

Abstract
The advent of contemporary proteomic technologies has ushered in definite advances to the field of auditory research and has provided the potential for a dramatic increase in applications in the near future. Two dimensional-differential gel electrophoresis (2D-DIGE) followed by matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS), antibody microarrays and tandem mass spectrometry have evolved as the major tools. Each of these techniques has unique features with distinct advantages. This review attempts to highlight the common as well as diverse characteristics of these methods and their suitability and application to different experimental conditions employed to investigate the auditory system. In addition a glimpse of the valuable scientific information that has been gained in the hearing field using a proteomic approach is given. Finally, a brief view of the directions that auditory proteomics research is headed for has been discussed.
Keywords: proteomics, DIGE, mass spectrometry, antibody microarray, pathway analysis, hearing
1. Introduction
The sequencing of complete genomes from model organisms including plants, humans and other animal models heralded a new perspective on biology and also brought new tools, methods, and language. In the past decade the disciplines of genomics, proteomics, and metabolomics have come into being and are still evolving. The meaning of proteomics is still evolving. To some, it means the general study of proteins and enzymes, while, to others, it implies the integrated study of the entire sum of all proteins produced by an organism or group of organisms. For the purpose of this review, we shall define proteomics as the large-scale study of multiple proteins of a particular biological sample.
The field of auditory proteomics is relatively new, but growing rapidly. Excellent reviews have been published on what we have learned from two dimensional electrophoresis approaches [40,41,49]. This review will consider what we have learned about auditory proteomics and what we could learn by combining contemporary bioinformatics methods with three approaches to handling proteins, two dimensional gels, tandem mass spectrometry, and antibody microarrays. We will attempt to give a perspective on the advantages, disadvantages and limitations of each method.
2. Why study proteomics?
The study of proteins is a natural extension of recent progress in molecular biology. Genomics and gene profiling has contributed immensely to understanding the molecular mechanisms involved in hearing function and pathology. Several hundred genes have been implicated in hereditary hearing loss [12,45]. With current advances in exon profiling of alternate splice isoforms, it is now possible to gain insights about specialized functions of encoded proteins [2,37]. There are, however, limitations to the type of information that can be gathered from gene profiling. For example, mRNA often poorly correlates with protein expression either due to degradation or inefficient translation [19]. Second, some genes may not be essential to the physiological processes in which they participate because their function may be compensated by other genes. Third, due to differential splicing of exons, there may be as many as 100,000 mRNAs in the transcriptome. Although many of these may have vital functional roles, many are still poorly understood. Each of these limitations points to a need to study the product of gene expression, proteins.
To complement genomic studies, a growing number of recent studies have focused on proteomics allowing for the study of translational and posttranslational regulation. Due to a variety of posttranslational modifications, there may be as many as 1,000,000 distinct proteins. By effecting enzymatic rates and strength of binding to other proteins and substrate metabolites, posttranslational modifications allow proteins to control and define the functional state of the cell. Unlike genomics, this type of control and regulation can be studied in compartments and in organelles (so called “sub proteomes”) that do not contain mRNA. Hence, proteomic data complements and extends the interpretation and application of genomic findings. In addition, proteins can be utilized as biomarkers for tissues, cell types, developmental stages, and disease states, as well as potential targets for drug discovery and interventional approaches.
3. How to study proteins with contemporary proteomic methods
Protein profiling
Protein chemistry has classically involved three steps, stimulation (including treatments, deprivation of treatments and aging), separation, and measurement. The first level of analysis is to characterize the abundance of a protein in a given tissue or cell type. This basic plan has not changed, but contemporary proteomics methods are now available to assay protein abundance and level changes on platforms that allow for assays of multiple proteins in the same experiment. This review will discuss three such methods, tandem mass spectrometry, 2D gel – mass finger print methodology, and antibody microarray with emphasis on the latter two.
Complex interactions
Many classical studies of protein function involved the use of assays for enzymatic activity. Subsequently, techniques evolved for the study of protein function that involved the concerted function of a group of proteins. Examples of this type of experiment, that often falls into a gray area between cell biology and biochemistry are cytoskeletal motility assays, cell adhesion assays, organelle assembly assays, membrane transport and ion channel assays. Methods to study complex interactions of proteins have ranged from recombining separated proteins in the study of ribosome function [15] to eliminating specific proteins from a more complex preparation by RNAi, specific inhibitors, dominant negative transfections, knockouts, and antibody pull-down. At present, there are no proteomic methods for the study of complex protein interactions on a high throughput platform.
Simple protein-protein interactions
Studies of binary protein-protein interactions, on the other hand, have evolved steadily from the classical methods of sedimentation and gel filtration. Advances include Immunoprecipitation [27], co-immunoprecipitation [11]. More recently fluorescent resonant energy transfer [20] and the yeast two hybrid system [47] have been used in the study of the inner ear. Methods for the high throughput analysis of protein-protein interactions are now available [3,33] and can be used with contemporary methods for the analysis of cellular signaling networks has been reviewed elsewhere [35]. Only one group has applied proteomic methods to the analysis of protein-protein interactions in the auditory system [24].
4. 2D-DIGE MALDI-TOF
Two-dimensional difference gel electrophoresis (2D-DIGE) is a differential method for comparing two protein samples. It combines conventional 2D gel electrophoresis with the sensitivity of fluorescent protein labeling for analytical gels and mass finger print analysis by mass spectrometry for preparative gels used for protein identification [44]. The landmark work of Patrick O’Farrell [29] made the use of 2D gels an attractive method of protein separation over the last 30 years. What distinguishes 2D-DIGE is the use of fluorescent dyes to label protein samples so that multiple samples can be run on the same gel [44]. The introduction of matrix-assisted laser desorption ionization – time of flight mass spectrometry (MALDI-TOF MS) for the identification of proteins from 2D gels [21] greatly advanced the use of 2D gels for proteometric analysis.
Combined use of 2D-DIGE and MALDI-TOF has been described in detail elsewhere [7,17,49]. The principle steps (Fig. 1) involve 1) labeling of proteins from control and experimental samples with fluorescent dyes followed by mixing differentially labeled protein samples, 2) separation of proteins by native charge on isoelectric focusing (IEF) gels, 3) separation of proteins by molecular weight on sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), 4) imaging control and experimental protein separated on the same gel using a scanner configured like a confocal microscope with multiple lasers, barrier filters and photomultiplier detectors, 5) off-line analysis of data sets from multiple gels for statistical analysis of changes in protein level, 6) robotic excision of protein spots and robotic proteolysis, typically by trypsin for cleavage at arginine and lysine or V8-protease for cleavage at glutamate and aspartate, 7) measurement of peptide masses by MALDI-TOF MS, and 8) off-line identification of proteins using a search engine(s) to compare measured mass lists with mass lists calculated from theoretical digests from that organisms genome database.
