

Abstract
Melioidosis is an infectious disease with a propensity for relapse, despite prolonged antibiotic eradication therapy for 12 to 20 weeks. A pharmacokinetic (PK) simulation study was performed to determine the optimal dosing of cotrimoxazole (trimethoprim-sulfamethoxazole [TMP-SMX]) used in current eradication regimens in Thailand and Australia. Data for bioavailability, protein binding, and coefficients of absorption and elimination were taken from published literature. Apparent volumes of distribution were correlated with body mass and were estimated separately for Thai and Australian populations. In vitro experiments demonstrated concentration-dependent killing. In Australia, the currently used eradication regimen (320 [TMP]/1,600 [SMX] mg every 12 h [q12h]) was predicted to achieve the PK-pharmacodynamic (PD) target (an area under the concentration-time curve from 0 to 24 h/MIC ratio of >25 for both TMP and SMX) for strains with the MIC90 of Australian strains (≤1/19 mg/liter). In Thailand, the former regimen of 160/800 mg q12h would not be expected to attain the target for strains with an MIC of ≥1/19 mg/liter, but the recently implemented weight-based regimen (<40 kg [body weight], 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; >60 kg, 320/1,600 mg q12h) would be expected to achieve adequate concentrations for strains with an MIC of ≤1/19 mg/liter. The results were sensitive to the variance of the PK parameters. Prospective PK-PD studies of Asian populations are needed to optimize TMP-SMX dosing in melioidosis.
Melioidosis, a serious human infectious disease caused by the gram-negative bacterium Burkholderia pseudomallei, is endemic in northern Australia and southeast Asia. The in-hospital mortality rate averages 20% in Australia and 40 to 50% in northeast Thailand. In survivors of acute disease, recurrence after apparent clinical response, despite appropriate antibiotic treatment, is reported at rates of between 13 and 23% (5, 9). Molecular typing has determined that the majority (75%) of recurrent disease is due to persistence and subsequent relapse of the original infecting strain, with 25% of cases being second infections (17). Current antibiotic recommendations are for an intensive intravenous phase (ceftazidime or a carbapenem) for 10 to 14 days followed by a prolonged eradication phase (trimethoprim-sulfamethoxazole [TMP-SMX], or cotrimoxazole, alone or in combination with doxycycline) (6).
Dosing regimens for the eradication phase of treatment vary between countries. In Australia, a TMP-SMX dose of 320/1,600 mg (two double-strength tablets) every 12 h (q12h) is recommended. In Ubon Ratchathani, Thailand, a TMP-SMX dose of 160/800 mg (two single-strength tablets) q12h (with doxycycline) has been used previously. However, a new weight-based dosing protocol (for patients of <40 kg [body weight], 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; and >60 kg, 320/1,600 mg q12h, plus doxycycline) is now used.
TMP-SMX is a commonly used synergistic antibiotic combination that acts on successive enzymes in the bacterial folate synthesis pathway. The appropriate pharmacokinetic-pharmacodynamic (PK-PD) parameter has not been defined for TMP-SMX. Limited data from children with pneumococcal otitis media suggested that clinical efficacy correlates with the proportion of time when antibiotic concentrations exceed the MIC of the infecting organism (8).
In this study, we performed time-kill studies to assess the likely PK-PD target and evaluated the TMP-SMX dosing regimens using a simulation model with Thai and Australian populations.
MATERIALS AND METHODS
Time-kill studies.
We performed time-kill studies using three Thai clinical isolates (strains 4861a [MIC, 4 μg/ml], 3126a [MIC, 1 μg/ml], and 3131a [MIC, 0.25 μg/ml]). These were identified as B. pseudomallei by the use of standard methods, and the MIC was determined by Etest. Time-kill studies were performed in duplicate in 30-ml bottles containing 10 ml of Mueller-Hinton broth (Oxoid, United Kingdom). A logarithmic-phase broth culture was diluted to give a starting inoculum of approximately 1 × 106 CFU/ml. Bottles were then incubated in a rotary shaker at 37°C in air with various concentrations of TMP-SMX corresponding to the MIC, 4 times the MIC, 6 times the MIC, and 10 times the MIC. Antibiotic-free broth was included as a control. Viability counts of an aliquot diluted in antibiotic-free broth were performed at 0, 4, 8, and 24 h and plotted on a logarithmic scale against time. Additional studies were performed using concentrations of 10 times, 20 times, and 40 times the MIC at 0, 15, 30, 45, and 60 min. Statistical analyses were performed on log-transformed data.
PK parameters.
