

Abstract
Immunocompromised patients are at risk of developing toxoplasmosis, and although chemotherapy is available, standard treatments are often complicated by severe side effects. Artemisinin is a new highly potent antimalarial drug that has activity against Toxoplasma gondii in vitro. However, artemisinin derivatives have previously been ineffective in vivo using a rat model of toxoplasmosis. In the present study, the efficacy of several new artemisinin derivates was investigated for treatment of mice infected with the parasite Toxoplasma gondii. Artemiside and artemisone displayed better inhibition than either artemisinin or artesunate against the parasite in vitro. Artemiside and artemisone treatment controlled parasite replication in vivo, and mice survived the acute infection. In a murine model of reactivated toxoplasmosis, both drugs increased survival, although artemiside was more effective. These results indicate that these newer derivatives of artemisinin may have potential for treatment of toxoplasmosis.
The protozoan parasite Toxoplasma gondii infects a large number of warm-blooded animals and is estimated to chronically infect up to one-third of the world's human population (11). The life cycle includes a sexual phase in the intestine of cats, leading to fecal shedding of a spore-like stage known as the oocyst (9). Asexual development takes place in a wide range of warm-blooded vertebrates and is characterized by an acute phase, during which the parasite propagates rapidly as tachyzoites, and a chronic phase, typified by tissue cysts containing slow-growing forms called bradyzoites (9). Humans can become infected by ingestion of oocysts that contaminate water or tissue cysts in undercooked meat. Following oral uptake, the parasite crosses the gut epithelium and disseminates throughout the body (5). The acute phase generally produces only mild symptoms in immunocompetent adults and is readily controlled by the immune response (29). The latent stage of infection is characterized by the presence of parasites within tissue cysts that form in the skeletal muscle and central nervous system (25).
Toxoplasmosis is of concern during pregnancy, due to the ability of the parasite to cross the placental barrier and cause infection in the developing fetus (21, 41). Additionally, immunocompromised patients are predisposed to development of reactivated toxoplasmosis due to cyst rupture and uncontrolled proliferation of tachyzoites (25). Patients at risk include those with underlying conditions, such as AIDS, or individuals who are therapeutically immunosuppressed due to organ transplantation or cancer chemotherapy. Toxoplasmic encephalitis is the most common clinical manifestation of reactivated disease in AIDS patients who do not receive highly active antiretroviral therapy or antiparasitic prophylaxis (25). If left untreated, reactivation leads to severe central nervous system (CNS) complications and may result in death.
Standard therapy for toxoplasmosis in humans includes combinations of pyrimethamine plus sulfadiazine. However, these drugs often cannot be tolerated due to severe side effects (22). Further, the current treatments control only actively proliferating parasites but have limited activity against bradyzoites within tissue cysts and hence do not eliminate the chronic infection; thus, prolonged treatment is necessary for immunocompromised patients (22). Atovaquone, originally developed for malaria, is also active against T. gondii both in vitro and in vivo, and it has been reported to have some activity against tissue cysts when administered for long periods of time (2). Atovaquone in combination with pyrimethamine or sulfadiazine was reported to prevent relapse of toxoplasmosis in AIDS patients (7). However, it has also been associated with breakthrough failures in other cases (4, 24). Hence, there is a need for the development of new therapeutics for treatment of toxoplasmosis.
Artemisinin (Fig. 1) is a sesquiterpene lactone produced by the plant Artemisia annua and is used for the treatment of fevers and chills as part of traditional Chinese herbal medicine (14). Artemisinin and its derivatives, including artesunate (Fig. 1), dihydroartemisinin (DHA), and artemether, are potent and rapidly acting antimalarial drugs that are currently used widely in combination therapies to treat malaria (3). The new artemisinin derivatives artemisone and artemiside, the thiomorpholine precursor of artemisone, have markedly superior in vitro antimalarial activity against chloroquine-sensitive and -resistant Plasmodium falciparum lines and also display greater activities against the rodent malaria Plasmodium berghei in vivo (40). The enhanced efficacy of artemisone is also apparent in studies involving an Aotus monkey model for P. falciparum malaria (13). Artemisone has greatly reduced neurotoxicity compared to the major circulating metabolite of artesunate, namely DHA, and as such it has a superior pharmacokinetic profile for humans (28).
FIG. 1.

Structure diagrams of artemisinin and related compounds. Artemisinin, 1; artesunate, 2; DHA, 3; artemether, 4; artemisone, 5; artemiside, 6; sulfamide derivative, 7; benzylpiperazine derivative, 8; pyrimidylpiperazine derivative, 9; and 4′-fluorophenyl derivative, 10.
The parent compound artemisinin is effective against T. gondii, although the 50% effective concentrations (EC50s) are ∼50-fold higher than those reported for P. falciparum (27). The mechanism of action of artemisinin and its derivatives is dependent upon the presence of the endoperoxide bridge (14), although the difference in potencies between P. falciparum and T. gondii suggests that different targets may be affected in these two organisms. The derivatives artesunate, artemether, and artemisone (27), as well as several other new derivatives (17), have also been shown to be more potent against T. gondii in vitro. Despite in vitro activity, previous studies tested artemether with a rat model with concurrent infection with Pneumocystis carinii and T. gondii and found that it was ineffective in controlling infection (6).
In the present study, we tested the antiparasitic efficacy of several new derivates of artemisinin against T. gondii cultured in vitro and in murine models of acute and reactivated toxoplasmosis.
