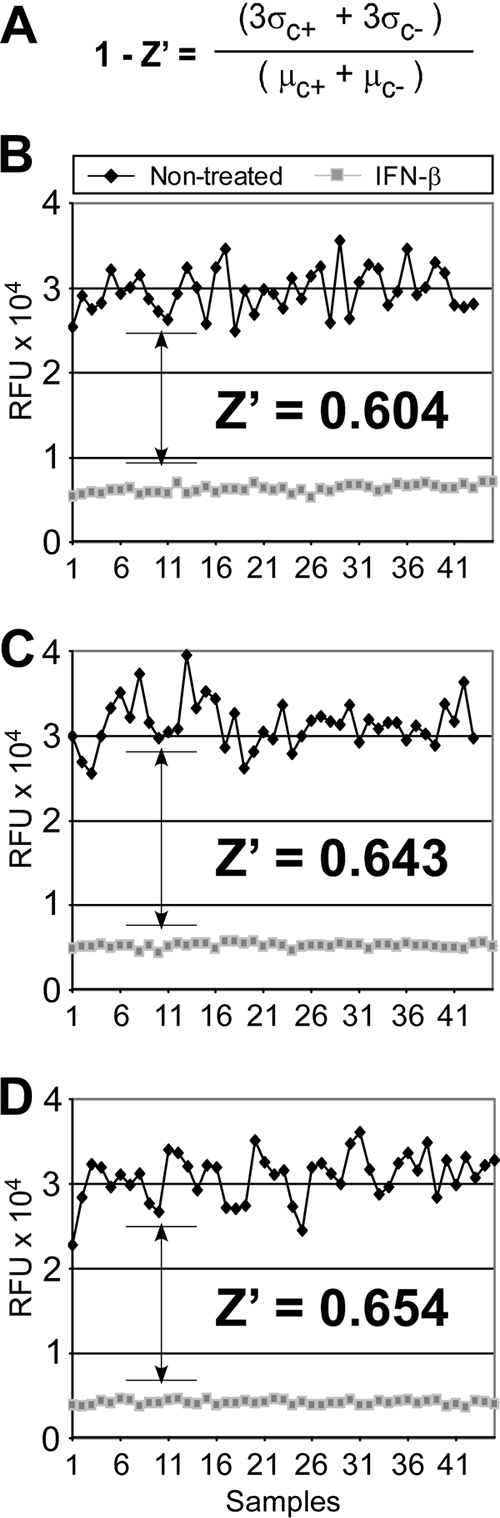
HCV FRET assay Z′ analysis. (A) The Z′ equation used to measure the distance between the standard deviations for the positive (signal) and negative (noise) controls of the assay (σ, standard deviation; μ, average; C+, positive controls; C−, negative controls). (B to D) Graphical representation of FRET signal (RFU) from three representative plates (n = 46 separate samples) used to calculate the Z′ per plate. HCV-infected 96-well plates containing untreated and treated samples were analyzed. IFN-β (250 U/ml) was used as a positive inhibitor of HCV replication, as it consistently and reproducibly reduced NS3 protease activity. The standard deviations and the number distribution between the end-point signals obtained for the controls (untreated) versus the background (IFN treated) were measured, and the Z′ statistic was calculated.
DISCUSSION
In this study we report the development of a FRET-based low-MOI HCVcc infection assay suitable for HTS of compound libraries. By combining the recently developed infectious HCV cell culture system (31, 50, 56) and novel nondividing Huh7 cell culture system (9, 46) with a sensitive FRET-based readout for an endogenous, virally encoded enzyme, we have established an HTS platform that offers several distinct advantages over previously reported HCV drug discovery strategies, as discussed in detail below.
Cell-based HCV enzymatic reporter assay.
A fundamental consideration for screening libraries of potential HCV antiviral compounds is the development of an assay that is sensitive, cost effective, and easily adapted to automation. Although analysis of HCV RNA levels by RTqPCR and infectious particle secretion by limited dilution assays are accurate determinants of HCVcc infection, these assays are not readily amenable to HTS. As such, HCV replicon and HCVcc constructs with exogenous foreign reporters, such as luciferase, secreted alkaline phosphatase, chloramphenicol transferase, beta-lactamase, or beta-galactosidase, have been developed widely as alternative ways of measuring HCV replicon replication (14, 18, 27, 39, 52, 53) and HCVcc infection (55) for HTS. Although many of these enzymatic reporter-based assays have been adapted for HCV replicon-based HTS campaigns, inherent issues such as specificity (i.e., effect of compound on reporter expression/function) and the effects that foreign sequences might have on HCV infection (e.g., decreases in HCV infection efficiency) can be even more problematic when developing an HCV-based enzymatic-reporter HTS assay. Therefore, alternatives to foreign enzymatic reporters would be preferable for the advancement of cell-based HCVcc HTS.