We used a single-compartment PK model with probabilistic sampled parameters for TMP and SMX separately. We considered regional variation in the MIC90 (the MIC required to inhibit the growth of 90% of bacterial isolates) and body weight and derived other PK parameters (including the coefficients of absorption and elimination, plasma protein binding, and bioavailability) from published literature. We considered four dosing regimens for TMP and SMX: 160/800 mg q12h, 160/800 mg q8h, 320/1,600 mg q12h, and 320/1,600 mg q8h. We also evaluated the weight-based protocol (<40 kg, 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; and >60 kg, 320/1,600 mg q12h).
In a published series, the PK variables associated with TMP and SMX are reported as means and standard deviations, suggesting that they are normally distributed (Table 1). Volume of distribution correlated with body mass. Volume of distribution/body mass was estimated from PK studies (performed mainly with European populations) and data from the 1995 United Kingdom Health Survey (13). For simulations involving Thai patients, published data on variation in body mass in Khon Kaen, northeast Thailand, were used in estimating parameters (21). For simulations involving Australian patients, published data were obtained from the 1995 Australian National Nutrition Survey (1).
TABLE 1.
Pharmacokinetic parameters used in this studyc
| Parameter | TMP
|
SMX
|
||
|---|---|---|---|---|
| Value(s) | Reference(s) | Value(s) | Reference(s) | |
| Bioavailability (F) | 0.875 (± 0.064) | 15, 18, 23, 25 | 0.95 (± 0.026) | 15, 23, 25 |
| Coefficient of absorption (ka)a | 1.63 (± 0.50) h−1 | 15, 18, 23-25, 29 | 1.13 (± 0.24) h−1 | 18, 23-25 |
| Coefficient of elimination (ke)b | 0.080 (± 0.010) h−1 | 15, 18, 23, 25, 29 | 0.080 (± 0.0054) h−1 | 15, 23, 25, 29 |
| Vol of distribution (Vd) | 110 liter (1.46 liter/kg) | 13, 18, 19, 23, 25, 29 | 16 liter (0.213 liter/kg) | 13, 19, 23, 25, 29 |
| Binding to plasma proteins | 50.0 ± 1.7% | 27 | 60.2 ± 3.7% | 27 |
| Thai adult body mass | 60.0 (± 10.2) kg | 21 | 60.0 (± 10.2) kg | 21 |
| Estimated vol of distribution in Thai adults | 88 (± 14.8) liter | 12.8 (± 2.2) liter | ||
| Australian adult body mass | 80.0 (± 14.5) kg | 1 | 80.0 (± 14.5) kg | 1 |
| Estimated vol of distribution in Australian adults | 116 (± 21.4) liter | 17 (± 3.1) liter | ||
Calculated from time to peak concentration and terminal half-life, where not reported directly.
Calculated from terminal half-life, where not reported directly.
In primary analysis, parameters were assumed to follow normal distribution, with mean and standard deviation shown.
PD parameters.
On the basis of time-kill studies demonstrating concentration-dependent killing, we defined the PK-PD target as 24-h drug exposure (free area under the concentration-time curve from 0 to 24 h [fAUC0-24 h]/MIC ratio of >25) in our analysis. We also assessed the dosing regimens against an alternative target in which TMP-SMX free-blood concentrations exceeded the MIC90 for at least 60% of the interdose period based on previous work with pneumococcal disease (8). The TMP-SMX MIC90 demonstrates geographic variation. Determination of the MIC (by Etest) of 80 Queensland isolates (20) and 170 clinical isolates from the Northern Territory (14) found that the MIC90 was 1/19 mg/liter, with only three isolates in the latter study having an MIC of 2/38 mg/liter. However, in Thailand, the MIC90 for 1,976 clinical isolates was 3/58 mg/liter by the Etest (30). We used primary data on the MIC from Thailand to determine the proportion of isolates for which concentrations of TMP-SMX were expected to be adequate. We performed two sensitivity analyses, the first assuming that PK parameters varied in a log-normal, rather than a normal, distribution and the second doubling the coefficient of variation in PK parameters to account for possible variation due to disease states.
Simulation.
PK formulae (Appendix) were programmed using MATLAB 7.1 (Mathworks, Natick, MA). All parameters contributing to the PK formulae were assumed to be independent, normally distributed random variables (or log-normal in the sensitivity analysis). A Monte Carlo integration method was used to determine the expected range of serum antibiotic concentrations over a 24-h dosing interval across each population. Values for each of the parameters were sampled from their distributions, and the expected serum concentrations over a 24-h period were calculated. The fAUC0-24 h/MIC ratio and the proportion of time over the MIC90 for each region were derived by integration. Ten thousand simulations were conducted, and the proportion of the population expected to achieve the PK-PD target was noted for each simulation. A sensitivity analysis examined the probability of attainment of an alternative target, a concentration exceeding the MIC for at least 60% of the interdose period.
RESULTS
Time-kill studies.
