

Abstract
Driven by findings that human papillomavirus (HPV)-induced degradation of p53 differs by a TP53 polymorphism at codon 72 (Pro72Arg), past studies of TP53 genetic variants and cervical cancer have focused on this nonsynonymous polymorphism, with mixed results. We analyzed common single nucleotide polymorphisms (SNPs) across the TP53 locus in a population-based nested case-control study in Guanacaste, Costa Rica. We evaluated 11 SNPs, including Pro72Arg (rs1042522), among 1,281 women: 465 with cervical intraepithelial neoplasia grade 3/cancer (CIN3+), 380 with HPV persistence (median: 25 months), and 436 random population controls. We combined HPV persistence and CIN3+ into one case group because they did not differ in TP53 genotypic frequencies and calculated odds ratios (OR) and 95%confidence intervals (CI) for individual SNPs and inferred haplotypes. We observed that proline at codon 72 was associated with increased risk of CIN3+/persistence compared to population controls. Relative to GG (Arg), the CG (Pro/Arg) and CC (Pro) genotypes had a 1.3-fold (95% CI, 0.99–1.6) and 1.8-fold (95% CI, 1.2–2.7) increased risk, respectively (p-trend<0.01). rs12951053 and rs1642785 were also associated with CIN3+/persistence (p-trend, 0.05 and 0.04, respectively), as was a haplotype containing the codon 72 variant (rs1042522), rs12951053, rs1642785, and rs12947788 (OR, 1.6; 95% CI, 1.1–2.3 versus the most common haplotype, which comprised the major alleles for all 11 SNPs). Although genetic variation in TP53 might affect the natural history of HPV and cervical cancer, further work is needed to elucidate the possible mechanism.
Keywords: TP53, polymorphisms, human papillomavirus, persistence, cervical neoplasia
Introduction
The TP53 gene encodes the p53 protein, which is involved in a series of pathways, such as apoptosis, transcription, DNA repair, maintenance of genomic stability, and cell cycle control (1). Human papillomavirus (HPV) specifically targets p53, along with phosphorylated Rb and other proteins, in order to maintain a cellular environment that promotes viral replication (2). By disrupting normal cell cycle control, HPV may establish persistent infection, a key step in cervical carcinogenesis (3, 4).
Polymorphisms in the TP53 gene have been hypothesized to modify the conformation of the p53 protein, affecting the ability of HPV to bind and degrade p53 and thus potentially acting early in cervical carcinogenesis. In particular, most studies have examined a common nonsynonymous SNP at codon 72, which results in a shift from arginine to proline. An initial laboratory study suggested that the HPV16 E6 protein was better able to degrade p53 containing arginine [G] at codon 72 than TP53 containing proline [C] at that position (5), prompting several epidemiological studies to evaluate the association between this specific polymorphism and risk of cervical cancer and its precursors. The results of these studies have been conflicting.
A meta-analysis of 45 studies published between 1998 and 2002 suggested that any effect of TP53 codon 72 polymorphisms is likely to be modest, with a small increased risk associated with arginine at codon 72 (6). The meta-analysis also found substantial heterogeneity between the estimates from these studies. Some of this heterogeneity was attributable to study populations with genotypic distributions that departed from Hardy-Weinberg equilibrium, which may suggest problems with genotyping/laboratory methods or study design (7, 8).
Our previous study across three populations found no evidence that arginine at TP53 codon 72 increased risk of cervical cancer or its precursors (9). In fact, there was a trend toward increased risk of disease associated with proline at codon 72. Other studies have also found increased risk associated with proline at codon 72 (10–12).
To our knowledge, no studies to date have evaluated both multiple TP53 polymorphisms and TP53 haplotypes and risk of cervical cancer and its precursors. An expanded analysis of TP53 polymorphisms may add insight since other SNPs in addition to that at codon 72 might also affect amino acid change. To address this gap, we extended our previous study by expanding the number of individuals evaluated and by evaluating a set of 11 SNPs (including the SNP that defines the codon 72 polymorphism) to determine whether TP53 codon 72 or other single nucleotide polymorphisms (SNPs) and/or haplotypes are associated with cervical cancer or its precursors within our population-based cohort study in Guanacaste, Costa Rica.
Materials and Methods
Study Population
The present study used a population-based cohort in Guanacaste, Costa Rica, where 10,049 women were recruited in 1993-4 and followed for seven years either actively at six-month to one-year intervals or passively, depending on risk of developing cervical intraepithelial neoplasia (CIN) grade 2 or higher (13, 14). Cervical samples were collected for HPV testing as previously described (13, 14). Human genetic material came from buffy coat specimens. We selected from the population-based cohort all women with histologically confirmed prevalent or incident cervical intraepithelial neoplasia grade 3 or cancer (CIN3+) (n=185); women with type-specific HPV persistence, defined as positivity for the same HPV type at two or more consecutive visits (n=432) (median length of persistence: 25 months, range: 5–93 months); and a random selection of controls with <CIN2 who were either HPV negative or had non-persistent HPV (n=492). Women with CIN2 and without persistent HPV were excluded because CIN2 has poor reproducibility (15) and many CIN2 cases regress (16). Women with CIN2 who also had type-specific HPV persistence were included in the persistent HPV group.