To address this issue, we adapted an NS3-based FRET assay originally reported by O'Boyle et al. in 2005 in which they used HCV NS3 protease activity as a “virally encoded reporter” in an HCV replicon-based system (43). However, rather than using NS3 protease activity as a means to quantify reduction in HCV steady-state levels analogous to the O'Boyle replicon system, we determined that NS3 protease activity parallels HCV infection kinetics (Fig. 1 and 5) and designed a low-MOI infection assay approach in which all aspects of the viral life cycle could be monitored by NS3 protease activity. To achieve the signal necessary for a 96-well or smaller format, we incorporated a more sensitive 5-FAM/QXL 520 NS3 FRET substrate, which offers increased fluorescence quantum yield, with reduced cell background autofluorescence. Using this substrate, we confirmed that the FRET signal that was produced was linear with the amount of NS3 present and that NS3 protease activity could be measured quantitatively throughout infection (Fig. 2 and 5), allowing us to conclude that NS3 protease activity is an acceptable endogenous reporter for assaying HCVcc infection in vitro.
Ability to detect compounds that inhibit any aspect of HCV infection.
Unlike HCV replicon systems that specifically assay viral RNA replication, the infectious HCV cell culture system recapitulates all aspects of the viral life cycle, such as binding, entry, uncoating, translation, replication, assembly, maturation, and egress. This is a considerable advantage, as it provides the opportunity to identify compounds that inhibit any step in the viral life cycle (Fig. 6). Notably, however, while in vitro virus screening assays are often performed at a high MOI (e.g., 10 to 20 FFU/cell) over a short period of time, resulting in a synchronized single-cycle round of viral replication, which maximizes signal and minimizes cell culture variability over time, this approach is biased, in that inhibitors of entry and replication are more readily identified than compounds that inhibit later viral infection processes, such as maturation, egress, and any subsequent cell-to-cell spread. To avoid this predisposition and capitalize on the opportunity that the infectious HCV cell culture system presents, our HTS campaign (Fig. 6A) is uniquely based on a low-MOI (e.g., 0.05 FFU/cell) 6-day infection assay during which multiple rounds of the complete viral life cycle are required for spread of HCV throughout the culture, corresponding with a proportional linear increase in NS3 protein accumulation (Fig. 5A and B). Importantly, HCVcc does not grow to extremely high titers in vitro; therefore, this approach also represents a very practical solution, in that less viral inoculum is needed to achieve a sufficiently high signal for detection.
Use of nondividing, synchronized cell cultures for HTS.
A critical key to the successful development and execution of our low-MOI, 6-day infection assay was the incorporation of nondividing Huh7 cells (Fig. 3). Not only does the growth-arrested steady-state nature of these cells virtually eliminate the complications inherent in cell-based HTS assays, such as cell culture-related variability from well to well (Fig. 4), it additionally allows for high reproducibility and robust HCV infection over an extended period of time rather than only a few days (Fig. 4B and 6), making it feasible to adapt longer-term HCV infection strategies for HTS, a feature not previously afforded by other HCV-based HTS assays (16, 19, 24, 55).
The use of these well-characterized (9, 46), nondividing Huh7 cell cultures also imparts several other potential advantages. Aside from the ease with which these ready-for-use cultures can be maintained and their inherent tolerability of the common compound library diluent DMSO, a more tangible benefit is that these cell cultures are resistant to many of the nonspecific effects some compounds can have on the growth and viability of actively dividing cell cultures, which routinely result in false-positive hits. While this certainly does not eliminate the need to screen for compound cytotoxicity, the fact that the cultures are maintained in a quiescent nonproliferating state does make additional secondary screens to monitor compound-mediated effects on cell growth unnecessary. Lastly, culturing Huh7 cells under these nondividing conditions also results in enhanced Huh7 cell differentiation, with the upregulation of liver-specific gene expression (9, 46) and hepatocyte-specific phase I and phase II drug metabolism functions (9). The use of metabolically active cells would prove highly beneficial when screening prodrug compounds, which need to be metabolized to an active form in order to exert their potential antiviral effect. Hence, while all the benefits that these more physiologically relevant nondividing Huh7 cell cultures may contribute to our HTS HCV drug discovery efforts are still unknown, it is clear that the advantages are numerous and significant.
