

Abstract
Epidermal Growth Factor Receptor (EGFR) targeting in non-small cell lung cancer (NSCLC) is an established treatment modality, yet only benefits a minority of patients. STAT3 (signal transducer and activator of transcription-3) plays an important role in the oncogenic signal transduction pathway of NSCLC. Inhibition of STAT3 results in NSCLC growth inhibition and apoptosis. We have previously shown that combined inhibition of EGFR and STAT3 by small molecules resulted in improved therapeutic efficacy as compared to blocking EGFR alone. However, the STAT3 protein has a number of endogenous negative regulators including PIAS3 (Protein Inhibitor of Activated STAT3). In this study we investigated for the first time the role of PIAS3 in modulating oncogenic EGFR-STAT3 signaling pathway in lung cancer and the antiproliferative effect of using PIAS3 in conjunction with EGFR blockade in NSCLC. We demonstrate that PIAS3 is expressed in variable degrees in all NSCLC cells. EGF and IL-6 stimulation resulted in the association of PIAS3 with STAT3. The PIAS3/STAT3 complex then bound the STAT3 DNA binding sequence resulting in STAT3 regulated gene expression. Over-expression of PIAS3, using a PIAS3 expression construct, decreases STAT3 transcriptional activity. Furthermore, over-expression of PIAS3 consistently decreased proliferation. EGFR blockade and PIAS3 over-expression in combination had significantly greater anti-proliferative effects as compared to either EGFR blockade or PIAS3 over-expression alone. In conclusion, PIAS3 is expressed in NSCLC cell lines and its over-expression decreased STAT3 transcriptional activity, decreased proliferation of NSCLC cells and when used in conjunction with EGFR inhibitors, increased the anti-proliferative effects.
INTRODUCTION
Lung cancer is the number one cause of cancer related death in the United States (1). Targeting Epidermal Growth Factor Receptor (EGFR) with small molecule inhibitors has emerged as a therapeutic option in lung cancer. Clinical studies however have shown response rates of only 10% with the majority of patients having progression of their cancer (2). Therefore to improve the therapeutic efficacy of EGFR inhibitors, these agents are being combined with other agents targeting persistently activated downstream proteins such as AKT and STAT3.
Signal Transducer and Activators of Transcription (STAT) are important cytoplasmic proteins that act as transcription factors to regulate gene expression. STAT proteins, especially STAT3, are important in the development and progression of cancers by either preventing apoptosis or promoting proliferation (3). Upon activation by upstream receptor tyrosine kinases, of which EGFR plays a dominant role (4), STAT3 is phosphorylated (p-STAT3) and forms a homo- or heterodimer that acts as a transcriptional factor on binding to promoter regions of genes that regulate cell cycle progression, apoptosis, angiogenesis, tumor invasion and metastasis (5).
In non-small cell lung cancer (NSCLC) cell lines that have constitutively active mutant EGFR, STAT3 is phosphorylated and is necessary for the proliferative effects associated with mutant EGFR (6). Furthermore, inhibiting STAT3 activity abrogates the transforming effects of EGFR activating mutations (4). In vitro data show that EGFR blockade decreased STAT3 activation. Similarly, cell lines resistant to EGFR inhibitors demonstrate persistent activation of STAT3 (8). Thus STAT3 is a key molecule in maintaining a transformed phenotype and inhibition of STAT3 has become a potential target for drug development in lung cancer (7). Indeed blockade of STAT3 results in extensive apoptosis of NSCLC cells (8). We have previously demonstrated that combined inhibition of EGFR and STAT3 using small molecules has synergistic anti-proliferative effects in a variety of NSCLC cell lines (9, 10) and similar data has recently been shown in head and neck cancer cell lines (11). Given the importance of the STAT3 signaling pathway and its potential for new drug development target, finding alternative methods to regulateSTAT3 are of interest.
STAT3 has several physiological negative regulators. Most of these negative regulators target events upstream of STAT3. For example Suppressor of Cytokine Signaling (SOCS) binds to TYK2 and JAK2 which in turn inhibits cytokine mediated activation of STAT proteins (3). Protein Inhibitor of STAT (PIAS) represents a group of 5 proteins (PIAS1, PIAS3, PIASxα, PIASxβ, and PIASy) which function to decrease DNA activation by blocking STAT DNA-binding activity (12). Protein Inhibitor of Activated STAT3 (PIAS3) plays a dominant role as a direct negative regulator of STAT3 activity. PIAS3 was first identified as a transcriptional repressor of activated STAT3, inhibiting STAT3’s DNA binding activity (13). PIAS3 is present in 2 forms, a 68 and a 85 KDa band correlating to the non-sumoylated and sumoylated form of PIAS3, reflecting its ability to function as E3 type small ubiquitin modifier (SUMO) ligases (14). Its transcriptional repressor effect does not, however, uniformly require sumoylation of its target protein (12). Northern blot analysis shows widespread distribution of PIAS3 gene expression in human tissue. A variety of cancers have increased expression of PIAS3 compared to normal tissue (15). For example, PIAS3 is expressed in prostate cancer cell lines and myeloma cell lines and functions as a transcriptional cofactor for the androgen and estrogen receptors respectively (16, 17). Its over-expression can induce apoptosis in prostate cancer cells both in vitro and in vivo (18). Although there are emerging data on the role of PIAS3 in other malignancies, no study has evaluated the role of PIAS3 in NSCLC.