Because the original cohort was a true random sample of the Guanacaste population, they arose from the same source population as cases included in the Costa Rican National Tumor Registry and National Cytology Laboratory. Thus, we identified 448 additional CIN3+ cases from Guanacaste who were not in the cohort but were diagnosed with CIN3+ from June 1993 through December 1994 by retrieving and reviewing pathology records. Of these, 56 (13%) were deceased, 18 refused (4%), 39 (8%) could not be found, 4 (1%) did not have linkable data, and 4 (1%) were sick or pregnant, leaving 327 supplemental cases (73%) who agreed to participate and gave 20 mL of peripheral blood. The supplemental cases were genotypically similar to the cohort cases (data not shown), and thus could be combined. Both NCI and Costa Rica Institutional Review Boards approved the study.
Laboratory Methods
DNA extraction
The PureGene purification kits/Autopure protocol (Gentra Systems) was used to extract DNA from the buffy coats at the University of Costa Rica in San Jose.
HPV testing
In the Guanacaste cohort study, cervical specimens were collected with a Cervex (Rovers, The Netherlands) cervix brush and stored in specimen transport medium (STM, Digene, Gaithersburg, MD). All specimens were tested for HPV DNA by PCR using a MY09/MY11 primer system (13, 14, 17, 18). Cervical specimens, and thus HPV testing results, were not available for supplemental cases.
TP53 Genotyping
Among the women selected from the cohort study, 454/492 (92%) random controls, 390/432 (90%) women with persistent HPV infection, and 149/185 (81%) CIN3+ cases had sufficient DNA for genotyping, along with 322/448 (72%) of the initially identified supplemental CIN3+ cases. We evaluated 11 SNPs by genotype assays across the TP53 gene that were developed using the SNP500Cancer data set, which includes 23 individuals of self-described Hispanic background as well as 79 individuals of African, Caucasian, and Pacific Rim 1. All SNPs were synonymous except the SNP at codon 72. Genotype assays were performed at the National Cancer Institute Core Genotyping Facility (Gaithersburg, MD) using Taqman (Foster City, CA) or Epoch (Bothell, WA) platforms with the sequence data and assay conditions 1. Previously, extensive resequence analysis had been performed on 23 Hispanic individuals from the SNP500Cancer project. The 11 SNPs that were genotyped as a part of this project tag 10 of a total of 12 bins in the 23 Hispanic individuals. These SNPs span the TP53 gene from the beginning of the first intron to the end of exon 11.
We interspersed blinded duplicate samples from 100 participants throughout all assays for quality control (QC). All assays had ≥99% agreement between QC duplicates. Each plate also included genotype-specific QC samples of homozygote wild-type (WT), heterozygote, homozygote variant, and DNA-negative controls. Genotyping was successful for 96% to 100% of DNA samples for all SNPs. For the controls, none of the SNPs demonstrated a deviation from fitness for Hardy-Weinberg proportion. Sixty-four percent of the TP53 SNPs had p>0.2, and the lowest p value was 0.06 for rs2909430 with 350 observed AA, 88 AG, and 1 GG genotypes and 353.61 expected AA, 80.77 AG, and 4.61 GG genotypes.
Final analytic population
We analyzed genotyping results from a total of 465 CIN3+ cases, 380 women with persistent HPV infection, and 436 random controls.
Statistical Analysis
Gene-disease associations
We used the most frequent homozygous genotype as referent for each genotype. Given that we found no differences in the genotype distribution for CIN3+ cases compared to women with persistent HPV (Table 1), we combined CIN3+ and persistent HPV into one outcome. We also conducted a sensitivity analysis restricting to women with type-specific infections lasting 2 or more years and found no notable differences. Odds ratios (OR) and 95% confidence intervals (95% CI) for CIN3+ cases only compared to random controls and for women with persistent HPV compared to random controls are provided in Supplemental Table 1. In an additional sensitivity analysis, we restricted analyses of women with CIN3+/persistence versus random controls to women in the Guanacaste cohort, excluding supplemental cases (Supplemental Table 2). The distribution of TP53 genotypes was not associated with cohort versus supplemental case status (data not shown).
Table 1.