Figure 1. Work flow schematic for implementation of 2D-DIGE.

After homogenization proteins are labeled with up to 3 fluorescent dyes. The third dye, is usually used as an intragel standard for alignment of gels and quanfication of spots. Proteins are separated on 2D gels by charge (IEF) and molecular weight (SDS-PAGE). Analytical gels are scanned by a confocal-like scanner and gray scale images of each color are analyzed by software special for 2D-gels. Proteins of interest are excised robotically and cleaved, typically by trypsin, before laser irradiation. Ionized peptides accelerate though a magnetic field to the MALDI-TOF photomultiplier detector. Masses of peptides are determined with a precision of less than 1 atomic mass unit by measuring time of flight between the laser burst and the incidence of detection.
Prior to off-line mass finger print analysis (step 8), three procedures are recommended to enhance the probability of protein identification and have been reported in detail elsewhere [7]. First, a careful internal calibration using auto digestion of trypsin provides the highest level of precision. Sequencing grade trypsin used to produce peptide fragments is modified by reductive methylation to minimize autolysis. However, limited autolysis of trypsin does occur. Two autolytic tryptic peptides, with masses 842.510 and 2211.105 Da, are present in most mass spectra. In this way, resolution to less than the mass of one neutron can be achieved for successful protein identification. Second, and for the same reason, use the monoisotopic mode of search with carefully selected peaks containing non-isotopic elements. This is important because MALDI-TOF MS is capable of distinguishing between the mass of peptides containing all non-isotopic elements and integral numbers of naturally occurring isotopes (e.g. all carbon-12 vs. one or more carbon-13). The difference in one atomic mass unit can be critical in proper protein identification. Third, masses from spurious peaks not related to the protein, e.g. from auto digestion of trypsin and from keratin contaminants, must be removed from the mass list prior to mass finger print analysis since the probabilistic identification score will be lowered by the presence of non-matching peaks. Fortunately, one or more of these contaminants occur in every sample and can be exploited for internal calibration and easily removed by software (“m/z”, Proteometrics; Peak Erazor, Lighthouse Data, Denmark, http://www.protein.sdu.dk).
Searches may be conducted for matching masses from theoretical tryptic or V-8 peptides from the NCBI and Swiss Protein. We have used the on-line search engine MASCOT (Matrix Sciences at: http://www.matrix-science.com/cgi/htsearch) with the following search parameters: monoisotopic masses, with a tolerance of 200 ppm or less (typically 75–100 ppm or 0.1–0.5 Da), 1 missed tryptic cleavage, a carbamidomethyl modification of cysteine (to account for iodoacetamide treatment) and variable modifications of oxidation of methionine and phosphorylation of serine, threonine or tyrosine. As alternatives to the Mascot search engine, we have also made use of ExPASY’s Aldente search engine (Gasteiger et al., 2005), Rockefeller and New York Universities’ Profound search engine and University of California San Francisco’s Protein Prospector (Clauser et al., 1999). Use of multiple search engines is recommended for proteins with border line probabilistic scores in order to generate additional candidates for confirmation by independent methods.
5. Antibody Microarrays
Microarray technology for performing the protein assays was developed by Ekins et al in the late 1980s [13]. Initially, 96-well microtiter plates were used [14]. Membrane based arrays were also used because of their superior binding capacity [4]. Later, arrays were developed by spotting the antibodies on a chip. A typical antibody microarray (Fig. 2) consists of a large number of antibodies distributed in rows and columns with spot sizes <250μm. The antibodies are either bound to a nitrocellulose membrane attached to a glass slide or covalently immobilized directly on the glass slide itself. Experimental implementation also varies for antibody microarrays (Fig. 3). Some investigators use an enzyme-linked immunosorbent assay ELISA-like sandwich composed of capture antibody spotted on the substrate, bound antigen and fluorescent labeled detection antibody. Others use single antibodies with bound antigen labeled with a radioactive isotope or a fluorescence dye [22]. Other variations include fluorescent detection with either single color dyes with a single sample per array or dual colored labeling with both experimental and control samples differentially labeled and bound to the same array. A comparison of some commercially available antibody microarrays, which may be used for any of these assays, is given in Table 1.
Figure 2. A typical antibody microarray.
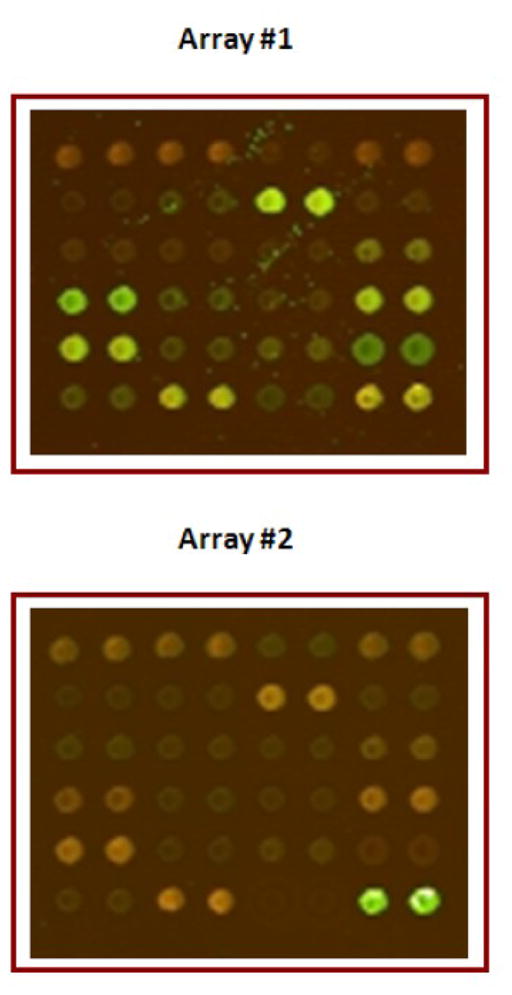
One block from an antibody microarray that consists of 725 antibodies spotted as duplicates in 32 blocks, each having 6 rows and 8 columns is shown. For array # 1, control proteins were labeled with Cy3 and proteins from experimental group with Cy5. For array # 2, the labeling dyes were swapped (controls labeled with Cy5 and experimental with Cy3). The pseudo-color arrays depict the ratio of fluorescence intensities of protein in control sample (scanned at 532 nm) versus the experimental sample (635nm) for array 1 and vice-versa for array 2.