We thus hypothesized that: 1) PIAS3 is expressed in NSCLC; 2) PIAS3 will interact with STAT3 upon ligand-induced STAT3 activation; 3) over-expression of PIAS3 will inhibit STAT3 transcriptional activity and NSCLC tumor growth; 4) EGFR blockade in conjunction with PIAS3 over-expression will augment the growth inhibitory effects of EGFR inhibitors.
MATERIALS AND METHODS
Cell Lines
Lung cancer cell lines utilized included adenocarcinoma lines A549, H1650, H522, H441, H1975, H827 and squamous cell carcinoma lines H1869, Calu1 and H520. All cell lines were purchased through ATCC (Manassas, VA) and maintained in DMEM/ Ham’s F12 media containing 1% glutamine, 10% fetal bovine serum, and 1% penicillin/ streptomycin in a humidified 5% CO2 environment. NuLi cells were maintained in Bronchial Epithelial Cell Growth Media (BEGM; Cambrex Corporation, East Rutherford, NJ).
Western Blotting
To obtain protein lysates, cells that were in log-phase growth (50-70% confluence) were lysed in buffer containing 1% Triton X-100, 0.15M sodium chloride, 50mM Tris, pH 7.4, and protease inhibitors (aprotinin 50μg/mL, pepstatin 50μg/mL, leupeptin 10μg/mL, EDTA 0.4mM, sodium orthovanadate 0.4mM, sodium fluoride 10mM, sodium pyrophosphate 10mM, phenylmethylsulfonyl fluoride 5mM). Protein concentrations were determined by the Bradford method (BioRad Protein Assay, Invitrogen, Carlsbad, CA). Fifteen micrograms of protein from each sample was separated on 10% SDS-PAGE, transferred to PVDF membrane (Immobilon, Millipore, Bedford, MA), and blotted with specific antibodies (1:200 dilution for all antibodies, Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For comparison of protein expression amongst cell lines we used the Scion Image software. This software uses the scanned image of a western blot film. The dense areas or bands are outlined and each band space is manually defined. The software then uses arbitrary units to quantitate the density of each band. Thus using the defined arbitrary units, increases or decreases in the protein expression level can be determined.
Immunoprecipitation
One hundred micrograms of protein lysate was complexed with 2μg anti-PIAS3 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) for 2 hours at 4°C, with rotation. Protein A-Sepharose beads (Zymed Laboratories, Inc., San Francisco, CA) were added, the solution was incubated for 30 minutes, washed with lysis buffer, centrifuged, and the supernatant removed. The bead-protein-antibody complex was then resuspended in 1X Laemmli buffer, boiled at 95°C for 5 minutes, and 10μL of the supernatant containing the protein-antibody complex was used for separation by SDS-PAGE and Western blotting.
Electrophoretic Mobility Shift Assay (EMSA) and Supershift Assay
Nuclear lysates were prepared using 10mM HEPES buffer containing 1.5mM magnesium chloride, 10mM potassium chloride, and 1mM dithiothreitol to lyse the cell membrane, and following centrifugation, 20mM HEPES buffer containing 1.5mM magnesium chloride, 42mM sodium chloride, 0.2mM EDTA, 25% glycerol and 1mM dithiothreitol to lyse the nuclear membrane. Supernatant was retained for assays after another centrifugation. Five to ten micrograms of nuclear protein was incubated at 18°C for 30 minutes with transcription factor probe, hSIE, which specifically binds activated STAT3 proteins with high affinity (EMSA “Gel Shift” Kit, Panomics, Inc., Redwood City, CA). Samples were then run on a 7.5% precast acrylamide gel (BioRad, Invitrogen, Carlsbad, CA) and transferred to a nylon membrane. Bound oligos were immobilized by baking the membrane at 85°C or by cross-linking the membrane in a UV Crosslinker for 3 minutes followed by blocking and staining of the membrane using a Streptavidin-HRP conjugate. Substrate solutions included in the Panomics kit were used for detection, and the membrane was exposed to Hyperfilm. Supershift assays were performed using the same procedure, but included the addition of 2μg antibody, either anti-PIAS3 or anti-STAT3, during initial incubation with hSIE nuclear extracts.