Distribution of TP53 genotypes among CIN3+ cases versus women with persistent HPV infection and corresponding odds ratios and 95% confidence intervals, adjusted for age.
| TP53 Polymorphism | Genotype | CIN3+ Cases (N=465) | Persistent HPV Controls (N=379) | OR* | 95% CI* | p-trend | |
|---|---|---|---|---|---|---|---|
| rs17887200† | AA | 397 | 334 | 1.0 | 0.23 | ||
| 22369bp 3' of STP A>G | AG | 58 | 43 | 1.2 | 0.78 | 1.9 | |
| GG | 2 | 0 | NA | ||||
| rs9894946† | CC | 396 | 316 | 1.0 | 0.28 | ||
| 22342bp 3' of STP T>C | CT | 59 | 60 | 0.76 | 0.51 | 1.1 | |
| TT | 4 | 3 | 1.2 | 0.25 | 5.6 | ||
| rs1614984† | CC | 249 | 221 | 1.0 | 0.18 | ||
| 21226bp 3' of STP C>T | CT | 175 | 131 | 1.2 | 0.90 | 1.6 | |
| TT | 38 | 27 | 1.3 | 0.74 | 2.2 | ||
| rs12951053† | TT | 321 | 260 | 1.0 | 0.69 | ||
| IVS7+92 T>G | GT | 133 | 105 | 1.1 | 0.78 | 1.5 | |
| GG | 7 | 12 | 0.49 | 0.19 | 1.3 | ||
| rs12947788† | CC | 321 | 262 | 1.0 | 0.68 | ||
| IVS7+72 T>C | CT | 137 | 106 | 1.1 | 0.79 | 1.5 | |
| TT | 4 | 11 | 0.34 | 0.11 | 1.1 | ||
| rs1625895† | GG | 348 | 282 | 1.0 | 0.88 | ||
| IVS6+62 A>G | AG | 104 | 90 | 0.96 | 0.69 | 1.3 | |
| AA | 10 | 7 | 1.1 | 0.39 | 2.9 | ||
| rs2909430† | AA | 366 | 292 | 1.0 | 0.42 | ||
| IVS4−91 A>G | AG | 85 | 81 | 0.84 | 0.59 | 1.2 | |
| GG | 7 | 5 | 1.0 | 0.31 | 3.4 | ||
| rs9895829† | TT | 416 | 333 | 1.0 | 0.60 | ||
| IVS4−125 T>C | CT | 30 | 25 | 0.98 | 0.56 | 1.7 | |
| CC | 2 | 3 | NA | ||||
| rs1042522 (Pro72Arg)† | GG | 206 | 182 | 1.0 | 0.29 | ||
| Ex4+119 C>G | CG | 191 | 144 | 1.2 | 0.91 | 1.7 | |
| CC | 61 | 50 | 1.2 | 0.75 | 1.8 | ||
| rs1642785† | GG | 217 | 166 | 1.0 | 0.66 | ||
| IVS2+38 C>G | CG | 190 | 167 | 0.88 | 0.65 | 1.2 | |
| CC | 51 | 43 | 0.99 | 0.62 | 1.6 | ||
| rs8079544 | GG | 433 | 355 | 1.0 | 0.67 | ||
| IVS1−112 G>A | AG | 30 | 23 | 1.1 | 0.61 | 1.9 | |
| AA | 2 | 1 | NA | ||||
OR, odds ratio. CI, confidence interval. Adjusted for age.
Numbers do not add up to totals because of missing values.
We used unconditional logistic regression with SAS version 8.2 (SAS Institute, Cary, NC) for crude analyses, age-adjusted analyses using tertiles of <30, 30–49, 50+ years, and p-trend analyses based on the three-level ordinal variable (0, 1, and 2) of homozygote common type, heterozygote, and homozygote variant. We also conducted analyses restricted to oncogenic HPV (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68) (n=249), HPV16 (n=62), and HPV18 (n=22) among women with persistent HPV infection and random controls. All CIN3+ cases were included in the oncogenic HPV analysis since oncogenic HPV has been shown to be present in nearly all CIN3+ (4), although only CIN3+ cases with HPV16 (n=55) or HPV18 (n=13) were included in additional analyses that restricted on these individual types.
Haplotype analysis
We examined haplotype structures defined by the genotyping of the 11 TP53 SNPs using Haploview version 3.11 (19), as well as an expectation-maximization algorithm to find the maximum-likelihood estimate (20). We assessed overall differences in haplotype distribution between CIN3+/persistent HPV cases and controls using the global score test (21) from the HaploStats program in R (version 2.0.1) (22). This program estimates additive risk estimates by fitting a logistic regression model using the posterior probabilities of the haplotypes as weights to estimate the regression coefficients in an iterative manner (21), adjusting for age.
To evaluate the potential for false-positive findings due to chance, we calculated the false discovery rate (FDR) using the Benjamini-Hochberg method (23), which provides the expected ratio of false-positive findings to the total number of significant findings. We applied the FDR method to the p-trend tests of the risk with each additional variant allele using both p < 0.05 and p < 0.20, as has been recommended for assessing the probability of false positives (24).