Figure 3. Antibody microarray assay methods.

Four methods of performing an antibody microarray experiment showing the use of fluorescently labeled protein samples (red and green), radioactively labeled proteins (*), and unlabeled proteins (open oval). The fluorescent ratiometric assay is the easiest to implement and most cost effective. The radioactive and fluorescent sandwich assays have the greatest dynamic range (max signal: noise floor on the order of 1000:1). Dye labeled protein assays are easier to implement with large arrays (200–700 antibodies spotted in duplicate) due to complications of detection spatial sensitivity (radioactive assays) and cross-reactions among detection antibodies (sandwich assay). Potential interactions among detection antibodies limit the number of antibodies per array to 20–40 for the sandwich assay.
Table 1.
Features of some commonly used antibody microarrays.
| Manufacturer* | Sigma | Clontech | ||||
|---|---|---|---|---|---|---|
| Array | XP725 | CSAA1 | PPAA4 | MPAA3 | GRAA2 | Ab Microarray 500 |
| No. of Antibodies | 725 | 224 | 112 | 84 | 112 | 500 |
| Functional category | Broad spectrum | Cell signaling | p53 pathways | MAPK &PKC pathways | Gene regulation | Protein kinases, apoptosis, cancer, cell cycle, neurobiology |
| ~ Cost per antibody | $3 | $4 | $6 | $7 | $6 | $3 |
| Antibody printing | Spotted onto nitrocellulose coated glass slide | Covalently immobilized to glass slide | ||||
| Recommended | >2 | 2–4 | ||||
| Dye/Protein ratio | ||||||
| Estimated time frame (Homogenization – Drying) | ~ 4–6 hours | <1 day | ||||
| Recommended detection chemistry | Cy3-Cy5 based | Cy3-Cy5 based | ||||
| Sensitivity | 3 ng/ml (for caspase 9) | 20 pg/ml (protein not specified) | ||||
| Recommended starting material (initial protein concentration) | 300 μg @ 1 mg/ml | 220 μg @ 1.1 mg/ml | ||||
| Protein assay for labeled proteins | Bradford | BCA (Bicinchoninic Acid Assay) | ||||
Distinctive features of different antibody microarrays marketed by two manufacturers are furnished in the table. These microarrays are meant to be used for dual colored ratiometric assay that facilitates dye swap analysis. Apart from these * Array It, Ray Biotech, Spring Biosciences, Fullmoon Biosystems are some of the other manufacturers who use single dye detection method with biotin –streptavidin conjugated Cy3.
Principle and general protocol
The general protocol for performing a dual color ratio-metric assay is described here. Proteins are extracted from the tissue by homogenizing with extraction buffer usually provided with the antibody micoarray kit. Other appropriate lysis buffers have also been reported for protein extraction. Then the protein concentration of the sample is determined and proteins from the control and experimental samples are labeled with fluorescent dyes according to the manufacturer’s instructions. Unincorporated dye is removed by gel or membrane filtration and dye incorporation is determined spectrophotometrically (at 552 nm for Cy3 and 650 nm for Cy5). Differentially labeled samples from the control and experimental group are mixed and applied simultaneously at equal protein concentrations on the array using a dye-swapping paradigm [7]. With dye-swapping, one array is incubated with control proteins labeled with Cy3 mixed together with experimental proteins labeled with Cy5. A second slide is incubated with control and experimental proteins labeled with the opposite dyes. Swapping dyes allows for normalization of differences in quantum efficiencies (brightness at the emission wavelength) between the two dyes and differences in labeling chemistries. Fluorescent signal intensities for each sample are then recorded using a microarray scanner. Spots that showed a uniform distribution with a positive signal to noise ratio are taken for analysis. The data obtained can be normalized by calculating the geometric mean of background-corrected fluorescence values [22] or with reference proteins such as actin, tubulin or GAPDH.
Considerations specific to microarray experiments
Protein extraction buffers are provided with commercially available arrays. These can include protease inhibitors, phosphatase inhibitors and benzonase (decreases the sample viscosity). One manufacturer recommends against the use of protease inhibitors because of potential interference with protein labeling. We have routinely used a cocktail of inhibitors and obtained good labeling [22]. An initial protein concentration of 1 to 1.5 mg/ml is required for optimum labeling efficiency. Fluorescent dyes (Cy3-Cy5, Alexa or DyLight dyes from GE Healthcare, Invitrogen or Thermo Scientific) are the preferred detection method as they are safe, simple, sensitive and give high resolution. These light sensitive dyes mixed in borate or bicarbonate buffers should be used at the appropriate pH (8.5–9.6) to attain good labeling of proteins. Protein concentration should be assayed using non-fluorescent methods such as Bradford and bicinchoninic acid assays. To determine the molar concentration of the dye, a portion of the labeled samples is diluted to obtain a maximum absorbance in the range of 0.5–1.0 AU at appropriate wavelengths for the given molar extinction coefficients. Optimum D/P ratio is 2–4. Higher dye labeling may interfere with antibody binding thereby reducing sensitivity. If D/P is < 2, it is better to either repeat the labeling procedure or prolong the array incubation time. After incubation and wash steps, the slides should be left to dry completely before reading. Use of a centrifuge to speed up drying is detrimental due to comet-like streaking effects. Similarly, any movement of the slides during drying process may also create artifacts. The dried slides should be scanned within 24 to 48 hours. We have begun experiments to optimize extraction and dye labeling in order to improve the dynamic range of the assay and to avoid the use of unknown chemicals in proprietary buffers.
Versatility of antibody microarrays
In the auditory field, the first use of antibody microarrays was reported in the study of cisplatin ototoxicity [22]. Subsequently, we have employed arrays to investigate a diverse spectrum of hearing pathologies in rats and chinchillas (Fig. 4). In these studies, proteins were extracted from whole cochlea, postnatal day 3 (P3) organ of Corti, P3 spiral ganglion and auditory cortex of rats and three discrete regions of chinchilla cochlea. For each experiment, antibody microarray analysis produced a signal detection rate of at least 80%, i.e. antigen-antibody binding resulted in a signal exceeding background for greater than 80% of the antibody spots. Although antibodies spotted on arrays are usually tested against human, rat or mouse, our experience with chinchillas indicates that proteomic responses in other animal models can also be studied using the antibody microarrays. It should be noted, however, that, in these cases, it is all the more important to evaluate antibody specificity (see section 8). These results collectively indicate the sensitivity and utility of antibody microarrays, which can be employed for proteomic studies of hearing physiology and pathology in a variety of animal models.
Figure 4. Detection range of cochlear and brain proteins by antibody microarrays.