Stimulation with EGF or IL-6
Cells were grown in DMEM/ Ham’s F12 media containing 1% glutamine, 10% fetal bovine serum, and 1% penicillin/ streptomycin in a humidified 5% CO2 environment to approximately 60-80% confluency, then growth media was changed to DMEM/ Ham’s F12 media containing 1% glutamine and 1% penicillin/ streptomycin, containing no serum. Cells were allowed to grow in this serumfree media for an additional 24 hours. Cells were then stimulated with either Epidermal Growth Factor (EGF) at 50ng/mL for 10 minutes or with Interleukin-6 (IL-6) at 30ng/mL for 20 minutes. Following stimulations, protein or nuclear extracts were collected as described above.
PIAS3 plasmid transfection
Plasmid PIAS3 cDNA in a pCMV5 expression vector was generously provided by Dr. Ken Shuai from the David Geffen School of Medicine, UCLA. The plasmid was transfected into DH5α Competent (E. coli) cells according to the manufacturer’s directions (Invitrogen Corporation, Carlsbad, CA). Bacteria containing the plasmid PIAS3 was expanded into 500mL flasks and Maxipreps were performed using the Qiagen Plasmid Maxi Kit (Qiagen Inc., Valencia, CA) to obtain plasmid DNA. DNA was quantified and endonuclease restriction performed with BamH1 to verify the presence of the PIAS3 gene. The PIAS3 expression vector was transfected into A549, H1650, Calu1 and H1975 cells using FuGENE 6 Transfection Reagent according to the manufacturer’s protocol (Roche Applied Science, Indianapolis, IN). Briefly, 1-3 × 105 cells were seeded into each well of a 6-well plate and allowed to proliferate at 37°C in a humidified 5% CO2 incubator overnight. Appropriate amounts of FuGENE 6 Transfection Reagent and plasmid DNA were added to serum-free media without antibiotics and added drop wise to cells. After 5-6 hours, media was replaced with DMEM/HF12 containing 10% fetal bovine serum and 1% antibiotics, allowed to grow for 36 hours, and cells were serum starved for overnight and were stimulated with 50ng/mL EGF for 10 minutes, washed once with sterile PBS, and lysed with 1X Laemmli buffer. Ten microliters of each sample was run on SDS-PAGE (10% Tris-HCl gel) and Western blotted for PIAS3.
Luciferase Assay
A549 and H1650 cells were seeded at 1-3 × 105,000 cells per well in 6-well plates. Cells were transfected using FuGENE 6 Transfection Reagent with either plasmid PIAS3, pSTAT3-TA-Luc plasmid, or pTA-Luc plasmid (Clontech Mercury Pathway Profiling System) or in combination, in DMEM/ HF12 media containing no serum and no antibiotics. After 4-5 hours incubation, media was replaced with DMEM/HF12 media containing 10 % fetal bovine serum and antibiotics. Cells were allowed to grow for next 36 hrs followed by overnight serum starvation, cells were stimulated with 50ng/mL EGF for 15 minutes, then washed one time with sterile PBS. Glo Lysis Buffer (Promega Corp.) was added and plates incubated at room temperature for 5 minutes and the lysed cell samples were collected and transferred into a microcentrifuge tube wherein cell debris was pelleted by centrifugation. Luciferase activity in an aliquot of the cleared lysed cell supernatants (25 μl) was determined by mixing it with 50 μl of luciferase assay reagent (Promega) and measuring the resulting luminescence in MicroLumat Plus LB 96V luminometer (Berthold Detection System, Oak Ridge, TN, USA).
PIAS3 plasmid transfection with EGFR blockade
A549, H1650, H1975 and Calu1 cells were seeded at approximately 5 × 103 cells per well in 96-well plates and allowed to proliferate overnight in a humid, 5% CO2 incubator at 37°C in DMEM/HF12 growth media containing 10% serum and 1% antibiotics. The following day the media was changed to contain no antibiotics. Cells were then treated with erlotinib (EGFR blockade) at 0.1μM or 1μM, DMSO at the same dilution as 1μM erlotinib dose, transfected with plasmid PIAS3 or mock transfected, or a combination thereof (plasmid PIAS3 transfection with each dose of erlotinib, mock transfection with each dose of erlotinib). Cell proliferation was then quantified by cell counts or MTS assay (Promega, Madison, WI).