Bacterial concentrations were similar in all groups at time zero (analysis of variance, P = 0.47) but were different between the relative concentrations of TMP-SMX at 4 h, 8 h, and 24 h of incubation (all P < 0.001; Table 2). This suggests concentration-dependent killing between 0 and 4 h and a bacteriostatic effect between 4 and 24 h (Fig. 1). At higher concentrations, there was complete killing at 30 min following incubation with TMP-SMX at 20 times the MIC and at 15 min following incubation with 40 times the MIC. In a log-linear regression model, increasing the TMP-SMX concentration was associated with a decreasing bacterial concentration, after adjusting for the duration of incubation (coefficient of −0.33 log, P < 0.001).
TABLE 2.
Log bacterial concentrations over time in strains 4861a, 3126a, and 3131a incubated with various concentrations of TMP-SMX relative to the strain MICa
| TMP-SMX concn | Bacterial concn (CFU/ml)
|
|||
|---|---|---|---|---|
| Baseline | 4 h | 8 h | 24 h | |
| Control | 6.01 ± 0.047 | 7.55 ± 0.32 | 8.48 ± 0.47 | 10.07 ± 0.84 |
| 1× MIC | 6.08 ± 0.066 | 5.76 ± 0.39 | 6.57 ± 0.59 | 7.33 ± 1.09 |
| 4× MIC | 6.04 ± 0.11 | 5.29 ± 0.43 | 5.20 ± 0.88 | 5.24 ± 1.76 |
| 6× MIC | 6.03 ± 0.046 | 5.12 ± 0.42 | 5.23 ± 0.51 | 4.57 ± 1.72 |
| 10× MIC | 6.08 ± 0.084 | 3.62 ± 0.20 | 3.66 ± 0.037 | 3.30 ± 1.16 |
Results represent geometric mean and standard deviation of bacterial concentrations of the three strains in studies performed in duplicate.
FIG. 1.

Log bacterial concentrations over time in strains 4861a, 3126a, and 3131a incubated with various concentrations of TMP-SMX relative to the strain MIC. Bars represent geometric mean and standard deviation of bacterial concentration (CFU/ml) of the three strains in assays performed in duplicate.
PK-PD target of an AUC0-24 h/MIC ratio of >25.
For Thai adults, TMP dosing of at least 160 mg q8h was required before the probability of PK-PD target attainment exceeded 80%, if infecting isolates had an MIC of 1 mg/liter. The PK-PD target could be achieved reliably only with high TMP dosing (320 mg three times a day) at the susceptibility breakpoint (2 mg/liter) and could not be achieved for any dosing regimen with a high probability for the MIC90 (3 mg/liter) in this region (Table 3) . For SMX, dosing regimens of 800 mg q8h or greater achieved adequate concentrations exceeding the susceptibility threshold (38 mg/liter), and 1,600-mg-q12h dosing achieved adequate concentrations exceeding the MIC90 (57 mg/liter) in the region (Table 3). The Thai weight-based protocol would be expected to achieve the PK-PD target for strains with an MIC of up to 1/19 mg/liter. Based on the distribution of the TMP-SMX MIC observed in a previous study (30), the weight-based protocol would be expected to achieve an AUC0-24 h/MIC ratio of >25 with 82% of Thai patients.
TABLE 3.
Predicted probability of target attainment (AUC0-24h/MIC ratio of >25) with Thai population
| Dosing regimend | Breakpoint (%)
|
Proportion with adequate concentrations (%) | ||
|---|---|---|---|---|
| 1/19 mg/litera | 2/38 mg/literb | 3/57 mg/literc | ||
| TMP | ||||
| 160 mg q12h | 18 | <0.1 | <0.1 | 44 |
| 160 mg q8h | 80 | 2 | <0.1 | 61 |
| 320 mg q12h | 99 | 17 | <0.1 | 72 |
| 320 mg q8h | 100 | 81 | 18 | 85 |
| Weight based | 100 | 59 | 2 | 82 |
| SMX | ||||
| 800 mg q12h | 100 | 39 | 1.8 | 80 |
| 800 mg q8h | 100 | 99 | 34 | 90 |
| 1,600 mg q12h | 100 | 100 | 92 | 94 |
| 1,600 mg q8h | 100 | 100 | 100 | 96 |
| Weight based | 100 | 100 | 100 | 95 |
1 (TMP)/19 (SMX) mg/liter represents the MIC90 for Australian isolates.
2/38 mg/liter represents the susceptibility threshold.
3/57 mg/liter represents the MIC90 for Thai isolates.
Weight-based dosing for patients was as follows: <40 kg (body weight), 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; and >60 kg, 320/1,600 mg q12h.