Results
Three common SNPs in TP53 were associated with risk of CIN3+ or HPV persistence compared to population controls (Table 2). In particular, rs12951053, rs1042522 (codon 72), and rs1642785 polymorphisms demonstrated increased risk for CIN3+/HPV persistence for each additional variant allele with p-trends of 0.05, 0.002, and 0.04, respectively. Specifically for rs1042522 (codon 72), the CG and CC genotypes (proline) conferred a 1.3-fold to 1.8-fold increased risk in CIN3+/HPV persistence compared to the common GG genotype (arginine). Findings were similar comparing CIN3+ alone to random population controls (Supplemental Table 1), comparing HPV persistence to random population controls (Supplemental Table 1), and restricting to women in the Guanacaste cohort (Supplemental Table 2), although the estimates were more precise in the combined analyses with the increased sample size (Table 2). Using p < 0.05, only rs1042522 (codon 72) remains significant after applying the FDR, but all three polymorphisms remained significant using p < 0.20.
Table 2.
Risk estimates (odds ratios) and 95% confidence intervals for TP53 polymorphisms in CIN3+/persistent HPV cases compared to random controls, adjusted for age.
| TP53 Polymorphism | Genotype | CIN3+/Persistent HPV Cases (N=844) | Population Controls (N=436) | OR* | 95% CI* | p-trend | |
|---|---|---|---|---|---|---|---|
| rs17887200† | AA | 731 | 385 | 1.0 | 0.52 | ||
| 22369bp 3' of STP A>G | AG | 101 | 45 | 1.2 | 0.82 | 1.7 | |
| GG | 2 | 2 | 0.55 | 0.08 | 3.9 | ||
| rs9894946† | CC | 712 | 366 | 1.0 | 0.82 | ||
| 22342bp 3' of STP T>C | CT | 119 | 68 | 0.91 | 0.65 | 1.3 | |
| TT | 7 | 2 | 1.9 | 0.38 | 9.0 | ||
| rs1614984† | CC | 470 | 244 | 1.0 | 0.99 | ||
| 21226bp 3' of STP C>T | CT | 306 | 152 | 1.1 | 0.82 | 1.3 | |
| TT | 65 | 36 | 0.93 | 0.60 | 1.4 | ||
| rs12951053† | TT | 581 | 323 | 1.0 | 0.05 | ||
| IVS7+92 T>G | GT | 238 | 101 | 1.3 | 1.0 | 1.7 | |
| GG | 19 | 8 | 1.3 | 0.58 | 3.1 | ||
| rs12947788† | CC | 583 | 324 | 1.0 | 0.09 | ||
| IVS7+72 T>C | CT | 243 | 104 | 1.3 | 1.0 | 1.7 | |
| TT | 15 | 8 | 1.1 | 0.45 | 2.6 | ||
| rs1625895† | GG | 630 | 332 | 1.0 | 0.18 | ||
| IVS6+62 A>G | AG | 194 | 97 | 1.1 | 0.80 | 1.4 | |
| AA | 17 | 2 | 4.4 | 1.0 | 19.0 | ||
| rs2909430† | AA | 658 | 345 | 1.0 | 0.34 | ||
| IVS4−91 A>G | AG | 166 | 86 | 1.0 | 0.76 | 1.4 | |
| GG | 12 | 1 | 6.1 | 0.79 | 47.5 | ||
| rs9895829† | TT | 749 | 379 | 1.0 | 0.98 | ||
| IVS4−125 T>C | CT | 55 | 31 | 0.89 | 0.57 | 1.4 | |
| CC | 5 | 1 | 2.5 | 0.29 | 21.4 | ||
| rs1042522 (Pro72Arg)b | GG | 388 | 234 | 1.0 | 0.002 | ||
| Ex4+119 C>G | CG | 335 | 160 | 1.3 | 0.99 | 1.6 | |
| CC | 111 | 37 | 1.8 | 1.2 | 2.7 | ||
| rs1642785† | GG | 383 | 213 | 1.0 | 0.04 | ||
| IVS2+38 C>G | CG | 357 | 183 | 1.1 | 0.85 | 1.4 | |
| CC | 94 | 31 | 1.7 | 1.1 | 2.6 | ||
| rs8079544† | GG | 788 | 404 | 1.0 | 0.70 | ||
| IVS1−112 G>A | AG | 53 | 29 | 0.93 | 0.58 | 1.5 | |
| AA | 3 | 2 | 0.77 | 0.13 | 4.6 | ||
OR, odds ratio. CI, confidence interval. Adjusted for age.
Numbers do not add up to totals because of missing values.