RESULTS
PIAS3 is expressed in NSCLC cell lines
We first sought to confirm the expression of PIAS3 in NSCLC cell lines. As shown in Figure 1 a mixture of adenocarcinoma (A549, H1650, H1975, H522, H441) and squamous cell carcinoma cell lines (Calu1, H520) were examined for PIAS3. PIAS3 expression was identified in each cell line examined, though expression was variable. In order to determine the relative protein expression in each cell line densitometry analysis of the protein bands was done which yielded the following arbitrary units: NuLi (8053), A549 (9246), H522 (8089), Calu1 (3776), H441 (4354), H1650 (3586), H1869 (5609), H827 (8004), H1975 (9516), H520 (5414). This analysis showed the highest expressing cell line (A549) had a 2.5-fold greater expression of PIAS3 compared to the lowest expressing cell line (H1650). Other cell lines with high expression include H1975, H522 and H827. For further studies we chose A549 and H1975 cells as representative of high PIAS3 expressors. Normal lung epithelial cells (NuLi) also expressed PIAS3.
Figure 1.
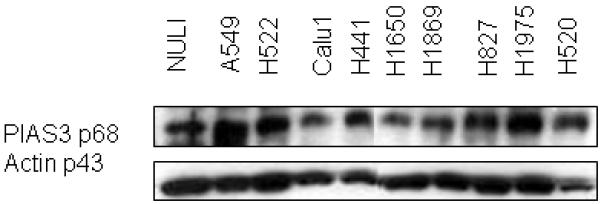
PIAS-3 protein expression by western blotting in NuLi cells and NSCLC cell lines. Identical amounts of protein from the cell lines was subjected to SDS-PAGE, transferred to nylon membrane, and probed with a PIAS3 and actin antibody. Variable degree of PIAS3 expression can be seen. Using densitometry analysis (see methods section), A549 and H1975 have the highest and H1650 and Calu1 the lowest expression. Actin is used as the loading protein control.
EGF and IL-6 stimulation results in association of PIAS3 with STAT3 and DNA binding
Given that STAT3 is activated by EGFR and IL-6, we selected two NSCLC cell lines (H1650 and A549) with different PIAS3 expression levels to determine the effect of EGFR and IL-6 stimulation on PIAS3 interaction with STAT3. In co-immunoprecipitation studies we first evaluated the binding of PIAS3 to STAT3 (Figure 2). In the absence of EGF or IL-6 and in serum-free state, no association of PIAS3 and STAT3 was seen. However, upon EGF or IL-6 stimulation, there is a clear association of PIAS3 and STAT3 resulting in co-immunoprecipitation. This interaction was blocked in cells pretreated with an EGFR tyrosine kinase inhibitor (AG1478) despite EGF stimulation. These data suggest that STAT3 activation is required for induction of this association. We also demonstrated that with STAT3 blockade (using siRNA targeting STAT3) there is no change in PIAS3 protein levels (data not shown). This suggests that STAT3 blockade does not cause a decrease in total cellular PIAS3.
Figure 2.

Ligand-induced PIAS3 -STAT3 binding in NSCLC. H1650 and A549 cell lines were exposed to EGF or IL-6 and extracts immunoprecipitated for PIAS3 followed by western blotting for STAT3. In the absence of EGF or other stimulation (serum-free state), PIAS3 is not associated with STAT3. With EGF stimulation (50 ng/mL × 10 minutes) or IL-6 (30 ng/mL × 20 minutes) PIAS3-STAT3 association is observed. The EGF-induced interaction is inhibited by the small molecule EGFR tyrosine kinase inhibitor, AG1478. Total PIAS3 levels for both cells lines are shown as a control.
With ligand-induced STAT3 activation required for PIAS3-STAT3 interaction, we also sought to determine the functional role of the PIAS3-STAT3 complex. This is a known transcriptional complex and studies were performed to evaluate nuclear translocation and DNA binding. After stimulation of A549 cells with EGF or IL-6, EMSA was performed on nuclear extracts with the STAT3 transcription factor probe, hSIE, which specifically binds activated STAT3 proteins with high affinity. As shown in Figure 3, EGF or IL-6 stimulation increased activated STAT3 in the nucleus evidenced by increased binding of hSIE. Further confirmation of STAT3 was performed by supershifting the hSIE band using a STAT3 antibody. PIAS3 was also identified in the complex through a supershift of hSIE with PIAS3 antibody. This EMSA assay also demonstrates that not all STAT3 supershifts with the anti-PIAS3 antibody, suggesting that some of the STAT3 that is bound to its consensus sequence is not bound to PIAS3. This in turn provides the opportunity to further manipulate STAT3 DNA transcriptional activity by increasing intracellular PIAS3.
Figure 3.