Cell-based HCVcc FRET assay performance.
Because a central feature of our HTS assay relates to the identification of inhibitors that target all steps in the viral life cycle, we not only compared the performance of the assay to those of standard RTqPCR and WB analyses to determine the equivalency between the assay methods, but also validated the assay by testing compounds that target HCV entry, replication, or egress. Importantly, inhibitors targeting all aspects of the HCV viral life cycle scored positively using the HCVcc FRET assay, and the level of inhibition achieved was comparable to that obtained by RTqPCR and/or WB analysis (Fig. 6B to D). As might be expected, we observed that the ability to detect inhibitors of HCV entry was more dependent on whether compounds were added coinfection or p.i. (i.e., 0, 2, and 4 versus 2 and 4 days p.i.) compared to that of inhibitors targeting postentry steps (replication and egress). Notably, however, the fact that inhibition was still observed when treatment with entry inhibitors was initiated 2 days p.i. further attests to the effectiveness of the low-MOI HTS approach we have adopted (see Fig. S2 in the supplemental material).
While the iminosugar NB-DNJ has been shown to inhibit HCVcc Jc1 spread by ∼80% in Huh7.5-infected cells presumably by affecting viral glycoprotein glycosylation, here we observed a more modest effect by RTqPCR and FRET analysis. This difference in potency may be due to differences in the basal activity levels of and/or intracellular competition for the related cellular processes in nondividing versus dividing Huh7 cell cultures or differences in the HCV clones utilized. If indeed, nondividing hepatocytes are less sensitive to perturbations in the protein glycosylation pathway, this might represent an example of how the use of nondividing Huh7 cells might be advantageous for identifying compounds more likely to be effective in vivo. Alternatively, because the Jc1 is an intragenotypic JFH-1 chimera that expresses envelope glycoproteins from the HCV J6 clone, it may be that strain-specific variability within the viral glycoproteins results in differential susceptibility to the inhibitory effects of NB-DNJ. The latter possibility highlights a fundamental issue inherent in any HCV HTS campaign and demonstrates why incorporation of secondary screens utilizing HCVcc chimeras expressing genotypically diverse glycoproteins into any HCVcc-based HTS assay would be prudent to ensure identification of inhibitors of all genotypes.
Lastly, we determined the accuracy of our HTS assay by measuring the Z′ factor. The Z′ factor is a value that measures the quality of an HTS assay by assessing the dynamic range of the assay as well as the variation within the assay by measuring the distance between the standard deviations for the positive (signal) and negative (noise) controls. This value also assesses the noise/error associated with the assay. For an HTS to be considered accurate, a Z′ value of greater than 0.5 is necessary. In our study, the Z′ values achieved using the HCVcc FRET assay were consistently greater than 0.6 (Fig. 7), which is particularly important for fluorescence-based assays which typically have lower signal-to-background ratios (54). Taken together, these data indicate that the HCVcc FRET HTS assay is an accurate and reproducible system with low variability and highly suitable for HTS.
In summary, our study describes the first cell-based HCVcc FRET assay, which incorporates nondividing Huh7 cells and allows for the identification of inhibitors that target all aspects of the viral life cycle. Since it is also highly compatible for measuring compound-mediated toxicity, this unique cell-based HCVcc FRET HTS assay should therefore facilitate the rapid identification and development of new and novel HCV therapeutics. Notably, with the design of alternative peptide substrates, this HTS strategy could also be readily applicable for the screening of antiviral compounds against other viruses which encode an endogenous protease (e.g., human immunodeficiency virus, dengue, enteroviruses, Norwalk virus, rhinovirus, severe acute respiratory syndrome coronavirus, rubella, and others).
Supplementary Material
Acknowledgments
We thank Peter Corcoran for outstanding technical assistance and the members of the Uprichard lab for helpful discussions.
This work was supported by the National Institutes of Health Public Health Service grant R01-AI070827 and the University of Illinois Chicago Council to support Gastrointestinal and Liver Disease (UIC GILD).