MATERIALS AND METHODS
Parasite growth assay.
The T. gondii strain RH (ATCC 50838), clone 2F (ATCC 50839), which expresses bacterial β-galactosidase, was used for in vitro growth assays (27). Parasites were maintained as tachyzoites in human foreskin fibroblast cells grown in Dulbecco's modified Eagle's medium with 10 mM HEPES, 44 mM sodium bicarbonate, 10% fetal bovine serum, 2 mM glutamine, and 10 μg/ml gentamicin. For growth inhibition assays, parasites were inoculated in 96-well plates containing monolayers of human foreskin fibroblast cells and were treated with different compounds at concentrations ranging from 0.010 μM to 1.0 μM for 72 h at 37°C. Following treatment, the culture medium was removed, and parasite growth was determined based on detection of β-galactosidase activity, as described previously (27). The end product of the reaction was read as absorbance at 570 nm, and mean values from three independent experiments were used to estimate the EC50s and EC90s by curve fitting using nonlinear regression with a variable slope in Prism 5.0 (GraphPad Software Inc.).
Mouse infections.
The luciferase expressing the PRU-Luc-GFP type II strain (kindly provided by J. Boothroyd, Stanford University School of Medicine, CA) of T. gondii was used for mouse infections. CD1 outbred female mice (Charles River Laboratories, Wilmington, MA) 8 to 10 weeks of age were used for all acute infection studies. CD1 mice are relatively resistant to infection by type II strains, so we first established the dose of tachyzoites that would cause lethal infection in a majority of mice (data not shown). Control and drug-treated mice were infected by intraperitoneal (i.p.) injection of 106 tachyzoites in 0.2 ml phosphate-buffered saline, compounds were injected (subcutaneously [s.c.] 10 mg/kg) every day for 8 days, and survival was followed for 25 days. Animals were maintained in an AAALAC-approved facility overseen by the Institutional Animal Care Committee at Washington University.
Parasite burdens.
For bioluminescence experiments, animals were imaged at 2-day intervals, following treatment with mild anesthesia using isofluorene and i.p. injection of 0.15 mg/kg body weight of firefly luciferin D (Biosynth AG, Staad, Switzerland). Animals were imaged using a Xenogen IVIS 100 (Caliper LifeSciences, Hopkinton, MA), and data are expressed in relative light units. As described previously, the output from luciferase imaging in vivo is not directly related to parasite numbers but rather provides a relative index of the severity of infection (30). Experiments contained three animals per group that were monitored for 25 days postinfection, after which chronic infections were confirmed by examining homogenates of the brain for tissue cysts. Briefly, animals were humanely sacrificed, and the brains were removed and homogenized in 1.0 ml of sterile phosphate-buffered saline, pH 7.2. Homogenates were examined by bright-field or phase-contrast microscopy at ×40 magnification, cysts were counted from four separate 20-μl aliquots, and counts were used to estimate the total number of cysts per brain.
Murine model of reactivated toxoplasmosis.
Inbred female gamma interferon (IFN-γ−/−) mice on a C57BL/6 background were maintained under specific pathogen-free conditions. Eight- to 10-week-old mice were orally infected with 20 tissue cysts of PRU-Luc-GFP T. gondii that were obtained from the brains of chronically infected wild-type mice. C57BL/6 mice are highly susceptible to infection and develop high tissue cyst burdens in the chronic phase (37). To control acute infection, mice were treated with sulfadiazine (Sigma-Aldrich, St. Louis, MO) in the drinking water (400 mg/liter) for 3 weeks beginning 2 days after infection. Two days after discontinuation of sulfadiazine, mice were treated s.c. with compounds (10.0 mg/kg [body weight]) every day for 8 days. Survival and parasite burdens were monitored at 2-day intervals for 25 days, as described above.
Compounds.
Structures of compounds used in this study are given in Fig. 1. Artemisinin was obtained from Sigma Aldrich. Artesunate was a gift from Knoll AG, Basel, Switzerland. The 10-amino-artemisinins artemisone, artemiside, the sulfamide derivative, the benzylpiperazine derivative, and the pyrimidylpiperazine derivative were prepared as previously described (10). The 4′-fluorophenyl derivative was prepared as described previously (12). Compounds were dissolved in dimethyl sulfoxide (DMSO) and stored at −80°C in single-use aliquots until used for in vitro studies. Alternatively, compounds were dissolved in DMSO and stored at 4°C for in vivo studies.
RESULTS
Testing the in vitro antiparasitic activity of new artemisinin derivates.
The antiparasitic activities of several new artemisinin derivates were monitored using a β-galactosidase assay for parasite growth in vitro (27). This assay relates enzyme activity from a transgenic T. gondii line expressing Escherichia coli β-galactosidase to parasite numbers and is highly linear over a wide range of cell numbers (33). We used a derivative of the RH strain for these experiments, as it represents the rapidly growing type I strains that are acutely virulent in the mouse model. Following treatment in vitro for 72 h, all of the compounds inhibited parasite growth, with similar EC50s ranging from ∼0.11 to 0.21 μM and with EC50s ranging from 0.17 to 0.37 μM (Fig. 2; Table 1). These activities compare favorably to those of artemisinin, which previous studies have indicated has an EC50 in vitro of ∼0.8 μM (27). The most potent compounds were artemiside and artemisone (Fig. 2; Table 1). Artemisone was somewhat less potent than previously reported (EC50 of ∼0.03 μM) (27), although further testing of these earlier stocks reveal inhibition consistent with that reported here (data not shown). This loss of potency may have resulted from freezing the stocks in DMSO and thawing prior to testing, a procedure we subsequently determined resulted in modest loss of activity. None of the compounds demonstrated overt toxicity to host cell monolayers (data not shown).