Table 3 presents associations with selected combinations of the 11 TP53 polymorphisms that were genotyped, rather than with each individual SNP. The haplotype with variant alleles for rs12951053, rs12947788, rs1042522 (codon 72), and rs1642785 conferred one of the highest risk estimates for CIN3+/persistent HPV (ORA-C-C-G-T-G-A-T-C-C-G, 1.6; 95% CI, 1.1–2.3), supporting the individual SNP results for rs12951053, rs1642785, and in particular, the G to C change in rs1042522 (codon 72). The R2 between the codon 72 polymorphism and the variant alleles for rs12951053, rs12947788, and rs1642785 were 0.28, 0.28, and 0.75, respectively. The codon 72 change was also prominent in another haplotype with a similar magnitude of effect (ORA-C-C-T-C-A-G-T-C-C-G, 1.7; 95% CI, 0.97–2.9). Interestingly, the three haplotypes with the highest ORs all include both the G to C change in rs1042522 (codon 72) and the G to C change in rs1642785 (Table 3), which is the nearest SNP from rs1042522 (codon 72). Some haplotypes with the rs1042522 (codon 72) and rs1642785 G to C change do not show increased risk (e.g., ORA-C-C-T-C-G-A-C-C-C-A, 0.98; 95% CI, 0.46–2.1), which is also true for the haplotype with only the rs1042522 (codon 72) G to C change (ORA-C-C-T-C-G-A-T-C-G-G, 1.0; 95% CI, 0.50–2.2). However, the prevalence of these haplotypes is low. Thus, power may be limited for these haplotypes.
Table 3.
Association of TP53 haplotypes with risk of HPV Persistence/CIN3+, adjusted for age.
| Haplotype | CIN3+/ Persistent HPV Cases (Frequency) | Population Controls (Frequency) | OR* | 95% CI* | |
|---|---|---|---|---|---|
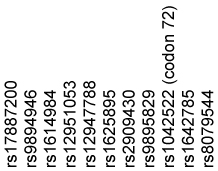 |
|||||
| A C C T C G A T G G G | 0.472 | 0.507 | 1.0 | ||
| A T C T C G A T G G G | 0.013 | 0.012 | 1.1 | 0.48 | 2.5 |
| A C T T C G A T G G G | 0.139 | 0.152 | 1.0 | 0.77 | 1.3 |
| A C C T C G A T C G G | 0.016 | 0.017 | 1.0 | 0.50 | 2.2 |
| A C C T C A G T C C G | 0.038 | 0.026 | 1.7 | 0.97 | 2.9 |
| A T C T C A G T C C G | 0.057 | 0.055 | 1.1 | 0.78 | 1.6 |
| A C C G T G A T C C G | 0.092 | 0.060 | 1.6 | 1.1 | 2.3 |
| A C T G T G A T C C G | 0.013 | 0.015 | 0.90 | 0.34 | 2.4 |
| G C T G T G A T C C G | 0.042 | 0.035 | 1.3 | 0.86 | 2.1 |
| A C C T C G A C C C A | 0.012 | 0.012 | 0.98 | 0.46 | 2.1 |
| A C T T C G A C C C A | 0.022 | 0.021 | 1.1 | 0.60 | 1.9 |
| Rare Haplotypes | 0.084 | 0.088 | 1.0 | 0.73 | 1.5 |
OR, odds ratio. CI, confidence interval. Adjusted for age.
Analyses restricted by HPV type further support the association between proline at codon 72 (rs1042522) and increased risk of CIN3+/HPV persistence (Table 4). Although rs12951053, rs1042522 (codon 72), and rs1642785 were associated with both oncogenic and non-oncogenic CIN3+/HPV persistence, the associations were more pronounced when restricted to oncogenic HPV. We had limited numbers of cases and controls for HPV16- and 18-specific analyses. However, the results similarly support an increased risk related to the presence of at least one allele with proline at codon 72.
Table 4.
ORs (95% CI)* for TP53 polymorphisms in CIN3+/persistent HPV cases† compared to random controls, stratified by HPV type (oncogenic HPV, HPV16, HPV18, and non-oncogenic HPV) and adjusted for age.
| HPV type | |||||
|---|---|---|---|---|---|
| TP53 Polymorphism | Oncogenic‡ (599 cases/74 controls) | HPV16 (96 cases/21 controls) | HPV18 (27 cases/8 controls) | Non-oncogenic§ (268 cases/95 controls) | |
| rs12951053 | |||||
| TT | 1.0 | 1.0 | 1.0 | 1.0 | |
| GT | 2.1 (1.1–4.1) | 0.86 (0.26–2.8) | 1.6 (0.26–9.7) | 1.4 (0.79–2.5) | |
| GG | NA | NA | NA | 1.5 (0.32'7.5) | |
| p-trend | 0.01 | 0.95 | 0.48 | 0.23 | |
| rs1042522 (Pro72Arg) | |||||
| GG | 1.0 | 1.0 | 1.0 | 1.0 | |
| CG | 1.8 (1.1–3.1) | 1.2 (0.41–3.3) | 6.7 (0.62–72.9) | 1.5 (0.92–2.6) | |
| CC | 3.1 (1.1–9.0) | 1.5 (0.16–14.3) | 1.4 (0.11–17.3) | 2.1 (0.85–4.9) | |
| p-trend | 0.01 | 0.68 | 0.33 | 0.04 | |
| rs1642785 | |||||
| GG | 1.0 | 1.0 | 1.0 | 1.0 | |
| CG | 1.3 (0.78–2.2) | 0.33 (0.1–1.1) | 3.7 (0.60–23.2) | 1.4 (0.85–2.3) | |
| CC | 4.6 (1.1–19.7) | 0.68 (0.1–7.0) | NA | 2.0 (0.77–5.1) | |
| p-trend | 0.03 | 0.17 | 0.09 | 0.08 | |
OR, odds ratio. CI, confidence interval. Adjusted for age.