For Australian adults, a TMP dose of 320 mg q12h had an 81% probability of achieving target attainment for isolates at the MIC90 of the region (1 mg/liter), and the weight-based protocol was predicted to have a high probability of achieving the target AUC/MIC ratio. No regimen could reliably achieve concentrations adequate to exceed the susceptibility threshold (2 mg/liter) (Table 4). All SMX dosing regimens achieved adequate concentrations exceeding the regional MIC90 (19 mg/liter), and SMX dosing regimens of 1,600 mg q12h or higher achieved adequate concentrations exceeding the susceptibility threshold (38 mg/liter).
TABLE 4.
Predicted probability of target attainment (AUC0-24h/MIC ratio of >25) with Australian patients
| Dosing regimend | Breakpoint (%)
|
||
|---|---|---|---|
| 1/19 mg/litera | 2/38 mg/literb | 3/57 mg/literc | |
| TMP | |||
| 160 mg q12h | 3 | <0.1 | <0.1 |
| 160 mg q8h | 36 | 0.2 | 0.1 |
| 320 mg q12h | 81 | 3 | 0.2 |
| 320 mg q8h | 100 | 36 | 3 |
| Weight based | 100 | 31 | 0.6 |
| SMX | |||
| 800 mg q12h | 98 | 7 | 0.2 |
| 800 mg q8h | 100 | 66 | 7 |
| 1,600 mg q12h | 100 | 98 | 42 |
| 1,600 mg q8h | 100 | 100 | 98 |
| Weight based | 100 | 100 | 98 |
1 (TMP)/19 (SMX) mg/liter represents the MIC90 for Australian isolates.
2/38 mg/liter represents the susceptibility threshold.
3/57 mg/liter represents the MIC90 for Thai isolates.
Weight-based dosing for patients was as follows: <40 kg (body weight), 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; and >60 kg, 320/1,600 mg q12h.
Sensitivity analyses.
With Australian adults, the probability of attainment of the AUC/MIC ratio target for the TMP-SMX 320-/1,600-mg regimen at the Australian MIC90 threshold of 1/19 mg/liter was reduced from 81% to 71%, assuming a higher variance in PK parameters (bioavailability, coefficients of absorption and elimination, and protein binding). The proportion of patients achieving the target AUC/MIC ratio remained high (97%), assuming higher variance in PK parameters. The probability of target attainment for strains with an MIC of 1/19 mg/liter was not significantly different (84%), assuming a log-normal distribution of PK parameters.
In Thai adults, the probability of attainment of the target AUC/MIC ratio of the 160-/800-mg-q8h regimen for strains with an MIC of 1/19 mg/liter was reduced to 72% if a higher variation in PK parameters was assumed. Both the 320-/1,600-mg-q12h and weight-based regimens reliably attained the target AUC/MIC ratio for isolates with an MIC of 1/19 mg/liter, assuming higher variation in PK parameters (1,600 mg twice daily, 94%; and weight-based, 87%). The probability of target attainment for strains with an MIC of 1/19 mg/liter was not changed significantly, assuming a log-normal distribution of PK parameters.
PK-PD target concentrations higher than MIC for >60% of interdose period.
In Thai adults, the TMP 320-mg-q12h regimen would be required to reliably achieve concentrations exceeding the MIC for >60% of the interdose period for isolates with an MIC up to 1 mg/liter (Table 5; Fig. 2). SMX dosing regimens of >800 mg q8h were adequate for susceptible strains (Table 5). The weight-based protocol was expected to attain the PK-PD target with 93% of Thai patients infected with a strain with an MIC of 1 mg/liter but with only 1.5% of patients infected with isolates at the susceptibility breakpoint (2 mg/liter). Based on the distribution of the TMP MIC from a previous study (30), the weight-based protocol would be expected to achieve TMP-SMX concentrations exceeding the MIC for at least 60% of the interdose interval with 81% of patients.
TABLE 5.
Predicted probability of target attainment (concentration exceeding MIC for >60% of interdosing interval) in Thai patients
| Dosing regimend | Breakpoint (%)
|
Proportion with adequate concentrations (%) | ||
|---|---|---|---|---|
| 1/19 mg/litera | 2/38 mg/literb | 3/57 mg/literc | ||
| TMP | ||||
| 160 mg q12h | 13 | <0.1 | <0.1 | 57 |
| 160 mg q8h | 79 | 2 | <0.1 | 70 |
| 320 mg q12h | 98 | 13 | <0.1 | 85 |
| 320 mg q8h | 100 | 79 | 17 | 97 |
| Weight based | 100 | 93 | 25 | 91 |
| SMX | ||||
| 800 mg q12h | 100 | 31 | 1 | 88 |
| 800 mg q8h | 100 | 99 | 40 | 93 |
| 1,600 mg q12h | 100 | 100 | 86 | 96 |
| 1,600 mg q8h | 100 | 100 | 100 | 97 |
| Weight based | 100 | 100 | 100 | 97 |
1 (TMP)/19 (SMX) mg/liter represents the MIC90 for Australian isolates.