Antibody microarrays detected more than 80% of the investigated proteins in the cochlea and auditory cortex of adult rats, two discrete regions of P3 rat cochlea and three regions of chinchilla cochlea. This high detection rate indicates that antibody microarrays can be used for investigating the proteomic changes in diverse tissue types of different animal models that are commonly used for auditory research.
6. Tandem Mass Spectrometry
Tandem spray mass spectrometry (MS-MS) provides, arguably, the most powerful and, indisputably, the most sophisticated technology of all contemporary proteomic methods. Using MS-MS a complex protein sample can be applied and hundreds of protein identifications can be achieved in relatively short times. The principle of operation is similar to 2D-DIGE – MALDI-TOF MS except that the instrumentation is contained in one platform (Fig. 5). Samples are first hydrolyzed by a selective protease, typically trypsin. Then peptides are separated by high performance liquid chromatography (HPLC) and routed through two mass spectrometers in series. The first mass spectrometer operates in the time of flight mode like that used for DIGE analysis (section 4) and provides mass information on each tryptic peptide. The second mass spectrometer disintegrates each peptide and provides a time dependent measure of the mass that is used to determine the amino acid that was removed and thereby provide amino acid sequence for each peptide. As with mass finger print analysis, analysis of MS-MS data is done off line using a search engine and mass data to identify peptides and to give a probabilistic identification of proteins in the sample. Details of the technology have been reviewed elsewhere [5]. Limitations and applications of the method are discussed in section 7 in comparison with 2D-DIGE and antibody microarray. MS-MS has recently been used for auditory proteomics experiments in the study of the proteins of the stereocilia [16,38]. These data are briefly discussed in section 9.
Figure 5. Typical tandem mass spectrometry system.

A high performance liquid chromatography initial stage (HPLC) is followed by tandem mass spectrometers MS1 and MS2. MS1 gives peptide mass and MS2 give amino acid sequence of individual peptides. Analysis software and search engine assign multiple peptide sequences in protein identification.
Tandem MS can be used in several ways. It can be used in conjunction with 2D-DIGE in place of MALDI-TOF for the identification of proteins. Although more complicated to learn and execute than MALDI-TOF, tandem MS offers distinct advantages of greater sensitivity and the ability to identify a greater number of proteins including proteins from organisms whose genome has not been fully sequenced. Tandem MS is also the method of choice for phospho-peptide mapping and mapping of other posttranslational modifications. In addition, several relatively recent labeling strategies have made it possible to use tandem MS platforms for analytical comparisons of protein expression between two samples. One method, isotope coded affinity tag-ICAT [18], uses differential labeling with radioactive tags for quantifying fold changes between two samples. Another, SILAC (stable isotope labeling by amino acids in cell culture), avoids the chemical modification and affinity purification steps of ICAT by using incorporation of stably labeled amino acids [10,30]. Compared to ICAT, this method is more convenient and inexpensive, but is limited to cell culture experiments whereas ICAT may be applied to in vivo experiments. A third method, ITRAQ (isobaric tag for relative and absolute quantification) uses chemical modification with tags that allow for simultaneous analysis of multiple samples. ITRAQ was originally developed using 4 distinct reporter ions [43], but has since been improved for 6-plex [9] and 8-plex [6] applications. The advantages for dose response and time course experiments are especially clear.
7. How to select a proteomic method
Selection of a proteomic method depends on the nature of the biological question, on the established expertise of a lab, and on time and funds budgeted for implementing a new method. In the last 10 years, an increasing number of universities have established proteomic core facilities or alternative ways to share costs and expertise among research faculties so that biological considerations may preside over cost. Table 2 provides a comparison of the three proteometric methods discussed above that may be useful in selecting an appropriate technique.
Table 2.
Comparison of three different proteomic research tools.
| 2D – DIGE | Antibody microarray | Tandem MS§ | |
|---|---|---|---|
| Preparation | Major proteins | Preselected proteins | Major proteins |
| Complexity of prep | No restrictions | No restrictions | Limited |
| Sensitivity | 10 μg per analytical sample 1 mg per preparative gel |
5 – 10 μg with custom micro-incubation chamber | nanogram |
| Dynamic Range | 103 | 103 | 103 |
| Availability | Depends on facility | Best | Depends on facility |
| Assay speed and analysis speed | Poor* | Excellent* | Good |
| Cost per identified protein | Highest | Lowest | Depends on facility fees |
| Cost of equipment | Moderate | Lowest | Highest |
| Training Required | Moderate | Lowest | Highest |
| Max level of analysis | Some posttranslational Inferences possible | Some antibodies against posttranslational modifications available | Best for mapping modified amino acids. |
| ~ No. of proteins per assay | 1200 – 2500 | 20 – 725 | 50 – 300 |
| Posttranslational modification detection capability | Phosphorylation Glycosylation |
Phosphorylation Methylation Acetylation Tyrosine nitration |
Phosphorylation Methylation Acetylation Nitration |
| Other isoforms detection capability | Proteolysis Splice variant translation products |
Restricted to antibody availability | Proteolysis Splice variant translation products |
2D-DIGE and antibody microarray are typically used for comparing two samples, ie fold change. Although tandem MS is used for the identification of peptides and proteins, we included it in this comparison as it can also be used for quantification when coupled with labeling techniques like ICAT, ITRAQ and SILAC.
A typical 2D-DIGE experiment takes 3–5 days for data collection and analysis and an additional 3–7 days for protein identification. On the other extreme, an antibody microarray experiment requires 1–2 days for the assay and analysis.
Abundance and complexity
As our understanding of the cell and its complex proteome increases, there is an increasing awareness of the critical importance of proteins of low abundance like receptors and other upstream signaling proteins. It has been estimated that abundant proteins, including structural proteins and metabolic enzymes, are expressed in the range of from 105 to 106 molecules per cell, while lower abundant proteins may be expressed with less than 100 molecules per cell [17]. The availability of the complete sequence of the human genome and those of several model systems has put data about poorly understood low abundance proteins within our grasp. It is estimated that each cell expresses between from 10–15,000 genes. Considering alternative splicing of RNA and a growing number of posttranslational modifications, there may be as many as 106 different protein isoforms in any given tissue. If a typical significant fold change for a protein is 2 to 10 fold for either total expression or posttranslational modification, then a proteomic system capable of assaying both highly abundant proteins (106 per cell) and low abundance proteins (<100 per cell) should have a dynamic range that exceeds 105 (five orders of dynamic range, 106per cell/102 per cell/10 folds of change). This is well within the capability of modern 16 and 32 digital processers. Photomultipliers exceeding 5 orders of dynamic range are also commercially available (Licel GbR, Berlin) and manufacturers of proteomic instrumentation advertise being able to achieve this goal. However, each of the proteomic methodologies discussed above is limited in dynamic range by one or more factors so that effective dynamic range of reported data is often on the order of 103. Each of the three proteometric methods discussed can benefit from the use of subcellular fractionation, a classical and time proven approach to working with low abundant proteins by enrichment for organelle specific proteins.