All CIN3+ cases were included, regardless of HPV testing.
Oncogenic HPV types included HPV types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68. HPV testing results were available for 65% of cases.
Non-oncogenic HPV types included HPV types 6, 11, 32, 40, 53, 54, 55, 61, 62, 67, 70, 71, 72, AE10 (74 variant), 81, 83, 84, 85, 89.
Discussion
We observed modest associations between TP53 polymorphisms and CIN3+/persistent HPV in a Latin American population. These results may suggest that TP53 polymorphisms affect both CIN3+ and persistent HPV infection. SNP associations were strengthened when restricted to women with detectable oncogenic HPV infections. The strongest effect was observed for codon 72 polymorphism, with proline at that position conferring risk (OR, 1.3; 95% CI, 0.99–1.6 for heterozygotes and OR, 1.8; 95% CI, 1.2–2.7 for homozygotes). The haplotype with the strongest risk estimate also included this nonsynonymous SNP. Thus, in our population we found that proline, not arginine, at codon 72 was associated with increased risk of CIN3+ and HPV persistence.
We also found evidence that additional DNA sequences in the TP53 gene may affect risk of CIN3+ and HPV persistence. For example, rs12951053 and rs1642785 were associated with increased risk of CIN3+/HPV persistence for each additional variant allele (p-trend 0.05 and 0.04, respectively). Although we did not see a more pronounced effect affect for haplotypes including the codon 72 polymorphism compared the codon 72 variant alone, we do note that one haplotype produced a similar increase in risk and also included polymorphism in rs12951053, rs12947788, and rs1642785. The codon 72 polymorphism was moderately correlated with rs1642785 but uncorrelated with rs12951053 and rs12947788. The three strongest haplotype ORs all included G to C changes in both rs1042522 and rs1642785.
The hypothesis that the genetic variation flanking codon 72 could affect the structure of p53 is not new. For example, Li et al found that E6 could only degrade p53 if the protein included amino acids 66 to 326 (25). It is also possible that proline at codon 72 is not the susceptibility allele but instead marks a haplotype in linkage disequilibrium with the true susceptibility allele, or that proline at codon 72 is the susceptibility allele and that the SNPs surrounding it are markers for that allele. The laboratory evidence supporting arginine at codon 72 as the susceptibility allele is mixed since the HPV E6 oncoprotein was better able to degrade p53 with arginine at position 72 than with proline in transfected Saos-2 cells but the rate of E6-mediated degradation of p53Pro and p53Arg was the same when the two proteins were tested in vitro (5).
If either multiple alleles affect susceptibility or codon 72 is only a marker for the actual susceptibility allele, it is reasonable to expect variation across populations with different ethnic backgrounds. Notably, there is substantial variation of the distribution of TP53 codon 72 polymorphisms across populations (26, 27). Further, haplotype structures within TP53 have also been shown to vary between ethnic groups (28). Meta-analysis has demonstrated substantial heterogeneity in results from different studies of TP53 codon 72 and cervical cancer, including ORs both above and below 1.0 (6). While this inconsistency across studies might reflect a lack of true association, it is also plausible that some of the heterogeneity may be due to differential haplotypic structure of TP53 codon 72 polymorphisms in different study populations with different ethnic backgrounds. In fact, some meta-analytic data suggest that the association between TP53 codon 72 polymorphism and invasive cervical cancer varies by country, with some countries showing a decreased risk of cervical cancer with homozygous arginine versus homozygous proline (29, 30). These results could potentially reflect differences affecting p53 protein structure in TP53 haplotypes across populations with different ethnic backgrounds. In addition, the distribution of HPV types and variants, which can vary by population (31, 32), may be important since the initial laboratory study suggested a stronger effect for HPV-18 E6 than HPV-16 E6 (5).
Bias may also help account for the heterogeneity between studies. One meta-analysis found that the most important study characteristic accounting for heterogeneity was whether the controls were in Hardy-Weinberg equilibrium, with studies in which controls were out of Hardy-Weinberg equilibrium producing higher ORs (6). Lack of fitness for Hardy-Weinberg proportion can indicate genotyping errors (7, 8) and/or study design that might lead to falsely elevated results. This meta-analysis also found evidence of funnel plot asymmetry (6), which can be due to publication bias, random error, or study characteristics associated with sample size (33). Thus, it is possible that some null or inverse findings were not reported since they were contradictory to the initial laboratory finding, that results reflected random error due to small study sizes, or that lack of Hardy-Weinberg equilibrium was associated with sample size. In our well-powered study, none of the TP53 SNPs deviated from fitness for Hardy-Weinberg proportion, and our results support and expand on that of our previous study (9).