EGF or IL-6 stimulation induces PIAS3-STAT3 complex binding to STAT3 consensus sequence. After EGF or IL-6 stimulation, nuclear extracts were made from A549 cells. The extracts were mixed with the STAT3 DNA binding consensus sequence hSIE and EMSA performed identifying activated STAT3 in the extracts. STAT3 was verified by incubating STAT3 antibody with the hSIE complex resulting in supershift of the band. PIAS3 was also identified in the complex through supershift of hSIE with a PIAS3 antibody. Arrows indicate supershift with anti-PIAS3 antibody.
Over-expression of PIAS3 results in decreased STAT3 transcriptional activity
To verify that the PIAS3-STAT3 interaction affects STAT3 transcriptional activity, both A549 and H1650 cells were transfected with a luciferase expression vector under the control of a STAT3-dependent promoter (pSTAT3-TA-Luc plasmid) (Figure 4). With EGF stimulation, luciferase activity increased significantly over control (pTA-luciferase vector not under the control of a STAT3 promoter) (A549, 4.1-fold increase p <0.001, H1650, 7.1-fold increase, p < 0.001, Figure 4). H1650 cells contain an EGFR activating mutation and are known to be highly responsive to EGF stimulation, resulting in a higher increase in luciferase activity as compared to the A549 cell line that has a wild-type EGFR sequence. Upon co-transfection with a PIAS3 expression construct a significant drop in luciferase expression was seen in both cell lines. With increased PIAS3 expression, luciferase activity in A549 cells fell 2.3-fold (p<0.001) and 4.8-fold in H1650 cell line (p<0.001).
Figure 4.
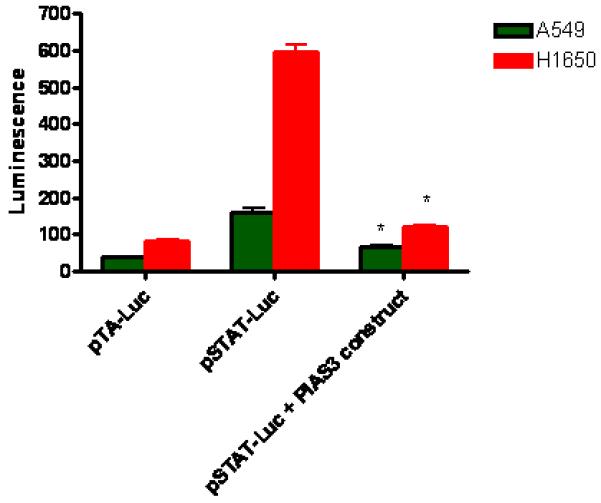
PIAS3 decreases STAT3 regulated gene expression. pTA that does not bind STAT3 is used as a negative control. EGF stimulation of A549 and H1650 transfected with a STAT3 promoter construct induced a significant increase in STAT3 regulated gene expression. Co-transfection with a PIAS3 expression construct decreased EGF stimulated STAT3 gene expression in both cell lines (* p<0.001 compared to control; ** p<0.001 compared to pSTAT-Luc). N=6, data reported as mean ± standard error.
PIAS3 over-expression decreases proliferation
To confirm a growth regulatory role for PIAS3, we next over-expressed PIAS3 in NSCLC cells. A549, H1650, Calu1 and H1975 cells were transiently transfected with a PIAS3 expression construct. As can be seen in Figure 5, transfection resulted in a significant increase in PIAS3 expression in the four cell lines. Transfection was also verified by the unique FLAG epitope tag, which was engineered into this PIAS3 cDNA. Over-expression of PIAS3 in all cell lines resulted in substantial growth inhibitory effect by both cell count and MTS assay (Figure 6, P<0.01 for all experiements). By cell count this ranged between 20% inhibition for Calu1 and >90% inhibition for H1975 cells.
Figure 5.
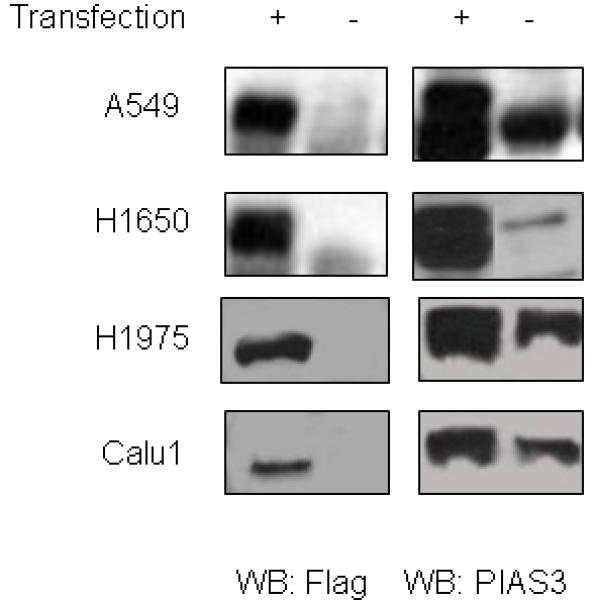
A549, H1650, Calu1 and H1975 cells were transfected with a PIAS3 expression construct. 48 hours after transfection cells were harvested and western blotting done for PIAS3 and Flag epitope. All four cell lines showed evidence of transfection with greater PIAS3 expression and appearance of the Flag epitope.