Footnotes
Published ahead of print on 20 July 2009.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Ahmed, A., and E. B. Keeffe. 1999. Treatment strategies for chronic hepatitis C: update since the 1997 National Institutes of Health Consensus Development Conference. J. Gastroenterol. Hepatol. 14(Suppl.):S12-S8. [DOI] [PubMed] [Google Scholar]
- 2.Alter, H. J., and L. B. Seeff. 2000. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis. 20:17-35. [DOI] [PubMed] [Google Scholar]
- 3.Bartosch, B., J. Dubuisson, and F. L. Cosset. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beames, B., D. Chavez, and R. E. Lanford. 2001. GB virus B as a model for hepatitis C virus. ILAR J. 42:152-160. [DOI] [PubMed] [Google Scholar]
- 5.Bianchi, E., C. Steinkuhler, M. Taliani, A. Urbani, R. D. Francesco, and A. Pessi. 1996. Synthetic depsipeptide substrates for the assay of human hepatitis C virus protease. Anal. Biochem. 237:239-244. [DOI] [PubMed] [Google Scholar]
- 6.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1975. [DOI] [PubMed] [Google Scholar]
- 7.Blight, K. J., J. A. McKeating, J. Marcotrigiano, and C. M. Rice. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181-3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourne, N., R. B. Pyles, M. Yi, R. L. Veselenak, M. M. Davis, and S. M. Lemon. 2005. Screening for hepatitis C virus antiviral activity with a cell-based secreted alkaline phosphatase reporter replicon system. Antivir. Res. 67:76-82. [DOI] [PubMed] [Google Scholar]
- 9.Choi, S., B. Sainz, Jr., P. Corcoran, S. L. Uprichard, and H. Jeong. 2009. Characterization of increased drug metabolism activity in dimethyl sulfoxide (DMSO)-treated Huh7 hepatoma cells. Xenobiotica 39:205-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choo, Q.-L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359-362. [DOI] [PubMed] [Google Scholar]
- 11.Dansako, H., M. Ikeda, K. Abe, K. Mori, K. Takemoto, Y. Ariumi, and N. Kato. 2008. A new living cell-based assay system for monitoring genome-length hepatitis C virus RNA replication. Virus Res. 137:72-79. [DOI] [PubMed] [Google Scholar]
- 12.Fattori, D., A. Urbani, M. Brunetti, R. Ingenito, A. Pessi, K. Prendergast, F. Narjes, V. G. Matassa, R. De Francesco, and C. Steinkuhler. 2000. Probing the active site of the hepatitis C virus serine protease by fluorescence resonance energy transfer. J. Biol. Chem. 275:15106-15113. [DOI] [PubMed] [Google Scholar]
- 13.Gastaminza, P., S. B. Kapadia, and F. V. Chisari. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 80:11074-11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goergen, B., S. Jakobs, P. Symmons, E. Hornes, K. H. Meyer zum Buschenfelde, and G. Gerken. 1994. Quantitation of HCV-replication using one-step competitive reverse transcription-polymerase chain reaction and a solid phase, colorimetric detection method. J. Hepatol. 21:678-682. [DOI] [PubMed] [Google Scholar]
- 15.Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487-5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao, W., K. J. Herlihy, N. J. Zhang, S. A. Fuhrman, C. Doan, A. K. Patick, and R. Duggal. 2007. Development of a novel dicistronic reporter-selectable hepatitis C virus replicon suitable for high-throughput inhibitor screening. Antimicrob. Agents Chemother. 51:95-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henry, S. D., H. J. Metselaar, R. C. Lonsdale, A. Kok, B. L. Haagmans, H. W. Tilanus, and L. J. van der Laan. 2006. Mycophenolic acid inhibits hepatitis C virus replication and acts in synergy with cyclosporin A and interferon-alpha. Gastroenterology 131:1452-1462. [DOI] [PubMed] [Google Scholar]
- 18.Hirowatari, Y., M. Hijikata, and K. Shimotohno. 1995. A novel method for analysis of viral proteinase activity encoded by hepatitis C virus in cultured cells. Anal. Biochem. 225:113-120. [DOI] [PubMed] [Google Scholar]