In addition to enrichment of low abundant proteins, subcellular fractionation also serves to reduce the complexity of protein samples. This consideration is especially important when considering using tandem mass spectrometry which is much more limited by complexity of the protein sample than either 2D-DIGE or antibody microarray. The limitation arises from the requirement for sophisticated algorithms to sort through the large and complex array of data. Another way around the complexity issue is to analyze narrow regions of single dimension SDS gels or spots from 2D gels. Tandem MS is, in fact, quickly replacing MALDI-TOF identification of 2D-DIGE proteins because of its high sensitivity to nanogram amounts of protein.
Advantages of antibody microarrays
There are several distinct advantages for antibody microarrays over other conventional techniques used for proteomic studies. 1) Antibody microarrays are fast, and are versatile with respect to their utility in multiple species. 2) Multiple proteins from two different conditions (control vs. experimental) can be analyzed simultaneously in one array. 3) Unlike 2D-DIGE, even low abundant proteins can be detected and hence microarrays are highly sensitive. 4) Commercial availability of antibodies makes it easier to validate microarray findings. 5) Availability of different types of arrays facilitates the evaluation of proteomic responses oriented toward specific groups of proteins of interest. 6) In addition to protein profiling, the presence of several antibodies against phosphorylated, dephosphorylated, methylated and nitrated proteins in the array, helps in the study of functional proteomics. 7) Finally, it is possible to construct custom antibody microarrays using robotic spotters [25] or to purchase custom arrays (Sigma-Aldrich). Although, the inability to detect novel proteins will remain as a major limitation for this technique, the numerous positive features listed here, clearly suggest a major role for antibody microarrays in future proteomic endeavors in the inner ear.
8. Confirmation and interpretation of proteomic results
Validation methods
A creed of genomics and proteomics investigators is that results from high throughput platforms are not valid until they are confirmed by an independent method. An example would be performing quantitative Western blots on proteins of interest from a proteomic screen. Most quantitative Western blotting involves a housekeeping control like actin or glyceraldehyde-3-phosphate dehydrogenase. This control provides a measure of equal loading of gel lanes and a quantitative means of normalizing for small perturbations from equal loading. Some economy of effort can be gained by stripping blots and reprobing with multiple antibodies.
Western blots pose two problems. First, they can be very time consuming depending on antigens or antibodies of interest. Second, Western blots are not all that independent a method when considering validating antibody microarray results. These barriers can be addressed in a number of ways. A faster and more efficient way of validating proteomic results for a larger list of proteins is by using ELISA or a dot or slot blot assay. The ELISA takes bit more development time, but can have a greater sensitivity and dynamic range than Western blots, dot blot or slot blot assays. Although development of a quantitative ELISA is relatively straight forward, it should be noted that there are consulting firms that specialize in assay development and that commercial ELISA kits are available for many proteins. In some cases, enzymatic assays and other bioassays might constitute an excellent choice as a validation method. For example, fluorescent substrate peptides are available for measuring caspase activity (Invitrogen – Molecular Probes). A host of kits are available for measuring other enzymes including antioxidant enzymes (Cayman Chemical, Dojindo) and protein kinases (EMD – Calbiochem, Invitrogen, Promega, Thermo Scientific – Pierce). Having suggested alternatives to the quantitative Western blot, it is important to point out that simple (non-quantitative) Western blots are the most efficient measure of antibody specificity and remain as irreplaceable and underutilized tools.
We have recently begun to use real time polymerase chain reaction (RT-PCR) assays as a time- and cost effective initial validation method. RT-PCR offers the advantage that it can be rapidly performed using custom-designed plates containing primers from genes of interest from tandem MS, 2D-DIGE or antibody microarray. It offers an additional advantage for use in parallel with microarray experiment in that, unlike immunoblots or ELISAs, it is truly an independent method of validation that does not depend on antibodies.
It should be noted that RT-PCR may only be considered as an initial validation method and is better regarded as a complementary tool. We have seen good agreement between the transcriptome and proteome response to ototoxic drugs for many genes and gene products at the same time point. However, the time lag between peaks in mRNA and protein synthesis can vary from 1h to 1 or more days and must be taken into consideration.
Finally, the importance of cellular and subcellular localization cannot be overemphasized. If two or more proteins identified by proteomic screening and validated by an independent method can be co-localized to same subcellular compartment, then the physiological relevance of any common networks associated with these proteins becomes much more credible. In a recent study of cisplatin-induced changes in protein expression, 3 out of 20 proteins of interest from a whole cochlea preparation are associated with the Golgi-centrosome region [22]. We have validated the presence of one of these using immunocytochemistry. Co-localization of all three proteins with Golgi-centrosome markers would provide a high degree of validation and also pose new questions about possible protein-protein interactions.
Pathway analysis
The enormity of information gathered with proteomic tools has led to the development of specialized methods to handle and interpret data. A global analysis of proteomic data obtained using high throughput techniques is facilitated by bioinformatics software platforms. These web based applications provide additional biological insights on the outcome of the research by helping to gain better interpretation of the available information. Basically, these platforms analyze the given protein data with reference to standard as well as customized databases that are generated by either human curation or language analysis software. Functional networks are built with the experimental data, indicating the relationships between the molecules identified in the experiments, which may help to formulate or corroborate hypotheses and may lead to better designing of future studies. Moreover these analyses also identify the canonical pathways that are likely to be linked with the analyzed proteins and grade them according to their significance level, which is calculated using a variety of statistical methods. The biological processes and disease phenotypes that are likely to be associated with the subsets of proteins are furnished by the software, which also indicates the subcellular location of the proteins. Overall these analytical tools augment the interpretation and characterization of large proteomic data sets. Among the several pathway analysis platforms that are available, few of them such as DAVID, Cytoscape and Biogrid (protein-protein interaction repository) are open source packages and are freely accessible, while others require license/contracts to access them. Some of the distinctive features of three commonly used software packages are furnished in Table 3.
Table 3.