Figure 6.
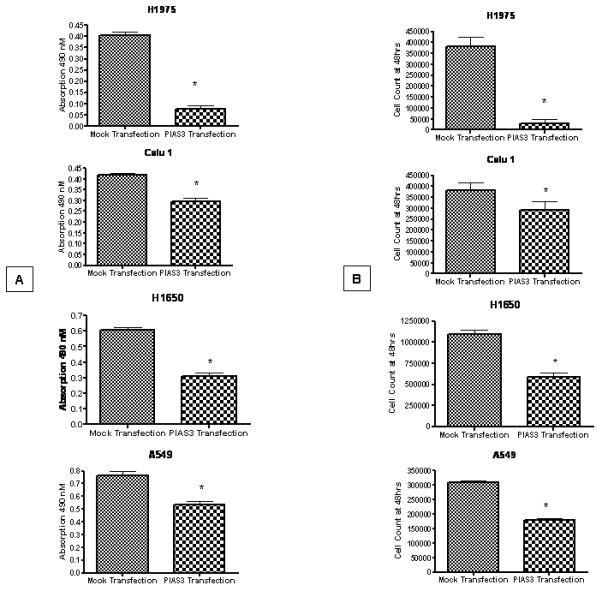
A549, H1650, Calu1 and H1975 cells were transfected with PIAS3 expression construct or a mock transfection. In all four cell lines, 48 hours after PIAS3 construct exposure a significant decrease in proliferation was seen both by MTS assay (A) (p<0.01 for all groups) or by cell count (B) (p<0.01 for all groups) compared to control (n=6, data reported as mean ± SE).
Combined PIAS3 transfection and EGFR blockade is more effective than single modality therapy
Based on our previous data that combined EGFR and STAT3 blockade improved the growth inhibitory activity of EGFR inhibitors A549, H1650, Calu1 and H1975 cells were exposed to erlotinib at the IC50’s of each cell lines (H1650 at 0.1 μM and A549 at 1 μM), transfected with a PIAS3 expression construct or a combination of both. In all four cell lines the combination resulted in a significant decrease in cell growth by both MTS and cell count methods as compared to erlotinib or PIAS3 transfection alone (Figure 7, p < 0.01 for all cell lines and methods).
Figure 7.
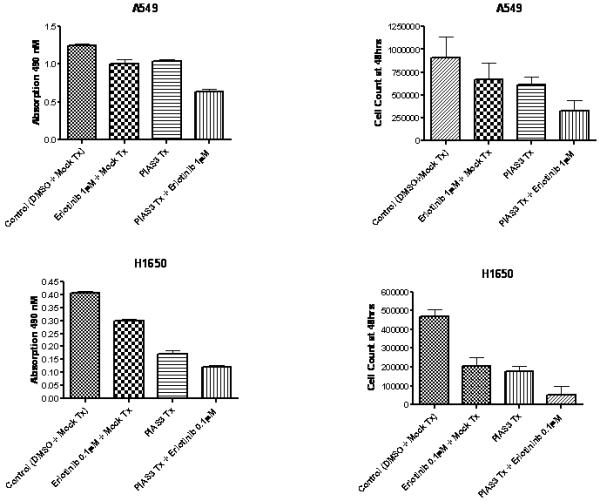
A549 and H1650 cells were exposed to erlotinib (1μM for A549 or 0.1μM for H1650), transfected with PIAS3 expression construct or exposed to both erlotinib and PIAS3. Both erlotinib and PIAS3 transfection resulted in growth inhibition compared to control (p< 0.01); however a combination of erlotinib and PIAS3 was significantly more effective than single treatment (p < 0.01). Proliferation was measured by MTS assay and cell count (n=6, data reported as mean ± SE).
DISCUSSION
Anti-EGFR therapy using small molecule inhibitors has become an established treatment strategy for NSCLC. Most patients, nevertheless, do not benefit from such an approach and responding patients ultimately develop resistance to these agents (2). Persistent activation of downstream STAT3 has been proposed as a putative mechanism by which cancer cells remain resistant to EGFR inhibitors. Therefore in an attempt to improve anti-EGFR therapy, concomitant targeting of STAT3 has been performed. Indeed our group, in a variety of cells lines, has demonstrated that STAT3 targeting increases the anti-tumor effects of EGFR inhibition (9, 10). Furthermore, similar data has been established for head and neck cancer (11), a disease with many similarities to lung cancer. Experimental methods by which STAT3 has been targeted include use of upstream JAK2 inhibitors (9), anti-sense therapies to STAT3 (8), use of a protein farnesyl transferase inhibitor that blocks STAT3 phosphorylation (10) and finally using a STAT3 decoy (11). In the current study we have taken a novel approach in which PIAS3 a naturally occurring inhibitor of STAT3 is used to alter the function of STAT3 in conjunction with EGFR blockade.