- 19.Huang, P., D. A. Goff, Q. Huang, A. Martinez, X. Xu, S. Crowder, S. D. Issakani, E. Anderson, N. Sheng, P. Achacoso, A. Yen, T. Kinsella, I. S. Darwish, R. Kolluri, H. Hong, K. Qu, E. Stauffer, E. Goldstein, R. Singh, D. G. Payan, and H. H. Lu. 2008. Discovery and characterization of substituted diphenyl heterocyclic compounds as potent and selective inhibitors of hepatitis C virus replication. Antimicrob. Agents Chemother. 52:1419-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda, M., M. Yi, K. Li, and S. M. Lemon. 2002. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 76:2997-3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kakiuchi, N., S. Nishikawa, M. Hattori, and K. Shimotohno. 1999. A high throughput assay of the hepatitis C virus nonstructural protein 3 serine proteinase. J. Virol. Methods 80:77-84. [DOI] [PubMed] [Google Scholar]
- 22.Kato, T., T. Date, M. Miyamoto, A. Furusaka, K. Tokushige, M. Mizokami, and T. Wakita. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808-1817. [DOI] [PubMed] [Google Scholar]
- 23.Kato, T., A. Furusaka, M. Miyamoto, T. Date, K. Yasui, J. Hiramoto, K. Nagayama, T. Tanaka, and T. Wakita. 2001. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J. Med. Virol. 64:334-339. [DOI] [PubMed] [Google Scholar]
- 24.Kim, S. S., L. F. Peng, W. Lin, W. H. Choe, N. Sakamoto, N. Kato, M. Ikeda, S. L. Schreiber, and R. T. Chung. 2007. A cell-based, high-throughput screen for small molecule regulators of hepatitis C virus replication. Gastroenterology 132:311-320. [DOI] [PubMed] [Google Scholar]
- 25.Konstantinidis, A. K., P. L. Richardson, K. A. Kurtz, R. Tripathi, C. M. Chen, P. Huang, J. Randolph, D. Towne, J. Donnelly, U. Warrior, T. Middleton, and W. M. Kati. 2007. Longer wavelength fluorescence resonance energy transfer depsipeptide substrates for hepatitis C virus NS3 protease. Anal. Biochem. 368:156-167. [DOI] [PubMed] [Google Scholar]
- 26.Koutsoudakis, G., A. Kaul, E. Steinmann, S. Kallis, V. Lohmann, T. Pietschmann, and R. Bartenschlager. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 80:5308-5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krieger, N., V. Lohmann, and R. Bartenschlager. 2001. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75:4614-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Law, M., T. Maruyama, J. Lewis, E. Giang, A. W. Tarr, Z. Stamataki, P. Gastaminza, F. V. Chisari, I. M. Jones, R. I. Fox, J. K. Ball, J. A. McKeating, N. M. Kneteman, and D. R. Burton. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25-27. [DOI] [PubMed] [Google Scholar]
- 29.Lee, J. C., Y. F. Shih, S. P. Hsu, T. Y. Chang, L. H. Chen, and J. T. Hsu. 2003. Development of a cell-based assay for monitoring specific hepatitis C virus NS3/4A protease activity in mammalian cells. Anal. Biochem. 316:162-170. [DOI] [PubMed] [Google Scholar]
- 30.Lee, J. C., M. C. Yu, T. W. Lien, C. F. Chang, and J. T. Hsu. 2005. High-throughput cell-based screening for hepatitis C virus NS3/4A protease inhibitors. Assay Drug Dev. Technol. 3:385-392. [DOI] [PubMed] [Google Scholar]
- 31.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623-626. [DOI] [PubMed] [Google Scholar]
- 32.Lindenbach, B. D., and C. M. Rice. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436:933-938. [DOI] [PubMed] [Google Scholar]
- 33.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 34.Mao, H. X., S. Y. Lan, Y. W. Hu, L. Xiang, and Z. H. Yuan. 2003. Establishment of a cell-based assay system for hepatitis C virus serine protease and its primary applications. World J. Gastroenterol. 9:2474-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao, S. S., J. DiMuzio, C. McHale, C. Burlein, D. Olsen, and S. S. Carroll. 2008. A time-resolved, internally quenched fluorescence assay to characterize inhibition of hepatitis C virus nonstructural protein 3-4A protease at low enzyme concentrations. Anal. Biochem. 373:1-8. [DOI] [PubMed] [Google Scholar]
- 36.Mathy, J. E., S. Ma, T. Compton, and K. Lin. 2008. Combinations of cyclophilin inhibitor NIM811 with hepatitis C Virus NS3-4A Protease or NS5B polymerase inhibitors enhance antiviral activity and suppress the emergence of resistance. Antimicrob. Agents Chemother. 52:3267-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCown, M. F., S. Rajyaguru, S. Le Pogam, S. Ali, W. R. Jiang, H. Kang, J. Symons, N. Cammack, and I. Najera. 2008. The hepatitis C virus replicon presents a higher barrier to resistance to nucleoside analogs than to nonnucleoside polymerase or protease inhibitors. Antimicrob. Agents Chemother. 52:1604-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mondal, R., G. Koev, T. Pilot-Matias, Y. He, T. Ng, W. Kati, and A. Molla. 2009. Development of a cell-based assay for high-throughput screening of inhibitors against HCV genotypes 1a and 1b in a single well. Antivir. Res. 82:82-88. [DOI] [PubMed] [Google Scholar]