2/38 mg/liter represents the susceptibility threshold.
3/57 mg/liter represents the MIC90 for Thai isolates.
Weight-based dosing for patients was as follows: <40 kg (body weight), 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; and >60 kg, 320/1,600 mg q12h.
FIG. 2.

Predicted concentration-time graphs for Thai adults.
With Australian adults, TMP dosing of 320 mg q12h would be expected to be adequate to achieve the PK-PD target with 73% of patients infected with isolates at the regional MIC90 (1 mg/liter). No TMP dosing regimen was able to reliably achieve the PK-PD target for isolates at the susceptibility breakpoint (Table 6; Fig. 3). SMX dosing at 800 mg q12h would be expected to be adequate for strains with the MIC90 (1 mg/liter), but 800 mg q8h or higher would be associated with a high probability of target attainment.
TABLE 6.
Predicted probability of target attainment (concentration exceeding MIC for >60% of interdosing interval) with Australian patients
| Dosing regimen | Breakpoint
|
||
|---|---|---|---|
| 1/19 mg/litera | 2/38 mg/literb | 3/57 mg/literc | |
| TMP | |||
| 160 mg q12h | 2 | <0.1 | <0.1 |
| 160 mg q8h | 35 | 0.2 | 0.1 |
| 320 mg q12h | 72 | 3 | 0.1 |
| 320 mg q8h | 100 | 35 | 3 |
| Weight based | 100 | 29 | 0.7 |
| SMX | |||
| 800 mg q12h | 96 | 5 | 0.2 |
| 800 mg q8h | 100 | 66 | 7 |
| 1,600 mg q12h | 100 | 96 | 34 |
| 1,600 mg q8h | 100 | 100 | 98 |
| Weight based | 100 | 100 | 98 |
1 (TMP)/19 (SMX) mg/liter represents the MIC90 for Australian isolates.
2/38 mg/liter represents the susceptibility threshold.
3/57 mg/liter represents the MIC90 for Thai isolates.
dWeight-based dosing for patients was as follows: <40 kg, 160/800 mg q12h; 40 to 60 kg, 240/1,200 mg q12h; and >60 kg, 320/1,600 mg q12h.
FIG. 3.

Predicted concentration-time graphs for Australian adults.
DISCUSSION
Despite prolonged eradication treatment courses, melioidosis is still associated with a significant relapse rate. In this study, we attempt to define an optimal dosing regimen of TMP-SMX that may reduce relapse. Limited data had suggested that for antibiotics with a time-dependent action (such as beta-lactams and TMP-SMX), improvements in clinical outcomes plateau when the time period in which the concentration exceeds the MIC90 is more than 60% (8). However, our data suggest that TMP-SMX exerts a concentration-dependent killing effect on B. pseudomallei within the blood concentration range achievable in vivo.
The assessment of TMP-SMX susceptibility is problematic. One study has suggested that TMP-SMX exerts a bactericidal action (2), but another suggests that it is bacteriostatic (11). Susceptibility testing for TMP-SMX has been difficult, with disc diffusion proving unreliable and MIC testing proving unreliable because of unclear end points (20, 30). Assessment of the TMP-SMX MIC using the Etest method has demonstrated lower susceptibility of B. pseudomallei isolates from Thailand than of those from Australia, with 38% of clinical isolates having an MIC of ≥1/19 mg/liter and 13% having an MIC of ≥2/38 mg/liter (30). In contrast, only 3 of 170 strains had an MIC of ≥1/19 mg/liter in one Australian study (14).
This study suggests that in Thailand, dosing regimens of TMP-SMX may be inadequate for strains at or around the susceptibility threshold. Susceptibility testing using the Etest provides more reliable end points and has recently been adopted by many major centers in northeast Thailand. This simulation study suggests that the new weight-based protocol (or a regimen of 320/1,600 mg twice daily) would be expected to achieve adequate serum exposure of TMP and SMX relative to the MICs of susceptible strains. In Australia, the currently used regimen of cotrimoxazole, 320/1,600 mg (two double-strength tablets) q12h, should be adequate for local isolates with an MIC of <1/19 mg/liter.
A previous study examined the dosing of coamoxiclav, suggesting that very-frequent-dosing regimens (six hourly or more frequent) are required to achieve adequate concentrations of these beta-lactams, based on checkerboard testing of clinical isolates (7). Inadequate dosing, as well as poorer tolerance and compliance, may explain why current dosing of coamoxiclav is associated with a relapse rate higher than those of TMP-SMX-based regimens in Thailand (16, 22). Similarly, the four-drug regimen of TMP-SMX, doxycycline, and chloramphenicol has been found to be antagonistic in vitro and poorly tolerated in vivo compared to TMP-SMX and doxycycline (3, 12). In Australia, TMP-SMX has been used alone, with few relapses documented (10). A trial of eradication therapy has recently started in Thailand using the weight-based dosing regimen of TMP-SMX (versus TMP-SMX and doxycycline).