FIG. 2.

Inhibition of in vitro growth of T. gondii by artemisinin derivatives. Parasite growth was monitored by measurement of β-galactosidase activity (absorbance at 570 nm) following 72 h of incubation with the different compounds (see Materials and Methods). Artemiside and artemisone showed slightly better activity in vitro compared to the other compounds. 7, sulfamide derivative; 8, benzylpiperazine derivative; 9, pyrimidylpiperazine derivative; and 10, 4′-fluorophenyl derivative.
TABLE 1.
Effective concentrations for inhibition of T. gondii growth in vitrob
| Compounda | EC50 (μM) | EC90 (μM) |
|---|---|---|
| Artemisone | 0.120 | 0.176 |
| Artemiside | 0.108 | 0.167 |
| Artesunate | 0.213 | 0.368 |
| Sulfamide derivative | 0.158 | 0.235 |
| 4′-Fluorophenyl derivative | 0.182 | 0.271 |
| Benzylpiperazine derivative | 0.213 | 0.339 |
| Pyrimidylpiperazine derivative | 0.156 | 0.263 |
Effect of artemisinin derivates on acute infection with T. gondii in the murine model.
We tested the antimicrobial activity of the most-active compounds using an in vivo assay for acute toxoplasmosis in the mouse. Mice were infected i.p. with tachyzoites of type II (PRU-Luc-GFP) T. gondii, compounds were injected every day for 8 days, and survival was monitored. While all control mice and most artemisinin-treated mice succumbed to infection, 60% of the artemiside-treated mice and more than 50% of the artemisone-treated mice survived the infection (Fig. 3A). To follow parasite replication, mice were infected as described above, and parasite numbers were detected by bioluminescence imaging. All control mice and the majority of artemisinin-treated mice developed high parasite numbers and died due to the uncontrolled infection (Fig. 2B). Artemiside- and artemisone-treated mice were able to control parasite replication and survived, indicating that these compounds effectively blocked replication and/or resulted in killing of parasites in vivo (Fig. 3B). To evaluate cyst formation in the chronic phase of the infection, brains were removed from chronically infected mice at 25 days postinfection, and cysts were counted microscopically. Artemiside- and artemisone-treated mice developed significantly lower cyst numbers in the brain compared to artemisinin-treated mice, confirming the antiparasitic efficacy of the compounds (Fig. 3C). This result likely reflects lower burdens of tachyzoites that developed in treated animals, rather than a direct effect on tissue cysts. Although cyst counts were not available from control mice, which succumbed to infection, separate studies using sublethal doses (about 10-fold lower than those used here) resulted in cyst burdens that ranged from 200 to 300 cysts per brain (data not shown).
FIG. 3.
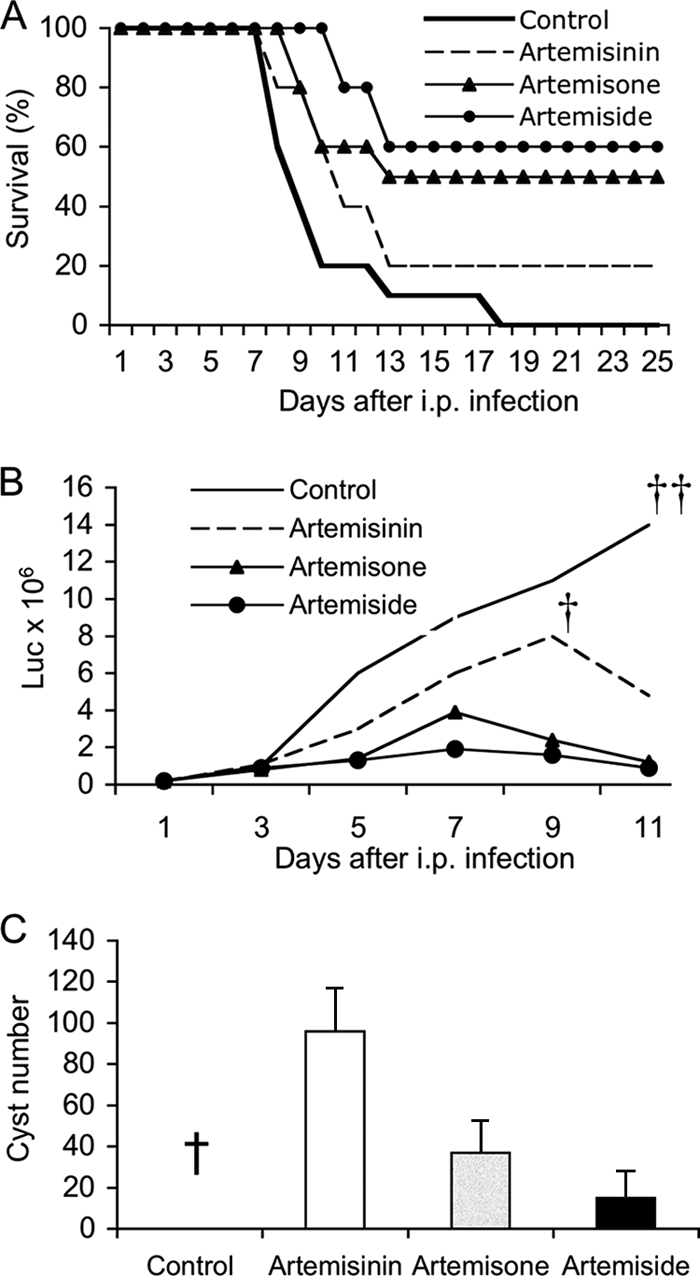
Inhibition of in vivo growth of T. gondii by artemisinin derivatives. (A) Survival of mice following oral challenge with T. gondii and administration of artemisinin derivates. CD1 mice were infected i.p. with 106 tachyzoites of type II (PRU-Luc-GFP) T. gondii, and compounds were injected (s.c. with 10 mg/kg) every day for 8 days. Survival of mice was monitored for 25 days. The majority of the artemiside- and artemisone-treated mice survived the infection, while artemisinin-treated and control mice died. Combined results from the three experiments, n = 3 or 4 animals per group, are shown. (B) Parasite burdens in mice during acute infection as measured by luciferase imaging. CD1 mice were