Although STAT proteins have a number of natural occurring negative regulators, including SOCS, JAK-activating binding protein (JAB) and STAT-induced STAT inhibitor (SSI), these inhibitors all act by inhibiting upstream JAK (14). However, STAT3 activation in lung cancer can occur without JAK activation and JAK inhibitors do not effect EGF induced STAT3 activation (8). PIAS3 on the other hand directly interacts with STAT3 and is a more specific inhibitor. Therefore we hypothesized that alterations of this negative regulator of STAT3 may be important in lung cancer by modulating STAT3 signaling.
We demonstrated for the first time that PIAS3 is expressed in NSCLC. We show also that association of PIAS3 to STAT3 is regulated by ligand-induced STAT3 activation. The observation that PIAS is associated with STAT only in cells stimulated by EGFR or IL-6, suggests that the PIAS-STAT interaction is regulated (14). This is consistent with the interaction of another protein of the PIAS family, PIAS1, which only associates with STAT1 under cytokine stimulation. This is also consistent with the ligand dependent interaction of other LXXLL motif-containing nuclear receptor co-regulators (14, 19). The functional effect of the PIAS3/STAT3 interaction is a negative effect on STAT3 transcriptional activity. Furthermore our data indicate that STAT3 transcriptional activity can be further decreased by increasing PIAS3 expression. PIAS3 thus becomes a potential therapeutic target by which one could attempt to increase expression of a negative regulatory protein in a potent oncogenic pathway. This approach has been taken for example in the case of SHIP (Src homology 2-containing inositol 5-phasphatases) and its effect on PI3K signaling where novel molecules have been developed to increase SHIP expression and decrease PI3K/AKT signaling (20).
When increased intracellular PIAS3 expression was combined with a small molecule inhibitor of EGFR, a significant decrease in cellular growth was seen as compared to EGFR inhibition or PIAS3 transfection alone. This further confirms our hypothesis that dual EGFR and STAT3 targeting is a therapeutic concept that can be used to treat lung cancer. We have recently demonstrated in a cohort of 44 resected NSCLC specimens the absence of PIAS3 expression by immunohistochemistry in 11% of adenocarcinomas and 62% of squamous cell carcinomas. In addition those expressing PIAS3 in general had only 1+ expression (defined as less than 5% of tumor cells) (21). Our preliminary data also shows that the decreased PIAS3 expression maybe under epigenetic control as exposure of H1650 lung cancer cell line to trichostatin A and 5-azacytidine leads to a significant increase in PIAS3 mRNA and protein expression (21). Thus this produces a potential clinical method by which one could increase intracellular PIAS3. This small study also failed to demonstrate in this cohort any relation between PIAS3 expression and pSTAT3 suggesting that PIAS3 downregulation is not an escape mechanism for
Although PIAS3 activity is clearly mediated through STAT3, it is conceivable that PIAS3 maybe working through other targets as well. For example PIAS3 has been shown to be a repressor of microphthalmia transcription factor (22) and it has been shown to interact with ATF1 (23), Smad3 (24), and androgen signaling (25). However none of these targets have been shown to have relevance in lung cancer. Further research into potential targets of PIAS3 is ongoing.
In conclusion we demonstrate for the first time the expression of PIAS3 in NSCLC and its interaction with the EGFR/STAT3 oncogenic pathway. We demonstrate that PIAS3 has growth inhibitory effects and increases the effectiveness of agents targeting EGFR.