Higher recurrence rates were documented in Thailand than in Australia before the recent implementation of the weight-based dosing regimen (5, 9). Possible explanations for this include the use of multiple (and possibly antagonistic) drug combinations (12), the higher rate of reinfection (rather than relapse) documented (17), and poor compliance rates (3, 4, 22). This study suggests that the previous dosing of TMP-SMX may have been suboptimal and may have been a contributing factor, explaining this regional difference in relapse rates. This study also has implications for laboratories in which TMP-SMX is used in postexposure prophylaxis following accidental exposure.
There are several limitations of this study. The apparent volumes of distribution used in the PK simulations were derived from anthropomorphic studies in each region, rather than the patients treated. To our knowledge, PK studies of TMP-SMX have not been performed with Asian populations. Other studies have defined important factors associated with relapse of melioidosis, such as tolerability and compliance, and higher doses of TMP-SMX may be associated with gastrointestinal and hematological intolerance. We used a simple one-compartment model; the site of persistence and the focus of later relapse for B. pseudomallei are not known, and the concentrations and TMP/SMX ratios in these compartments may be important. Some studies have demonstrated that B. pseudomallei in biofilm has an MIC 200 times higher than that of planktonic cells of B. pseudomallei; the clinical significance of this is not yet known (28). We did not use checkerboard testing in this study, as previous studies have defined the optimal ratio of TMP/SMX as 1:20, as used in susceptibility testing, and there is considerable uncertainty in defining the actual ratio at the site of action (23). Finally, no data on the appropriate PK-PD target are available from clinical studies, and our choice of a target AUC/MIC ratio of 25 is arbitrary.
Renal disease is common in patients with melioidosis; in addition to acute renal impairment related to sepsis, risk factors for melioidosis include diabetes mellitus (present in over half the patients with melioidosis) and nephrolithiasis secondary to renal tubular acidosis (26). Reduced renal clearance of TMP-SMX results in higher accumulation, and a dose reduction may be required. We did not model the effect of renal disease in this study. Although acute sepsis is usually associated with an increased variability in PK parameters, in melioidosis, TMP-SMX is used primarily for eradication therapy following treatment of acute disease with intravenous ceftazidime. However, the results of the simulation were sensitive to the variance in PK parameters, highlighting this as an issue in future PK-PD studies.
In this study, we have demonstrated that TMP-SMX is associated with concentration-dependent killing effects. Simulation suggests that the dosing regimen previously used in Thailand may not be sufficient for strains at or above the susceptibility threshold. In addition to other factors, including compliance with and the tolerability of the prolonged course of eradication therapy, the high MIC found in Thailand compared to Australia, and the higher reinfection rate, TMP-SMX dosing may account for the higher relapse rate documented in Thailand. The current weight-based regimen is predicted to achieve adequate concentrations of TMP and SMX. Studies are needed to verify the pharmacokinetics of TMP-SMX in Thai populations, to determine the tolerability of higher doses of TMP-SMX, and to validate the correlation between pharmacokinetics and clinical outcome in melioidosis.
Acknowledgments
E.M. is supported by a Capacity Building Grant from the Australian National Health and Medical Research Council (NH&MRC). A.C. is supported by a Health Professionals Fellowship from the NH&MRC. S.P. is supported by the Wellcome Trust. This study was funded by the Wellcome Trust of Great Britain.
No conflicting interests are declared.
APPENDIX
The coefficient of elimination, ke, is calculated from the terminal half-life, t1/2, by the formula:
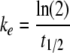 |
(1) |
The time to maximum concentration, Tmax, is given by the following expression; the coefficient of absorption, ka, can be found numerically.
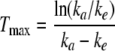 |
(2) |
For multiple dosing, the total concentration, C, at time t is given by the following expression, where repeated doses, D, with bioavailability F are administered every τ hours.
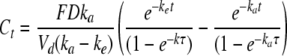 |
(3) |
The free, or unbound, concentration of drug, Cf, is calculated from the total drug concentration, Ct, and protein binding, B, as follows: Cf = B·Ct.
Footnotes
Published ahead of print on 20 July 2009.
REFERENCES
- 1.Australian Bureau of Statistics. 1997. 4802.0-National Nutrition Survey, 1995. Australian Bureau of Statistics, Canberra, Commonwealth of Australia.