This study has limitations. Supplemental cases diagnosed outside the Guanacaste cohort were retrospectively ascertained and DNA could not be obtained for deceased cases, which might have introduced survival bias. However, it is unlikely that survival would differ by TP53 status. Although there is extensive a priori evidence for the comprehensive evaluation of TP53, we cannot exclude the possibility that some of our results are false positives or false negatives given the number of comparisons we made. However, it is encouraging that all three associated SNPs had FDR p-trend values below our a priori notable threshold of 0.2 after taking into account all SNPs tested. Finally, we may have missed associations if the 11 SNPs that we evaluated did not provide adequate coverage of the TP53 gene. Even so, this study is to our knowledge the first to evaluate risk of CIN3+/HPV persistence associated with polymorphisms across the TP53 gene and the first to identify associations between TP53 polymorphisms and HPV persistence in addition to CIN3+. In addition, the study design is stronger than that of many previous studies since it is population-based. It is one of the largest studies to date and includes more cancers than previously reviewed studies of the codon 72 polymorphism (6). While other study results might be influenced by socioeconomic status due to differential access to health care, our cohort is not affected by this bias, and the results from the cohort alone support the results of the analyses that included supplemental cases. Our analysis also avoids the potential for substantial outcome misclassification by excluding women with CIN2 who did not have persistent HPV infection from both case and control groups. While not a true validation of our previous study (9) given the overlap in study populations, these results support and expand upon our earlier results.
In conclusion, we found that three common SNPS inTP53 were associated with increased risk of CIN3+ and HPV persistence and that the strongest risk was for the proline allele at codon 72, rather than arginine. Our haplotype analyses support these findings and provide evidence that additional variants in linkage disequilibrium with the codon 72 SNP could influence risk. These combinations may vary by ethnicity and may help explain the disparity between our results and those of other studies, although it is also possible that there is no true association. Given the differences in the genetic structure of TP53 by ethnic groups, combined with the inconsistent results from epidemiological studies to date, any future epidemiologic studies of this topic should evaluate TP53-disease associations by haplotypes across ethnic groups.
Supplementary Material
Acknowledgements
This research was supported by the Intramural Research Program of the NIH and the National Cancer Institute. Robert Welch contributed as an author on this manuscript until the time of his death in 2008. Jill Koshiol is now a Research Fellow in the Infections and Immunoepidemiology Branch.
Footnotes
http://snp500Cancer.nci.nih.gov. Packer BR, Yeager M, Burdett L, et al. SNP500Cancer: a public resource for sequence validation, assay development, and frequency analysis for genetic variation in candidate genes. Nucleic Acids Res 2006;34:D617-21.
References
- 1.Hussain SP, Harris CC. p53 biological network: at the crossroads of the cellular-stress response pathway and molecular carcinogenesis. J Nippon Med Sch. 2006;73:54–64. doi: 10.1272/jnms.73.54. [DOI] [PubMed] [Google Scholar]
- 2.Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007;98:1505–1511. doi: 10.1111/j.1349-7006.2007.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koshiol J, Lindsay L, Pimenta JM, et al. Persistent Human Papillomavirus Infection and Cervical Neoplasia: A Systematic Review and Meta-Analysis. Am J Epidemiol. 2008 doi: 10.1093/aje/kwn036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 5.Storey A, Thomas M, Kalita A, et al. Role of a p53 polymorphism in the development of human papillomavirus-associated cancer. Nature. 1998;393:229–234. doi: 10.1038/30400. [DOI] [PubMed] [Google Scholar]
- 6.Koushik A, Platt RW, Franco EL. p53 codon 72 polymorphism and cervical neoplasia: a meta-analysis review. Cancer Epidemiol Biomarkers Prev. 2004;13:11–22. doi: 10.1158/1055-9965.epi-083-3. [DOI] [PubMed] [Google Scholar]
- 7.Gomes I, Collins A, Lonjou C, et al. Hardy-Weinberg quality control. Ann Hum Genet. 1999;63:535–538. doi: 10.1017/S0003480099007824. [DOI] [PubMed] [Google Scholar]
- 8.Hosking L, Lumsden S, Lewis K, et al. Detection of genotyping errors by Hardy-Weinberg equilibrium testing. Eur J Hum Genet. 2004;12:395–399. doi: 10.1038/sj.ejhg.5201164. [DOI] [PubMed] [Google Scholar]