infected with 106 tachyzoites of type II (PRU-Luc-GFP) parasites, treated with compounds (10 mg/kg s.c.) every day for 8 days, and imaged every second day. Artemiside- and artemisone-treated mice were able to control the infection. Experiments were repeated four times; n = 3 or 4 animals per group. Graph shows one representative experiment. † indicates 4 of 5 animals succumbed to infection; thus, the average luciferase (Luc) value dropped after this point. †† indicates all animals succumbed to infection. Luc values represent relative light output. (C) Chronic infection in mice as monitored by cyst formation in the brains of CD1 mice. Artemiside- and artemisone-treated mice developed lower cyst numbers in the brain compared to artemisinin-treated mice. Data shown here are combined results from three experiments; n = 2 animals per group. Numbers represent an estimate of the total number of cysts per brain. Numbers are not available for control mice, which succumbed to infection (†).
Effect of artemisinin derivates on reactivation of latent T. gondii infection.
To simulate reactivation of latent infection, we used the murine model of reactivated toxoplasmosis in IFN-γ−/− mice, similar to that described previously (38). IFN-γ−/− mice are highly susceptible to both acute (39) and chronic reactivation (36) of toxoplasmosis. In order to test the efficacy of compounds during reaction, mice were challenged with parasites and treated with sulfadiazine to suppress acute infection and allow development of tissue cysts, which occurs during the first few weeks (Fig. 4A). Treatment was then removed to allow reactivation, which occurs by a process of stage conversion from bradyzoite to tachyzoite. Because IFN-γ−/− mice cannot control proliferation of tachyzoites, they rapidly succumb to infection following removal of sulfadiazine. We compared the efficacy of the most potent artemisinin derivatives using this in vivo model. Following the removal of sulfadiazine, treatment with artemiside prolonged survival in 80% of the mice, while treatment with artemisone prolonged survival in 60% of mice (Fig. 4B). In contrast, all of the control mice died within 10 days (Fig. 4B). Although these two compounds were able to control the initial phases of reactivated infection, they did not result in a complete cure. Rather, when the drugs were discontinued, mice began dying at day 18, and all mice eventually died by day 25 (data not shown). This result indicates that artemisinin compounds were not able to eradicate the chronic infection, and hence, they likely do not act on bradyzoites directly. Similar outcomes are seen with treatment with other drugs that are nonetheless capable of controlling replication of tachyzoites, such as atovaquone, which has previously been tested using a similar model (10, 32).
FIG. 4.
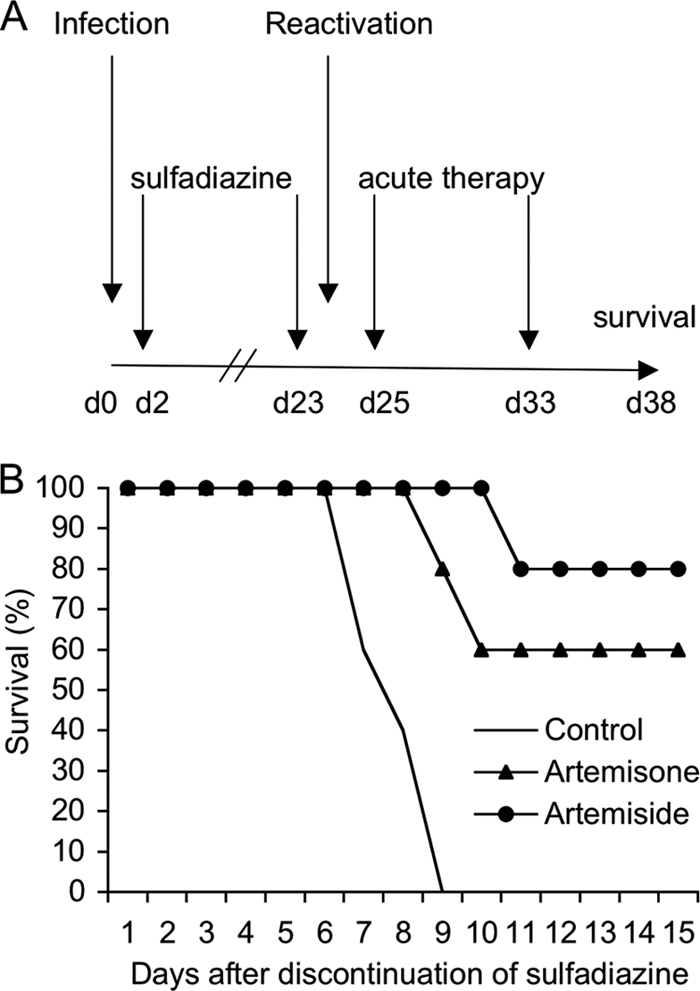
Inhibition of reactivation of chronic toxoplasmosis by artemisinin derivatives. (A) Murine model of reactivated toxoplasmosis. At day 0, IFN-γ−/− mice were infected orally with 20 cysts of PRU-Luc-GFP, and 2 days later sulfadiazine (400 mg/kg) was administered for 3 weeks in the drinking water. Two days after discontinuation of the drug, compounds were administered for 8 days (s.c., 10 mg/kg), and survival was monitored. (B) Treatment with artemiside or artemisone resulted in increased survival following reactivation. Data shown are combined results from three individual experiments; n = 3 to 5 animals per group.