- 2.Bassett, D. C. 1971. The sensitivity of Pseudomonas pseudomallei to trimethoprim and sulphamethoxazole in vitro. J. Clin. Pathol. 24:798-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaowagul, W., W. Chierakul, A. J. Simpson, J. M. Short, K. Stepniewska, B. Maharjan, A. Rajchanuvong, D. Busarawong, D. Limmathurotsakul, A. C. Cheng, V. Wuthiekanun, P. N. Newton, N. J. White, N. P. Day, and S. J. Peacock. 2005. Open-label randomized trial of oral trimethoprim-sulfamethoxazole, doxycycline, and chloramphenicol compared with trimethoprim-sulfamethoxazole and doxycycline for maintenance therapy of melioidosis. Antimicrob. Agents Chemother. 49:4020-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaowagul, W., A. J. Simpson, Y. Suputtamongkol, M. D. Smith, B. J. Angus, and N. J. White. 1999. A comparison of chloramphenicol, trimethoprim-sulfamethoxazole, and doxycycline with doxycycline alone as maintenance therapy for melioidosis. Clin. Infect. Dis. 29:375-380. [DOI] [PubMed] [Google Scholar]
- 5.Chaowagul, W., Y. Suputtamongkol, D. A. Dance, A. Rajchanuvong, J. Pattara-arechachai, and N. J. White. 1993. Relapse in melioidosis: incidence and risk factors. J. Infect. Dis. 168:1181-1185. [PubMed] [Google Scholar]
- 6.Cheng, A. C., and B. J. Currie. 2005. Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18:383-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chierakul, W., J. Wangboonskul, T. Singtoroj, W. Pongtavornpinyo, J. M. Short, B. Maharjan, V. Wuthiekanun, D. A. Dance, P. Teparrukkul, N. Lindegardh, S. J. Peacock, N. P. Day, W. Chaowagul, and N. J. White. 2006. Pharmacokinetic and pharmacodynamic assessment of co-amoxiclav in the treatment of melioidosis. J. Antimicrob. Chemother. 58:1215-1220. [DOI] [PubMed] [Google Scholar]
- 8.Craig, W. A., and D. Andes. 1996. Pharmacokinetics and pharmacodynamics of antibiotics in otitis media. Pediatr. Infect. Dis. J. 15:255-259. [DOI] [PubMed] [Google Scholar]
- 9.Currie, B. J., D. A. Fisher, N. M. Anstey, and S. P. Jacups. 2000. Melioidosis: acute and chronic disease, relapse and re-activation. Trans. R. Soc. Trop. Med. Hyg. 94:301-304. [DOI] [PubMed] [Google Scholar]
- 10.Currie, B. J., D. A. Fisher, D. M. Howard, J. N. Burrow, D. Lo, S. Selva-Nayagam, N. M. Anstey, S. E. Huffam, P. L. Snelling, P. J. Marks, D. P. Stephens, G. D. Lum, S. P. Jacups, and V. L. Krause. 2000. Endemic melioidosis in tropical northern Australia: a 10-year prospective study and review of the literature. Clin. Infect. Dis. 31:981-986. [DOI] [PubMed] [Google Scholar]
- 11.Dance, D. A., V. Wuthiekanun, W. Chaowagul, and N. J. White. 1989. The antimicrobial susceptibility of Pseudomonas pseudomallei. Emergence of resistance in vitro and during treatment. J. Antimicrob. Chemother. 24:295-309. [DOI] [PubMed] [Google Scholar]
- 12.Dance, D. A., V. Wuthiekanun, W. Chaowagul, and N. J. White. 1989. Interactions in vitro between agents used to treat melioidosis. J. Antimicrob. Chemother. 24:311-316. [DOI] [PubMed] [Google Scholar]
- 13.Department of Health. 1997. Health Survey for England 1995. The Stationary Office, London, United Kingdom.