Common commercial pathway analysis platforms.
| Metacore | Ingenuity | Ariadne | |
|---|---|---|---|
| Curation method | Human curated | Human curated | Language analysis software |
| Analytical filters | Disease, tissue, species, subcellular localization, interactions, metabolites | Disease, tissue, species, cell type, relationships | Diseases, cell processes, organelles, relationships |
| No. of pathways | ~ 683 maps of canonical pathways | ~ 183 canonical pathways | ~ 227 signaling &21 cell process pathways (ResNet Mammalian Database) |
| Scores | p-value, Z-score & G-score (networks) | p-value (network & functions) | p-value (networks) |
| Supported species | Mouse, rat, worm, fly, yeast & dog | Human, mouse, rat & canine | Human, rat &mouse (ResNet Mammalian Database) |
Characteristic features of three pathway analysis software are furnished as representative samples. However, it has to be noted that several other platforms are freely available, like DAVID (NIH), which is also widely used for pathway analysis of large data sets.
Criteria for Physiologically Significant Changes in Protein Expression
In the 1970’s Nimmo and Cohen [28] and Krebs and Beavo [26] established a set of criteria that must be met for a phosphorylation to be considered physiological. These criteria have served well to establish the physiological relevance of changes in the phosphoproteome. A similar set of criteria may be useful in the analysis of contemporary proteomic data. In order for a proteomic screening result to be considered relevant, we suggest the following criteria, modeled after those of Krebs and Beavo.
Demonstrate by independent methods that a protein level changes significantly with a given stimulus or treatment (or absence/mutation of a specific gene).*
Demonstrate that the stimulus or treatment changes the rate of synthesis or rate of degradation of the protein.
Demonstrate that the change in protein level correlates with an altered physiological measure.
Demonstrate that the change in protein level occurs in a cell type that can be shown to participate in the physiological phenotype.
Demonstrate in vivo that the protein is involved in the physiological response.
*The fold change that will produce a physiologically significant change in the state of the cell or tissue depends on many factors including the individual protein characteristics and the method of analysis.
9. What we have learned from auditory proteomics
Proteomic studies in normal inner ear
Application of proteomic methods to investigate the auditory system began as early as 1980 when two-dimensional electrophoretic separations of proteins from the organ of Corti of guinea pig were reported [42]. The studies that followed, attempted to identify the proteins in a normal ear. Consistent with their use of electrophoresis, these studies were the first to identify highly abundant proteins of the cochlea including, organ of Corti protein I and II (OCPI and OCPII) and oncomodulin. Subsequently, the same group identified ototconial proteins by MS [41]. Unique proteins in perilymph that were not found in plasma were identified in guinea pigs and humans, using two-dimensional gel electrophoresis [32]. Proteins in the hair bundle were investigated by different groups. Shotgun MS was used to identify many of the most abundant bundle proteins in chicken vestibular hair bundles [38]. The tip link antigen of the hair bundle was identified as protocadherin-15 by MS [1]. The secreted acidic cysteine rich glycoprotein (Sparc), thought to be necessary for normal otolith growth was recently discovered using a tandem MS technique [23]. The expression of chloride intracellular channel protein CLIC5 in stereocilia of chicken utricle was also studied by MS [16].
Proteomic investigation of cochlear pathologies
Proteomic methods are also being employed to investigate auditory pathologies. For example, application of proteomic methods like 2D-DIGE and MS has been suggested as experimental tools to investigate mouse models for deafness using a novel discovery strategy [49]. The potential utility of using mouse mutants like Pou4f3 mouse mutant to study the protein expression profiles of a specific tissue, hair cells in this case, has been discussed. Moreover, drug induced ototoxicity has also been studied using proteomic methods. 2D-DIGE followed by MALDI TOF MS was used to investigate the cisplatin induced proteomic changes in P3 rat cochlea [7]. Cisplatin-induced changes (greater than 1.5-fold) in expression of 22 cochlear proteins were reported. Later the same group reported the utility of antibody microarrays to analyze the cisplatin induced proteomic changes in cochlea from adult rats [22]. Among the 19 cochlear proteins whose expression levels either increased to ≥ 1.5 fold or decreased to ≤ 0.6 fold, after cisplatin treatment, 15 were identified for the first time in cisplatin-induced ototoxicity. These studies highlight the value of using a proteomic approach for investigating cochlear pathologies.
Proteomic research in central hearing
So far, two studies have used a proteomic approach to investigate the central auditory apparatus. 2D-DIGE and MALDI TOF MS were used to study the protein expression in the vestibular nucleus during vestibular compensation [31]. In this study, 26 proteins were significantly altered in the medial vestibular nucleus of rats, one week after unilateral labrynthectomy. Functional characteristics of some of these proteins were reported to correlate with vestibular system plasticity. In another study, profiling of experience-regulated proteins by 2D-DIGE and tandem MS was done in the auditory forebrain of song-bird [34]. Several proteins that could be classified as metabolic enzymes, cytoskeletal proteins, neurotransmitter secretory proteins and calcium binding proteins were identified. Based on these findings, it has been suggested that the auditory processing in song-birds is regulated by a calcium level dependent protein network. These studies give an insight into the scope and application of proteomic methods to study the physiological as well as pathological state of the central auditory system. Hence it could be foreseen that the proteomic approach is more likely to be widely employed to investigate both central and peripheral auditory systems, which in turn may help to unravel the mechanisms underlying a diverse spectrum of otopathologies.
10. Future directions for auditory proteomics
The scope of proteomic studies is likely to widen further to cover various facets of auditory research. The number of applications are expected to grow due to the enormity of the data that may be generated and the importance that may be associated with their findings. Screening of inner ear proteins with custom designed arrays could be a classical example for this scenario. Proteomic profiling helps to obtain a comprehensive overview of the cellular or tissue proteome which in turn facilitates the characterization of functional activity and their perturbations. Especially, for a highly differentiated tissue with several distinct cell types and varied functional roles, like the inner ear, protein profiling is likely to play a major role in investigating these specialized tissues. Recently, the cochlear protein profiles of three different rat strains with normal hearing function were analyzed using a broad spectrum antibody microarray [Jamesdaniel et al., manuscript submitted].
Investigation of protein-protein and protein-DNA/RNA interactions is an area of great interest and will continue to grow. Protein-protein interactions are among the vital components that regulate cellular function. The combination of proteomic methods especially 2D-DIGE and MS preceded by co-immunoprecipitation is an excellent experimental approach to study protein-protein interactions [24]. Other emerging methods include binary interactome mapping with high throughput yeast two-hybrid screening and co-complex interactome mapping with high throughput coaffinity purification coupled with MS [46,48]. In addition, there has been an emerging shift from data gathering to data handling as exemplified in literature-curated protein interaction data sets [8]. Worm and yeast researchers have made extensive use of combining these methods to make electronic repositories with searchable interactome databases [36,39]. At least one of these databases (http://www.thebiogrid.org/index.php) contains a limited number of mammalian protein interaction datasets and is of immediate value for analysis of auditory proteomic data. These techniques are expected to rapidly evolve and to become widely employed by auditory researchers to understand the cellular mechanisms underlying the physiological or pathological conditions of inner ear.