DISCUSSION
Previous studies have shown that although artemisinin is somewhat active against T. gondii in vitro, it requires near-micromolar levels to inhibit in vitro growth by 50% (EC50 is 0.8 μM) (27). Several artemisinin derivatives, including artemether, artesunate, and artemisone, have been shown to be more effective against T. gondii in vitro (27). In comparison, artemisinin derivatives tested here demonstrate that artemisone and artemiside are the most potent analogs to date in terms of inhibiting growth of T. gondii in vitro. We tested these derivatives only against the rapidly growing type I strain RH, and it is conceivable that they might have different potencies against slower-growing type II strains, which account for most human infections (1, 15). Moreover, the efficacy of artemisinins against more diverse strains of T. gondii, which have recently been recognized as common in regions such as South America (18, 20), remains to be established. Nonetheless, our studies demonstrate that several artemisinin derivatives show promise for inhibiting T. gondii in vitro and in vivo.
Artemisone is a 10-alkylaminoartemisinin that was developed as a potent antimalarial drug (13). It demonstrated excellent pharmacodynamics and pharmacokinetics, and it was well tolerated by subjects in phase I trials (28). Artemisone displayed greater efficacy than artesunate (approximately threefold) in phase II trials with nonsevere-malaria patients (19). Artemisone also possesses negligible neurotoxicity both in vitro and in vivo (28). In contrast, the neurotoxicity of the current clinically used derivatives of artemisinins, such as artesunate or DHA, present a concern if protracted dose loadings are to be used against targets other than malaria. Although artemisinin and its derivatives are partially effective in inhibiting T. gondii in vitro, few studies have been performed to evaluate in vivo efficacies. A previous study using artemether, an analogue that has slightly lower activity in vitro, failed to show efficacy in a rat model of combined infection with T. gondii and P. carinii (6). This study used relatively high doses of compounds (18 and 100 mg/kg), and the failure to observe antitoxoplasmal activity may relate to characteristics of the rat model, a host that is highly resistant to toxoplasmosis. Here, we chose to explore the activities of several artemisinin derivatives in more susceptible murine models for acute and chronic toxoplasmosis.
We have previously found that the more lipophilic aminoartemisinin derivatives tend to be more active as antimalarial drugs in vitro. This correlation is also seen in vivo, where more lipophilic compounds, such as artemiside, exhibit at least threefold greater activity against both chloroquine-sensitive and -resistant strains of P. berghei and Plasmodium yoelii (10). To be effective in treating reactivation of chronic toxoplasmosis, compounds need to penetrate the blood-brain barrier. Therefore, we have selected compounds with a range of lipophilicities in order to establish how important this is in relation to in vivo activities. The log P values (log10 of the partition coefficient) for selected compounds are given in Fig. 1 and reflect their relative hydrophobicity as previously determined (10). However, it is clear from the in vitro data shown in Fig. 2 that there is no simple relationship between lipophilicity and efficacy. Rather, the studies demonstrate that artemisone (log P = 2.49) and artemiside (log P = 4.97) are the most active of the compounds. We observed that artemiside and artemisone were approximately equipotent and were about eightfold more effective than artemisinin and about twofold more active than artesunate. They were also slightly better than a series of semisynthetic alkylated derivatives of artemisinin that have been further described elsewhere (13). Other reports comparing artemisinin and artemether and several synthetic analogues report much higher EC50s (i.e., 8 uM for artemisinin [17] versus 0.8 uM for our studies [27]); although the reasons for this discrepancy are unknown.
Based on their in vitro efficacy and favorable pharmacokinetic and pharmacodynamic properties, we tested artemisone and artemiside using in vivo models for toxoplasmosis. Both of these compounds were effective in reducing mortality during acute challenge in the mouse model. Furthermore, there was an accompanying reduction in the chronic burden of tissue cysts in the CNS. Our studies do not suggest that either compound acts directly on bradyzoites within tissue cysts. Rather, the protective outcome is likely due to the overall reduced parasite burden due to inhibition and/or killing of tachyzoites in vivo. Although the doses of compounds necessary to achieve this effect are quite high, this may reflect pharmacokinetic parameters of the murine model (i.e., more-rapid turnover and/or clearance). Hence, the effective concentrations used here cannot be directly extrapolated to other hosts, including humans.