- 14.Jenney, A. W., G. Lum, D. A. Fisher, and B. J. Currie. 2001. Antibiotic susceptibility of Burkholderia pseudomallei from tropical northern Australia and implications for therapy of melioidosis. Int. J. Antimicrob. Agents 17:109-113. [DOI] [PubMed] [Google Scholar]
- 15.Kremers, P., J. Duvivier, and C. Heusghem. 1974. Pharmacokinetic studies of co-trimoxazole in man after single and repeated doses. J. Clin. Pharmacol. 14:112-117. [DOI] [PubMed] [Google Scholar]
- 16.Limmathurotsakul, D., W. Chaowagul, W. Chierakul, K. Stepniewska, B. Maharjan, V. Wuthiekanun, N. J. White, N. P. Day, and S. J. Peacock. 2006. Risk factors for recurrent melioidosis in northeast Thailand. Clin. Infect. Dis. 43:979-986. [DOI] [PubMed] [Google Scholar]
- 17.Maharjan, B., N. Chantratita, M. Vesaratchavest, A. Cheng, V. Wuthiekanun, W. Chierakul, W. Chaowagul, N. P. Day, and S. J. Peacock. 2005. Recurrent melioidosis in patients in northeast Thailand is frequently due to reinfection rather than relapse. J. Clin. Microbiol. 43:6032-6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nolte, H., and H. Buttner. 1973. Pharmacokinetics of trimethoprim and its combination with sulfamethoxazole in man after single and chronic oral administration. Chemotherapy 18:274-284. [DOI] [PubMed] [Google Scholar]
- 19.Patel, R. B., and P. G. Welling. 1980. Clinical pharmacokinetics of co-trimoxazole (trimethoprim-sulphamethoxazole). Clin. Pharmacokinet. 5:405-423. [DOI] [PubMed] [Google Scholar]
- 20.Piliouras, P., G. C. Ulett, C. Ashhurst-Smith, R. G. Hirst, and R. E. Norton. 2002. A comparison of antibiotic susceptibility testing methods for cotrimoxazole with Burkholderia pseudomallei. Int. J. Antimicrob. Agents 19:427-429. [DOI] [PubMed] [Google Scholar]
- 21.Pongchaiyakul, C., V. Kosulwat, N. Rojroongwasinkul, S. Charoenkiatkul, K. Thepsuthammarat, M. Laopaiboon, T. V. Nguyen, and R. Rajatanavin. 2005. Prediction of percentage body fat in rural Thai population using simple anthropometric measurements. Obes. Res. 13:729-738. [DOI] [PubMed] [Google Scholar]
- 22.Rajchanuvong, A., W. Chaowagul, Y. Suputtamongkol, M. D. Smith, D. A. B. Dance, and N. J. White. 1995. A prospective comparison of co-amoxiclav and the combination of chloramphenicol, doxycycline, and co-trimoxazole for the oral maintenance treatment of melioidosis. Trans. R. Soc. Trop. Med. Hyg. 89:546-549. [DOI] [PubMed] [Google Scholar]
- 23.Reeves, D. S., and P. J. Wilkinson. 1979. The pharmacokinetics of trimethoprim and trimethoprim/sulphonamide combinations, including penetration into body tissues. Infection 4(7 Suppl.):S330-S341. [DOI] [PubMed] [Google Scholar]
- 24.Rieder, J., D. E. Schwartz, M. Fernex, T. Bergan, E. K. Brodwall, A. Blumberg, P. Cottier, and W. Scheitlin. 1974. Pharmacokinetics of the antibacterial combination sulfamethoxazole plus trimethoprim in patients with normal or impaired kidney function. Antibiot. Chemother. 18:148-198. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz, D. E., and J. Rieder. 1970. Pharmacokinetics of sulfamethoxazole plus trimethoprim in man and their distribution in the rat. Chemotherapy 15:337-355. [DOI] [PubMed] [Google Scholar]
- 26.Suputtamongkol, Y., W. Chaowagul, P. Chetchotisakd, N. Lertpatanasuwun, S. Intaranongpai, T. Ruchutrakool, D. Budhsarawong, P. Mootsikapun, V. Wuthiekanun, N. Teerawatasook, and A. Lulitanond. 1999. Risk factors for melioidosis and bacteremic melioidosis. Clin. Infect. Dis. 29:408-413. [DOI] [PubMed] [Google Scholar]
- 27.Varoquaux, O., D. Lajoie, C. Gobert, P. Cordonnier, C. Ducreuzet, M. Pays, and C. Advenier. 1985. Pharmacokinetics of the trimethoprim-sulphamethoxazole combination in the elderly. Br. J. Clin. Pharmacol. 20:575-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vorachit, M., K. Lam, P. Jayanetra, and J. W. Costerton. 1993. Resistance of Pseudomonas pseudomallei growing as a biofilm on silastic discs to ceftazidime and co-trimoxazole. Antimicrob. Agents Chemother. 37:2000-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welling, P. G., W. A. Craig, G. L. Amidon, and C. M. Kunin. 1973. Pharmacokinetics of trimethoprim and sulfamethoxazole in normal subjects and in patients with renal failure. J. Infect. Dis. 128(Suppl.):556-566. [DOI] [PubMed] [Google Scholar]
- 30.Wuthiekanun, V., A. C. Cheng, W. Chierakul, P. Amornchai, D. Limmathurotsakul, W. Chaowagul, A. J. Simpson, J. M. Short, G. Wongsuvan, B. Maharjan, N. J. White, and S. J. Peacock. 2005. Trimethoprim/sulfamethoxazole resistance in clinical isolates of Burkholderia pseudomallei. J. Antimicrob. Chemother. 55:1029-1031. [DOI] [PubMed] [Google Scholar]