Characterization of the proteome of inner ear using different mouse mutants [49] is also expected to evolve as a useful strategy to study the various models of deafness. Moreover, proteomic studies of the inner ear may also contribute to drug discovery. For example, some proteins that are abundantly expressed in discrete cellular types under specific pathological condition could be exploited as potential targets for interventional approaches. In addition, identification of the common and unique responses, in terms of protein expression, in different otopathologies, is likely to give a greater insight into the general as well as precise underlying mechanisms. Given the unique characteristics of the auditory system and significant information that these proteomic tools are capable of unearthing, it could be foreseen that proteomics may have major share in determining the directions of future auditory research.
Acknowledgments
This review was inspired by previous applications of 2D gel electrophoresis to inner ear development and physiology, particularly the work of Dr. Isolde Thalmann. We are grateful for thoughtful conversations with Dr. Kumar Alagramam. We wish to acknowledge funding to DC from the National Organization of Hearing Research Foundation and Deafness Research Foundation and to RS from NIH grants R01DC006630 and R01DC00909101. Finally, we are appreciative of the depth and thoroughness of the reviewers’ comments that have greatly improved the manuscript.
Abbreviations
- 2D-DIGE
two dimensional-differential gel electrophoresis
- MALDI-TOF
matrix assisted laser desorption ionization time of flight
- MS
mass spectrometry
- IEF
isoelectric focusing
- SDS-PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
- BCA
bicinchoninic acid assay
- ELISA
enzyme-linked immunosorbent assay
- D/P
Dye to protein molar ratio
- HPLC
high performance liquid chromatography
- RT-PCR
real time polymerase chain reaction
- CLIC5
chloride Intracellular Channel Protein 5
- OCP
organ of Corti protein
- P3
postnatal day 3
- ITRAQ
isobaric tag for relative and absolute quantitation
- SILAC
stable isotope labeling by amino acids in cell culture
- ICAT
isotope coded affinity tag
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, Burgess SM, Lilley KS, Wilcox ER, Riazuddin S, Griffith AJ, Frolenkov GI, Belyantseva IA, Richardson GP, Friedman TB. The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci. 2006;26:7022–7034. doi: 10.1523/JNEUROSCI.1163-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bemmo A, Benovoy D, Kwan T, Gaffney DJ, Jensen RV, Majewski J. Gene expression and isoform variation analysis using Affymetrix Exon Arrays. BMC Genomics. 2008;9:529. doi: 10.1186/1471-2164-9-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berggard T, Linse S, James P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 2007;7:2833–2842. doi: 10.1002/pmic.200700131. [DOI] [PubMed] [Google Scholar]
- 4.Bussow K, Cahill D, Nietfeld W, Bancroft D, Scherzinger E, Lehrach H, Walter G. A method for global protein expression and antibody screening on high-density filters of an arrayed cDNA library. Nucleic Acids Res. 1998;26:5007–5008. doi: 10.1093/nar/26.21.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen CH. Review of a current role of mass spectrometry for proteome research. Anal Chim Acta. 2008;624:16–36. doi: 10.1016/j.aca.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 6.Choe L, D’Ascenzo M, Relkin NR, Pappin D, Ross P, Williamson B, Guertin S, Pribil P, Lee KH. 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Proteomics. 2007;7:3651–3660. doi: 10.1002/pmic.200700316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coling DE, Ding D, Young R, Lis M, Stofko E, Blumenthal KM, Salvi RJ. Proteomic analysis of cisplatin-induced cochlear damage: methods and early changes in protein expression. Hear Res. 2007;226:140–156. doi: 10.1016/j.heares.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Cusick ME, Yu H, Smolyar A, Venkatesan K, Carvunis AR, Simonis N, Rual JF, Borick H, Braun P, Dreze M, Vandenhaute J, Galli M, Yazaki J, Hill DE, Ecker JR, Roth FP, Vidal M. Literature-curated protein interaction datasets. Nat Methods. 2009;6:39–46. doi: 10.1038/nmeth.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, Sanchez JC. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 10.de Godoy LM, Olsen JV, de Souza GA, Li G, Mortensen P, Mann M. Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biol. 2006;7:R50. doi: 10.1186/gb-2006-7-6-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreyfuss G, Swanson MS, Pinol-Roma S. Heterogeneous nuclear ribonucleoprotein particles and the pathway of mRNA formation. Trends Biochem Sci. 1988;13:86–91. doi: 10.1016/0968-0004(88)90046-1. [DOI] [PubMed] [Google Scholar]
- 12.Eisen MD, Ryugo DK. Hearing molecules: contributions from genetic deafness. Cell Mol Life Sci. 2007;64:566–580. doi: 10.1007/s00018-007-6417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekins R, Chu F, Biggart E. Multispot, multianalyte, immunoassay. Ann Biol Clin (Paris) 1990;48:655–666. [PubMed] [Google Scholar]
- 14.Emili AQ, Cagney G. Large-scale functional analysis using peptide or protein arrays. Nat Biotechnol. 2000;18:393–397. doi: 10.1038/74442. [DOI] [PubMed] [Google Scholar]
- 15.Frank J. The ribosome--a macromolecular machine par excellence. Chem Biol. 2000;7:R133–141. doi: 10.1016/s1074-5521(00)00127-7. [DOI] [PubMed] [Google Scholar]
- 16.Gagnon LH, Longo-Guess CM, Berryman M, Shin JB, Saylor KW, Yu H, Gillespie PG, Johnson KR. The chloride intracellular channel protein CLIC5 is expressed at high levels in hair cell stereocilia and is essential for normal inner ear function. J Neurosci. 2006;26:10188–10198. doi: 10.1523/JNEUROSCI.2166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorg A, Weiss W, Dunn MJ. Current two-dimensional electrophoresis technology for proteomics. Proteomics. 2004;4:3665–3685. doi: 10.1002/pmic.200401031. [DOI] [PubMed] [Google Scholar]