Although current antibiotic therapy is available for toxoplasmosis, it suffers from problems of intolerance to sulfa drugs combined with a requirement for long-term maintenance, owing to a lack of efficacy against bradyzoites within tissue cysts (22). Although artemisinin derivatives do not directly address this deficiency, they may offer some potential for extending the spectrum of therapies available for toxoplasmosis. The ability to control tachyzoite replication may have utility for treatment of either primary acute toxoplasmosis or reactivation of chronic infection. Ocular toxoplasmosis is generally recognized to result from reactivation of congenital infection, yet in some regions, notably southern Brazil, ocular infections can occur following newly acquired infections in immunocompetent adults (16, 34). Ocular infections are often severe in these regions, thus representing patient populations for which new therapies with reduced toxicity and fewer side effects may be particularly valuable.
New drugs capable of controlling acute replication would also be valuable for use during pregnancy, when maternal infection may threaten to spread to the developing fetus. At present, spiramycin is used for such treatment in Europe, despite the lack of controlled trials showing efficacy (26). Spiramycin does not cross the placental barrier and so is not effective in treating infection in the fetus. Treatment with pyrimethamine and sulfadiazine (with folic acid supplementation) is recommended in cases of confirmed fetal infection; however, this treatment can be given only after the first trimester (26). Pyrimethamine-sulfadiazine treatment has been shown to improve outcomes in some cases (23); however, it carries the added risk of adverse drug reactions. Hence, more effective treatments that have fewer side effects would be beneficial. Artemisone has been shown to be safe in human phase I trials, and previous studies have shown that it is less neurotoxic and antiangiogenic than DHA (8). Despite this improved safety in humans, artemisone has shown fetal toxicity in a rat pregnancy model, although rats are more susceptible to these effects than rabbits (31). Development of less fetotoxic derivatives of artemisinin may be of use for preventing congenital infections.
We also tested both artemisone and artemiside in a murine model for reactivation that simulates the situation in immunocompromised patients. By treating IFN-γ−/− mice with sulfadiazine, it is possible to suppress active replication of tachyzoites but allow development of tissue cysts (38). Removal of the drug allows reactivation to occur due to cyst rupture and spontaneous conversion to tachyzoites. In the absence of IFN-γ, this leads to uncontrolled parasite replication (36). This model mimics the situation with severely immunocompromised patients that lack the T cells required to effectively control the parasite. Previous studies have used a very similar model based on knockout of the interferon consensus sequence binding protein (ICSBP) to demonstrate the efficacy of atovaquone for controlling reactivation in the mouse model of toxoplasmosis (10, 32). In the present study, artemiside showed slightly better protection in the murine reactivation model, and mice were also partially protected from lethal reactivation by treatment with artemisone. This greater potency might reflect the higher lipophilic nature of artemiside, perhaps allowing better access to the CNS. Although artemisone is a relatively polar artemisinin derivative, biodistribution studies of radiolabeled artemisone in rats indicate moderate penetration of the CNS after oral dosing (35). The corresponding studies for artemiside have not been carried out, but as it is substantially more lipophilic than artemisone, blood-brain penetration is expected to be higher.
The efficacies of artemisone and artemiside compare favorably with that of atovaquone (32), which has been used to decrease relapse of toxoplasmosis in AIDS patients (7). Combined treatments with artemisone or artemiside and pyrimethamine or clindamycin might also prove effective, and such regimens could provide an alternative for treatment in patients that fail to tolerate sulfa drugs.
In summary, the potent antimalarial compounds artemisone and artemiside show reasonable efficacy against toxoplasmosis in the murine model, raising the possibility that they may also be effective in controlling human toxoplasmosis.
Acknowledgments
This work was supported in part by a grant from the NIH (AI071299 and AI059176 to L.D.S.). I.R.D. was partially supported by a fellowship from the DFG, Germany.
Work at Hong Kong University of Science and Technology was carried out in the Open Laboratory of Chemical Biology of the Institute of Molecular Technology for Drug Discovery and Synthesis through financial support from the government of the HKSAR, University Grants Committee Areas of Excellence Fund, projects no. AoE P/10-01/01-02-I and AOE/P-10/01-2-II, and the University Grants Council grants no. HKUST 6493/06 M and 600507.
Footnotes
Published ahead of print on 27 July 2009.
REFERENCES
- 1.Ajzenberg, D., N. Cogné, L. Paris, M. H. Bessieres, P. Thulliez, D. Fillisetti, H. Pelloux, P. Marty, and M. L. Dardé. 2002. Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis and correlation with clinical findings. J. Infect. Dis. 186:684-689. [DOI] [PubMed] [Google Scholar]
- 2.Araujo, F. G., J. Huskinson, and J. S. Remington. 1991. Remarkable in vitro and in vivo activities of the hydroxynaphthoquinone 566C80 against tachyzoites and tissue cysts of Toxoplasma gondii. Antimicrob. Agents Chemother. 35:293-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashley, E. A., and N. J. White. 2005. Artemisinin-based combinations. Curr. Opin. Infect. Dis. 18:531-536. [DOI] [PubMed] [Google Scholar]
- 4.Baatz, H., A. Mirashahi, J. Puchta, H. Gumbel, and L. O. Hattenbach. 2006. Reactivation of toxoplasma retinochoroiditis under atovaquone therapy in an immunocompetent patient. Ocul. Immunol. Inflamm. 14:185-187. [DOI] [PubMed] [Google Scholar]
- 5.Barragan, A., and L. D. Sibley. 2003. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol. 11:426-430. [DOI] [PubMed] [Google Scholar]
- 6.Brun-Pascaud, M., F. Chau, F. Derouin, and P. M. Girard. 1996. Lack of activity of artemether for prophylaxis and treatment of Toxoplasma gondii and Pneumocystis carinii infections in rat. Parasite 3:187-189. [DOI] [PubMed] [Google Scholar]
- 7.Chirgwin, K., R. Hafner, C. Leport, J. Remington, J. Andersen, E. M. Bosler, C. Roque, N. Rajicic, V. McAuliffe, P. Morlat, D. T. Jayaweera, J. L. Vilde, B. J. Luft, et al. 2002. Randomized phase II trial of atovaquone with pyrimethamine and sulfadiazine treatment of toxoplasmic encephalitis in patients with acquired immunodeficiency syndrome: ATCC 237/ANRS 039 Study. Clin. Infect. Dis. 34:1243-1250. [DOI] [PubMed] [Google Scholar]
- 8.D'Alessandro, S., M. Gelati, N. Basilico, E. A. Parati, R. K. Haynes, and D. Taramelli. 2007. Differential effects on angiogenesis of two antimalarial compounds, dihydroartemisinin and artemisone: implications for embryotoxicity. Toxicology 241:66-74. [DOI] [PubMed] [Google Scholar]
- 9.Dubey, J. P. 2007. The life cycle of Toxoplasma gondii, p. 3-16. In J. W. Ajioka and D. Soldati (ed.), Toxoplasma: molecular and cellular biology. Horizon Bioscience, Norfolk, United Kingdom.