- 39.Murray, E. M., J. A. Grobler, E. J. Markel, M. F. Pagnoni, G. Paonessa, A. J. Simon, and O. A. Flores. 2003. Persistent replication of hepatitis C virus replicons expressing the beta-lactamase reporter in subpopulations of highly permissive Huh7 cells. J. Virol. 77:2928-2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahmias, Y., J. Goldwasser, M. Casali, D. van Poll, T. Wakita, R. T. Chung, and M. L. Yarmush. 2008. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 47:1437-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa, M., N. Sakamoto, N. Enomoto, Y. Tanabe, N. Kanazawa, T. Koyama, M. Kurosaki, S. Maekawa, T. Yamashiro, C. H. Chen, Y. Itsui, S. Kakinuma, and M. Watanabe. 2004. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 313:42-47. [DOI] [PubMed] [Google Scholar]
- 42.Nelson, H. B., and H. Tang. 2006. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence-based inhibition of HCV RNA and protein expression. J. Virol. 80:1181-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Boyle, D. R., II, P. T. Nower, J. A. Lemm, L. Valera, J. H. Sun, K. Rigat, R. Colonno, and M. Gao. 2005. Development of a cell-based high-throughput specificity screen using a hepatitis C virus-bovine viral diarrhea virus dual replicon assay. Antimicrob. Agents Chemother. 49:1346-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phuong do, T., C. M. Ma, M. Hattori, and J. S. Jin. 2009. Inhibitory effects of antrodins A-E from Antrodia cinnamomea and their metabolites on hepatitis C virus protease. Phytother Res. 23:582-584. [DOI] [PubMed] [Google Scholar]
- 45.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75:1252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sainz, B., Jr., and F. V. Chisari. 2006. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J. Virol. 80:10253-10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinmann, E., T. Whitfield, S. Kallis, R. A. Dwek, N. Zitzmann, T. Pietschmann, and R. Bartenschlager. 2007. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 46:330-338. [DOI] [PubMed] [Google Scholar]
- 48.Tscherne, D. M., C. T. Jones, M. J. Evans, B. D. Lindenbach, J. A. McKeating, and C. M. Rice. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 80:1734-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uprichard, S. L., J. Chung, F. V. Chisari, and T. Wakita. 2006. Replication of a hepatitis C virus replicon clone in mouse cells. Virol. J. 3:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Windisch, M. P., M. Frese, A. Kaul, M. Trippler, V. Lohmann, and R. Bartenschlager. 2005. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 79:13778-13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi, M., F. Bodola, and S. M. Lemon. 2002. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein. Virology 304:197-210. [DOI] [PubMed] [Google Scholar]
- 53.Yi, M., and S. Lemon. 2002. Replication of subgenomic hepatitis A virus RNAs expressing firefly luciferase is enhanced by mutations associated with adaptation of virus to growth in cultured cells. J. Virol. 76:1171-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang, J. H., T. D. Chung, and K. R. Oldenburg. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67-73. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, Y., P. Weady, R. Duggal, and W. Hao. 2008. Novel chimeric genotype 1b/2a hepatitis C virus suitable for high-throughput screening. Antimicrob. Agents Chemother. 52:666-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102:9294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong, J., P. Gastaminza, J. Chung, Z. Stamataki, M. Isogawa, G. Cheng, J. A. McKeating, and F. V. Chisari. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082-11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zuck, P., E. M. Murray, E. Stec, J. A. Grobler, A. J. Simon, B. Strulovici, J. Inglese, O. A. Flores, and M. Ferrer. 2004. A cell-based beta-lactamase reporter gene assay for the identification of inhibitors of hepatitis C virus replication. Anal. Biochem. 334:344-355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.