- 18.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 19.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallworth R, Currall B, Nichols MG, Wu X, Zuo J. Studying inner ear protein-protein interactions using FRET and FLIM. Brain Res. 2006;1091:122–131. doi: 10.1016/j.brainres.2006.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc Natl Acad Sci U S A. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamesdaniel S, Ding D, Kermany MH, Davidson BA, Knight PR, 3rd, Salvi R, Coling DE. Proteomic analysis of the balance between survival and cell death responses in cisplatin-mediated ototoxicity. J Proteome Res. 2008;7:3516–3524. doi: 10.1021/pr8002479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang YJ, Stevenson AK, Yau PM, Kollmar R. Sparc protein is required for normal growth of zebrafish otoliths. J Assoc Res Otolaryngol. 2008;9:436–451. doi: 10.1007/s10162-008-0137-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kathiresan T, Harvey MC, Sokolowski BH. The use of 2-D gels to identify novel protein-protein interactions in the cochlea. Methods Mol Biol. 2009;493:269–286. doi: 10.1007/978-1-59745-523-7_16. [DOI] [PubMed] [Google Scholar]
- 25.Knight PR, Sreekumar A, Siddiqui J, Laxman B, Copeland S, Chinnaiyan A, Remick DG. Development of a sensitive microarray immunoassay and comparison with standard enzyme-linked immunoassay for cytokine analysis. Shock. 2004;21:26–30. doi: 10.1097/01.shk.0000101668.49265.19. [DOI] [PubMed] [Google Scholar]
- 26.Krebs EG, Beavo JA. Phosphorylation-dephosphorylation of enzymes. Annu Rev Biochem. 1979;48:923–959. doi: 10.1146/annurev.bi.48.070179.004423. [DOI] [PubMed] [Google Scholar]
- 27.Merisko EM, Farquhar MG, Palade GE. Coated vesicle isolation by immunoadsorption on Staphylococcus aureus cells. J Cell Biol. 1982;92:846–857. doi: 10.1083/jcb.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nimmo HG, Cohen P. Hormonal control of protein phosphorylation. Adv Cyclic Nucleotide Res. 1977;8:145–266. [PubMed] [Google Scholar]
- 29.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 30.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 31.Paterson JM, Short D, Flatman PW, Seckl JR, Aitken A, Dutia MB. Changes in protein expression in the rat medial vestibular nuclei during vestibular compensation. J Physiol. 2006;575:777–788. doi: 10.1113/jphysiol.2006.112409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paugh DR, Telian SA, Disher MJ. Identification of perilymph proteins by two-dimensional gel electrophoresis. Otolaryngol Head Neck Surg. 1991;104:517–525. doi: 10.1177/019459989110400416. [DOI] [PubMed] [Google Scholar]
- 33.Phizicky EM, Fields S. Protein-protein interactions: methods for detection and analysis. Microbiol Rev. 1995;59:94–123. doi: 10.1128/mr.59.1.94-123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinaud R, Osorio C, Alzate O, Jarvis ED. Profiling of experience-regulated proteins in the songbird auditory forebrain using quantitative proteomics. Eur J Neurosci. 2008;27:1409–1422. doi: 10.1111/j.1460-9568.2008.06102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Preisinger C, von Kriegsheim A, Matallanas D, Kolch W. Proteomics and phosphoproteomics for the mapping of cellular signalling networks. Proteomics. 2008;8:4402–4415. doi: 10.1002/pmic.200800136. [DOI] [PubMed] [Google Scholar]
- 36.Reguly T, Breitkreutz A, Boucher L, Breitkreutz BJ, Hon GC, Myers CL, Parsons A, Friesen H, Oughtred R, Tong A, Stark C, Ho Y, Botstein D, Andrews B, Boone C, Troyanskya OG, Ideker T, Dolinski K, Batada NN, Tyers M. Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae. J Biol. 2006;5:11. doi: 10.1186/jbiol36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schweighoffer F, Ait-Ikhlef A, Resink AL, Brinkman B, Melle-Milovanovic D, Laurent-Puig P, Kearsey J, Bracco L. Qualitative gene profiling: a novel tool in genomics and in pharmacogenomics that deciphers messenger RNA isoforms diversity. Pharmacogenomics. 2000;1:187–197. doi: 10.1517/14622416.1.2.187. [DOI] [PubMed] [Google Scholar]
- 38.Shin JB, Streijger F, Beynon A, Peters T, Gadzala L, McMillen D, Bystrom C, Van der Zee CE, Wallimann T, Gillespie PG. Hair bundles are specialized for ATP delivery via creatine kinase. Neuron. 2007;53:371–386. doi: 10.1016/j.neuron.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonis N, Rual JF, Carvunis AR, Tasan M, Lemmens I, Hirozane-Kishikawa T, Hao T, Sahalie JM, Venkatesan K, Gebreab F, Cevik S, Klitgord N, Fan C, Braun P, Li N, Ayivi-Guedehoussou N, Dann E, Bertin N, Szeto D, Dricot A, Yildirim MA, Lin C, de Smet AS, Kao HL, Simon C, Smolyar A, Ahn JS, Tewari M, Boxem M, Milstein S, Yu H, Dreze M, Vandenhaute J, Gunsalus KC, Cusick ME, Hill DE, Tavernier J, Roth FP, Vidal M. Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat Methods. 2009;6:47–54. doi: 10.1038/nmeth.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thalmann I. Inner ear proteomics: a fad or hear to stay. Brain Res. 2006;1091:103–112. doi: 10.1016/j.brainres.2006.01.099. [DOI] [PubMed] [Google Scholar]
- 41.Thalmann I, Hughes I, Tong BD, Ornitz DM, Thalmann R. Microscale analysis of proteins in inner ear tissues and fluids with emphasis on endolymphatic sac, otoconia, and organ of Corti. Electrophoresis. 2006;27:1598–1608. doi: 10.1002/elps.200500768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thalmann I, Rosenthal HL, Moore BW, Thalmann R. Organ of Corti-specific polypeptides: OCP-I and OCP-II. Arch Otorhinolaryngol. 1980;226:123–128. doi: 10.1007/BF00455126. [DOI] [PubMed] [Google Scholar]
- 43.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 44.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 45.Van Camp G, Smith R. Hereditary Hearing Loss Homepage. 2009 http://webh01.ua.ac.be/hhh/
- 46.Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A, Vinayagam A, Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabasi AL, Vidal M. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wenthold RJ, Safieddine S, Ly CD, Wang YX, Lee HK, Wang CY, Kachar B, Petralia RS. Vesicle targeting in hair cells. Audiol Neurootol. 2002;7:45–48. doi: 10.1159/000046863. [DOI] [PubMed] [Google Scholar]
- 48.Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, Vidal M. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322:104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng QY, Rozanas CR, Thalmann I, Chance MR, Alagramam KN. Inner ear proteomics of mouse models for deafness, a discovery strategy. Brain Res. 2006;1091:113–121. doi: 10.1016/j.brainres.2006.02.069. [DOI] [PMC free article] [PubMed] [Google Scholar]