- 10.Dunay, I. R., M. M. Heimesaat, F. N. Bushrab, R. H. Muller, H. Stocker, K. Arasteh, M. Kurowski, R. Fitzner, K. Borner, and O. Liesenfeld. 2004. Atovaquone maintenance therapy prevents reactivation of toxopasmic encepahalitis in the murine model of reactivated toxoplasmosis. Antimicrob. Agents Chemother. 48:4848-4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall, S., K. A. Ryan, and D. Buxton. 2001. The epidemiology of toxoplasma infection, p. 58-124. In D. H. Joynson, and T. J. Wreghitt (ed.), Toxoplasmosis: a comprehensive clinical guide. Cambridge University Press, Cambridge, United Kingdom.
- 12.Haynes, R. K., H. O. Chan, M. K. Cheung, S. T. Chung, W. L. Lam, H. W. Tsang, A. Voerste, and I. D. Williams. 2003. Stereoselective preparation of 10α- and 10β-aryl derivatives of dihydroartemisinin. Eur. J. Org. Chem. 2003:2098-2114. [Google Scholar]
- 13.Haynes, R. K., B. Fugmann, J. Stetter, K. Rieckmann, H. D. Heilmann, H. W. Chan, M. K. Cheung, W. L. Lam, H. N. Wong, S. L. Croft, L. Vivas, L. Rattray, L. Stewart, W. Peters, B. L. Robinson, M. D. Edstien, B. M. Kotecka, D. E. Kyle, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke, U. Wollborn, K. Schmeer, and A. S. Romer. 2006. Artemisone: a highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. Engl. 45:1-8. [DOI] [PubMed] [Google Scholar]
- 14.Haynes, R. K., and S. Krishna. 2004. Artemisinins: activities and actions. Microb. Infect. 6:1339-1346. [DOI] [PubMed] [Google Scholar]
- 15.Howe, D. K., and L. D. Sibley. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis. 172:1561-1566. [DOI] [PubMed] [Google Scholar]
- 16.Jones, J. L., C. Muccioli, R. Belfort, Jr., G. N. Holland, J. M. Roberts, and C. Silveira. 2006. Recently acquired Toxoplasma gondii infection, Brazil. Emerg. Infect. Dis. 12:582-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones-Brando, L., J. D'Angelo, G. H. Posner, and R. H. Yolken. 2006. In vitro inhibition of Toxoplasma gondii by four new derivatives of artemisinin. Antimicrob. Agents Chemother. 50:4206-4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan, A., B. Fux, C. Su, J. P. Dubey, M. L. Darde, J. W. Ajioka, B. M. Rosenthal, and L. D. Sibley. 2007. Recent transcontinental sweep of Toxoplasma gondii driven by a single monomorphic chromosome. Proc. Natl. Acad. Sci. USA 104:14872-14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krudsood, S., P. Wilairatana, K. Chalermrut, W. Leowattana, B. Voith, B. Hampel, and S. Looareesuwan. 2005. Artemifone, a new anti-malarial artemisinin derivative: open pilot trial to investigate the antiparasitic activity of bay 44-9585 in patients with uncomplicated P. falciparum malaria, p. 142. In Medicine and Health in the Tropics. Proceedings of the XVI International Congress for Tropical Medicine and Malaria. Marseilles, France.
- 20.Lehmann, T., P. L. Marcet, D. H. Graham, E. R. Dahl, and J. P. Dubey. 2006. Globalization and the population structure of Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 103:11423-11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, Q., F. Wei, S. Gao, L. Jiang, H. Lian, B. Yuan, Z. Yuan, Z. Xia, B. Liu, Z. Xu, and X. Q. Zhu. 2009. Toxoplasma gondii infection in pregnant women in China. Trans. R. Soc. Trop. Med. Hyg. 103:162-166. [DOI] [PubMed] [Google Scholar]
- 22.McCabe, R. E. 2001. Antitoxoplasma chemotherapy, p. 319-359. In D. H. M. Joynson, and T. G. Wreghitt (ed.), Toxoplasmosis: a comprehensive clinical guide. Cambridge University Press, Cambridge, United Kingdom.