- 9.Hildesheim A, Schiffman M, Brinton LA, et al. p53 polymorphism and risk of cervical cancer. Nature. 1998;396:531–532. doi: 10.1038/25040. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharya P, Duttagupta C, Sengupta S. Proline homozygosity in codon 72 of p53: a risk genotype for human papillomavirus related cervical cancer in Indian women. Cancer Lett. 2002;188:207–211. doi: 10.1016/s0304-3835(02)00430-5. [DOI] [PubMed] [Google Scholar]
- 11.Comar M, Molin GD, Guaschino S, Campello C. p53 at codon 72 polymorphism, human papillomavirus infection and cervical lesions: a cross-sectional study from northeastern Italy. Eur J Obstet Gynecol Reprod Biol. 2004;114:210–214. doi: 10.1016/j.ejogrb.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Kim JW, Lee CG, Park YG, et al. Combined analysis of germline polymorphisms of p53, GSTM1, GSTT1, CYP1A1, and CYP2E1: relation to the incidence rate of cervical carcinoma. Cancer. 2000;88:2082–2091. doi: 10.1002/(sici)1097-0142(20000501)88:9<2082::aid-cncr14>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 13.Herrero R, Hildesheim A, Bratti C, et al. Population-based study of human papillomavirus infection and cervical neoplasia in rural Costa Rica. J Natl Cancer Inst. 2000;92:464–474. doi: 10.1093/jnci/92.6.464. [DOI] [PubMed] [Google Scholar]
- 14.Herrero R, Schiffman MH, Bratti C, et al. Design and methods of a population-based natural history study of cervical neoplasia in a rural province of Costa Rica: the Guanacaste Project. Rev Panam Salud Publica. 1997;1:362–375. doi: 10.1590/s1020-49891997000500005. [DOI] [PubMed] [Google Scholar]
- 15.Carreon JD, Sherman ME, Guillen D, et al. CIN2 is a much less reproducible and less valid diagnosis than CIN3: results from a histological review of population-based cervical samples. Int J Gynecol Pathol. 2007;26:441–446. doi: 10.1097/pgp.0b013e31805152ab. [DOI] [PubMed] [Google Scholar]
- 16.Ostor AG. Natural history of cervical intraepithelial neoplasia: a critical review. Int J Gynecol Pathol. 1993;12:186–192. [PubMed] [Google Scholar]
- 17.Herrero R, Castle PE, Schiffman M, et al. Epidemiologic profile of type-specific human papillomavirus infection and cervical neoplasia in Guanacaste, Costa Rica. J Infect Dis. 2005;191:1796–1807. doi: 10.1086/428850. [DOI] [PubMed] [Google Scholar]
- 18.Schiffman M, Herrero R, Hildesheim A, et al. HPV DNA testing in cervical cancer screening: results from women in a high-risk province of Costa Rica. Jama. 2000;283:87–93. doi: 10.1001/jama.283.1.87. [DOI] [PubMed] [Google Scholar]
- 19.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 20.Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 21.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2004 [Google Scholar]
- 23.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 24.Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–442. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Coffino P. High-risk human papillomavirus E6 protein has two distinct binding sites within p53, of which only one determines degradation. J Virol. 1996;70:4509–4516. doi: 10.1128/jvi.70.7.4509-4516.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beckman G, Birgander R, Sjalander A, et al. Is p53 polymorphism maintained by natural selection? Hum Hered. 1994;44:266–270. doi: 10.1159/000154228. [DOI] [PubMed] [Google Scholar]
- 27.Sjalander A, Birgander R, Kivela A, Beckman G. p53 polymorphisms and haplotypes in different ethnic groups. Hum Hered. 1995;45:144–149. doi: 10.1159/000154275. [DOI] [PubMed] [Google Scholar]
- 28.Bonnen PE, Wang PJ, Kimmel M, Chakraborty R, Nelson DL. Haplotype and linkage disequilibrium architecture for human cancer-associated genes. Genome Res. 2002;12:1846–1853. doi: 10.1101/gr.483802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jee SH, Won SY, Yun JE, et al. Polymorphism p53 codon-72 and invasive cervical cancer: a meta-analysis. Int J Gynaecol Obstet. 2004;85:301–308. doi: 10.1016/j.ijgo.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 30.Sousa H, Santos AM, Pinto D, Medeiros R. Is the p53 codon 72 polymorphism a key biomarker for cervical cancer development? A meta-analysis review within European populations. Int J Mol Med. 2007;20:731–741. [PubMed] [Google Scholar]
- 31.de Sanjose S, Diaz M, Castellsague X, et al. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: a meta-analysis. Lancet Infect Dis. 2007;7:453–459. doi: 10.1016/S1473-3099(07)70158-5. [DOI] [PubMed] [Google Scholar]
- 32.Smith JS, Lindsay L, Hoots B, et al. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer. 2007;121:621–632. doi: 10.1002/ijc.22527. [DOI] [PubMed] [Google Scholar]
- 33.Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta-analysis: power of statistical tests and prevalence in the literature. J Clin Epidemiol. 2000;53:1119–1129. doi: 10.1016/s0895-4356(00)00242-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.