Acknowledgments
Supported by K23 CA109348-01 (to AD) and R01 HL 075422 (to JAK) from the National Institutes of Health and the Coburn Haskill endowment (to AD) from University Hospitals Case Medical Center
REFERENCES
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabárbara P, Seymour L, National Cancer Institute of Canada Clinical Trials Group Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 3.Levy DE, Darnell JE., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–8. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 5.Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle. 2005;4:1131–3. doi: 10.4161/cc.4.9.1985. [DOI] [PubMed] [Google Scholar]
- 6.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 7.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54. [PubMed] [Google Scholar]
- 8.Song L, Turkson J, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22:4150–65. doi: 10.1038/sj.onc.1206479. [DOI] [PubMed] [Google Scholar]
- 9.Dowlati A, Nethery D, Kern JA. Combined inhibition of epidermal growth factor receptor and JAK/STAT pathways results in greater growth inhibition in vitro than single agent therapy. Mol Cancer Ther. 2004;4:459–63. [PubMed] [Google Scholar]
- 10.Dowlati A, Kluge A, Nethery D, Halmos B, Kern JA. SCH66336, inhibitor of protein farnesylation, blocks signal transducer and activators of transcription 3 signaling in lung cancer and interacts with a small molecule inhibitor of epidermal growth factor receptor/human epidermal growth factor receptor 2. Anticancer Drugs. 2008;19:9–16. doi: 10.1097/CAD.0b013e3282f1a908. [DOI] [PubMed] [Google Scholar]
- 11.Boehm AL, Sen M, Seethala R, Gooding WE, Freilino M, Wong SM, Wang S, Johnson DE, Grandis JR. Combined targeting of eGFR, STAT3, and Bcl-XL enhances antitumor effects in squamous cell carcinoma of the head and neck. Mol Pharmacol. 2008;73:1632–42. doi: 10.1124/mol.107.044636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharrocks AD. PIAS proteins and transcriptional regulation--more than just SUMO E3 ligases? Genes Dev. 2006;20:754–8. doi: 10.1101/gad.1421006. [DOI] [PubMed] [Google Scholar]
- 13.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 14.Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19:2638–44. doi: 10.1038/sj.onc.1203522. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Banerjee S. Differential PIAS3 expression in human malignancy. Oncol Rep. 2004;11:1319–24. [PubMed] [Google Scholar]
- 16.Gross M, Liu B, Tan J, French FS, Carey M, Shuai K. Distinct effects of PIAS proteins on androgen-mediated gene activation in prostate cancer cells. Oncogene. 2001;20:3880–7. doi: 10.1038/sj.onc.1204489. [DOI] [PubMed] [Google Scholar]
- 17.Wang LH, Yang XY, Mihalic K, Xiao W, Li D, Farrar WL. Activation of estrogen receptor blocks interleukin-6-inducible cell growth of human multiple myeloma involving molecular cross-talk between estrogen receptor and STAT3 mediated by co-regulator PIAS3. J Biol Chem. 2001;276:31839–44. doi: 10.1074/jbc.M105185200. [DOI] [PubMed] [Google Scholar]
- 18.Junicho A, Matsuda T, Yamamoto T, Kishi H, Korkmaz K, Saatcioglu F, Fuse H, Muraguchi A. Protein inhibitor of activated STAT3 regulates androgen receptor signaling in prostate carcinoma cells. Biochem Biophys Res Commun. 2000;278:9–13. doi: 10.1006/bbrc.2000.3753. [DOI] [PubMed] [Google Scholar]
- 19.Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci U S A. 1998;95:10626–31. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang L, Williams DE, Mui A, Ong C, Krystal G, van Soest R, Andersen RJ. Synthesis of pelorol and analogues: activators of inositol 5-phosphatase SHIP. Organic Letters. 2005;7:1073–76. doi: 10.1021/ol047316m. [DOI] [PubMed] [Google Scholar]
- 21.Kluge A, Nethery D, Kern JA, Eisenberg R, Halmos B, Ma P, Dowlati A. Protein inhibitor of activated STAT3 is expressed in human lung cancer and is under epigenetic control. Proc Am Assoc Cancer Res. 2008 abstract 3455. [Google Scholar]
- 22.Sonnenblick A, Levy C, Razin E. Interplay between MITF, PIAS3, and STAT3 in mast cells and melanocytes. Mol Cell Biol. 2004;24:10584–92. doi: 10.1128/MCB.24.24.10584-10592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nojiri S, Joh T, Miura Y, Sakata N, Nomura T, Nakao H, Sobue S, Ohara H, Asai K, Ito M. ATBF1 enhances the suppression of STAT3 signaling by interaction with PIAS3. Biochem Biophys Res Commun. 2004;314:97–103. doi: 10.1016/j.bbrc.2003.12.054. [DOI] [PubMed] [Google Scholar]
- 24.Long J, Wang G, Matsuura I, He D, Liu F. Activation of Smad transcriptional activity by protein inhibitor of activated STAT3 (PIAS3) Proc Natl Acad Sci U S A. 2004;101:99–104. doi: 10.1073/pnas.0307598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junicho A, Matsuda T, Yamamoto T, Kishi H, Korkmaz K, Saatcioglu F, Fuse H, Muraguchi A. Protein inhibitor of activated STAT3 regulates androgen receptor signaling in prostate carcinoma cells. Biochem Biophys Res Commun. 2000;278:9–13. doi: 10.1006/bbrc.2000.3753. [DOI] [PubMed] [Google Scholar]