- 23.McLeod, R., K. Boyer, T. Karrison, K. Kasza, C. Swisher, N. Roizen, J. Jalbrzikowski, J. Remington, P. Heydemann, A. G. Noble, M. Mets, E. Holfels, S. Withers, P. Latkany, P. Meier, and Toxoplasmosis Study Group. 2006. Outcome of treatment for congenital toxoplasmosis, 1981-2004: the National Collaborative Chicago-Based, Congenital Toxoplasmosis Study. Clin. Infect. Dis. 42:1383-1394. [DOI] [PubMed] [Google Scholar]
- 24.Megged, O., I. Shalit, I. Yaniv, J. Stein, S. Fisher, and I. Levy. 2008. Breakthrough cerebral toxoplasmosis in a patient receiving atovaquone prophylaxis after a hematopoietic stem cell transplant. Pediatr. Transplant. 12:902-905. [DOI] [PubMed] [Google Scholar]
- 25.Montoya, J. G., and O. Liesenfled. 2004. Toxoplasmosis. Lancet 363:1965-1976. [DOI] [PubMed] [Google Scholar]
- 26.Montoya, J. G., and J. S. Remington. 2008. Management of Toxoplasma gondii infection during pregnancy. Clin. Infect. Dis. 47:554-566. [DOI] [PubMed] [Google Scholar]
- 27.Nagamune, K., S. N. J. Moreno, and L. D. Sibley. 2007. Artemisinin-resistant mutants of Toxoplasma gondii have altered calcium homeostasis. Antimicrob. Agents Chemother. 51:3816-3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagelschmitz, J., B. Voith, G. Wensing, A. Roemer, B. Fugmann, R. K. Haynes, B. M. Kotecka, K. H. Rieckmann, and M. D. Edstein. 2008. First assessment in humans of the safety, tolerability, pharmacokinetics, and ex vivo pharmacodynamic antimalarial activity of the new artemisinin derivative artemisone. Antimicrob. Agents Chemother. 52:3085-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petersen, E., and O. Liesenfeld. 2008. Clinical disease and diagnostics, p. 81-100. In L. M. Weiss and K. Kim (ed.), Toxoplasma gondii. The model Apicomplexan: perspectives and methods. Academic Press, Amsterdam, The Netherlands.
- 30.Saeij, J. P., J. P. Boyle, M. E. Grigg, G. Arrizabalaga, and J. C. Boothroyd. 2005. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73:695-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmuck, G., A. M. Klaus, F. Krotlinger, and F. W. Langewische. 2009. Developmental and reproductive toxicity studies on artemisone. Birth Defects Res. B 86:131-143. [DOI] [PubMed] [Google Scholar]
- 32.Scholer, N., K. Krause, O. Kayser, R. H. Muller, K. Borner, H. Hahn, and O. Liesenfeld. 2001. Atovaquone nanosuspensions show excellent therapeutic effect in a new murine model of reactivated toxoplasmosis. Antimicrob. Agents Chemother. 45:1771-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seeber, F., and J. C. Boothroyd. 1996. Escherichia coli β-galactosidase as an in vitro and in vivo reporter enzyme and stable transfection marker in the intracellular protozoan parasite Toxoplasma gondii. Gene 169:39-45. [DOI] [PubMed] [Google Scholar]
- 34.Silveira, C., R. Belfort, Jr., C. Muccioli, M. T. Abreu, M. C. Martins, C. Victora, R. B. Nussenblatt, and G. N. Holland. 2001. A follow-up study of Toxoplasma gondii infection in southern Brazil. Am. J. Ophthalmol. 131:351-354. [DOI] [PubMed] [Google Scholar]
- 35.Steinke, W., B. Beckermann, H. Fischer, M. Gerisch, C. Kohlsdorfer, A. Römer, and G. Schmuck. 2005. Artemifone, a new anti-malarial artemisinin derivative: pharmacokinetics in rats and dogs, p. 143. In Medicine and Health in the Tropics. Proceedings of the XVI International Congress for Tropical Medicine and Malaria. Marseilles, France.
- 36.Suzuki, Y., F. K. Conley, and J. S. Remington. 1989. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J. Immunol. 143:2045-2050. [PubMed] [Google Scholar]
- 37.Suzuki, Y., and K. Joh. 1994. Effect of the strain of Toxoplasma gondii on the development of toxoplasmic encephalitis in mice treated with antibody to interferon-gamma. Parasitol. Res. 80:125-130. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki, Y., H. Kang, S. F. Parmley, S. Lim, and D. Park. 2000. Induction of tumor necrosis factor-α and inducible nitric oxide synthase fails to prevent toxoplasmic encephalitis in the absence of interferon-γ in genetically resistant BALB/c mice. Microb. Infect. 2:455-462. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki, Y., M. A. Orellana, R. D. Schreiber, and J. S. Remington. 1988. Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science 240:516-518. [DOI] [PubMed] [Google Scholar]
- 40.Vivas, L., L. Rattray, L. B. Stewart, B. L. Robinson, B. Fugmann, R. K. Haynes, W. Peters, and S. L. Croft. 2007. Antimalarial efficacy and drug interactions of the novel semi-synthetic endoperoxide artemisone in vitro and in vivo. J. Antimicrob. Chemother. 59:658-665. [DOI] [PubMed] [Google Scholar]
- 41.Wong, S., and J. S. Remington. 1994. Toxoplasmosis in pregnancy. Clin. Infect. Dis. 18:853-862. [DOI] [PubMed] [Google Scholar]