

Abstract
In higher organisms dependent on the regenerative ability of tissue stem cells to maintain tissue integrity throughout adulthood, the failure of stem cells to replace worn out, dead, or damaged cells is seen as one mechanism that limits lifespan. In these organisms, tumor suppressors such as p53 are central participants in the control of longevity because they regulate stem cell proliferation. Several recent reports have identified p53 as a longevity gene in organisms such as C. elegans and Drosophila melanogaster, which lack proliferative stem cells in all but the germline and have relatively short lifespans. This has forced us to reevaluate the role of p53 in the control of lifespan. We discuss how p53 might regulate longevity in both long- and short-lived species by controlling the activity of insulin-like molecules that operate in proliferating and non-proliferating compartments of adult somatic tissues. We also discuss the hierarchical structure of lifespan regulation where loss of p53 has lifespan extending effects. Finally, we suggest a molecular mechanism by which p53 might facilitate the response to severe nutrient deprivation that allows metabolically active cells to survive periods of starvation. Paradoxically, loss of p53 function in these cells would compromise lifespan.
Insulin/IGF signaling in C. elegans, Drosophila, and mice
The insulin (INS)/insulin-like growth factor (IGF) signaling axis is critical for control of growth and proliferation. Reduced function of this axis causes growth retardation in humans (Vaessen et al., 2002) and small size and hypocellularity in experimental organisms such as flies and mice (Clancy et al., 2001; Liu et al., 1993; Tatar et al., 2001). This axis also controls lifespan and aging in animals ranging from worms to mammals (Larsson et al., 2005). However, in contrast to its deleterious effects on growth and proliferation, reduced insulin/IGF signaling has beneficial effects on longevity, extending lifespan in all animals in which it has been tested (Piper et al., 2008). In fact, a cohort study of elderly Dutch citizens has revealed that genetic variations which cause reduced INS/IGF signaling are beneficial for old age survival, providing evidence that the IGF axis controls lifespan in humans as well (van Heemst et al., 2005). Thus, the same level of insulin/IGF activity that is beneficial early in life is detrimental later on (Fig. 1A). We have proposed that examples of antagonistic pleiotropy (Williams, 1957) such as this might be linked to genes that alter the activity of p53 (Ungewitter, in press). We now propose that the divergent effects of the insulin/IGF axis on growth and proliferation on the one hand and longevity on the other can be explained by the ability of p53 control insulin/IGF activity. In tumor cells, p53 suppresses growth and proliferation by blocking IGF-1 action, which has the desirable outcome of prolonging life. This same activity in normal stem cells, however, can lead to tissue deterioration and aging, reduced viability, and shortened lifespan.
Fig. 1. IGF-1 signaling cascade.
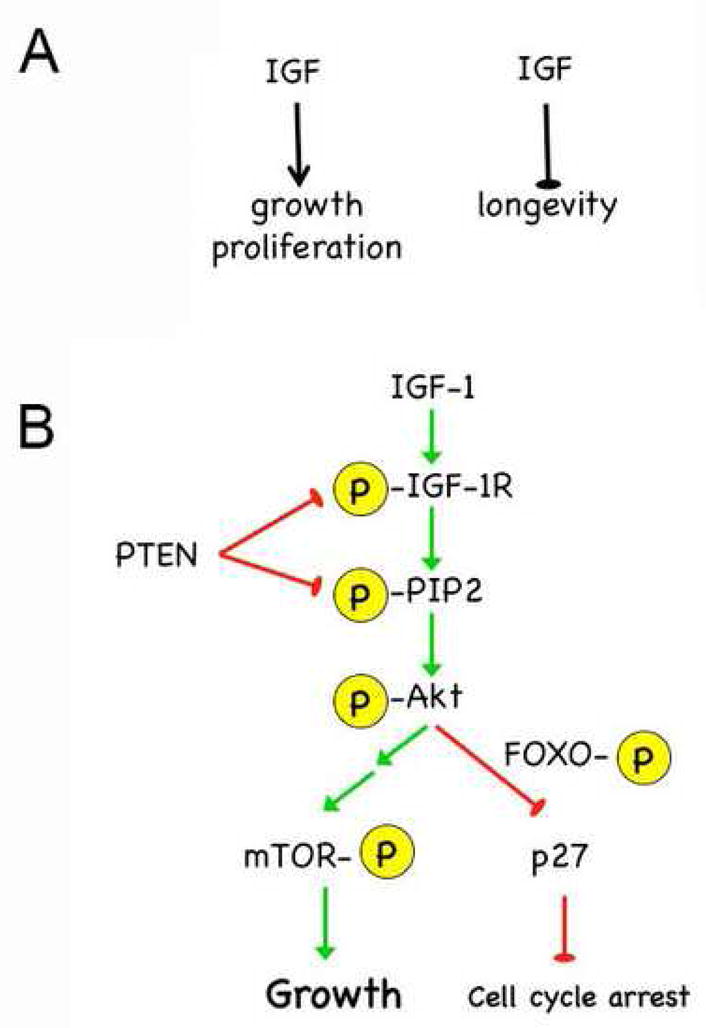
A. The paradox of INS/IGF activity: high activity promotes growth but limits longevity.
B. Signaling is regulated by phosphorylation (yellow symbols) through a highly conserved pathway, which is shown here for mammalian IGF-1. Receptor activation by ligand binding generates PIP3 and can be blocked by PTEN. AKT phosphorylation turns on a growth pathway that acts through mTOR and is blocked by 4E-BP (not shown) and turns off FOXO activity in the nucleus, which otherwise causes cell cycle arrest.
An outline of the insulin/IGF signaling pathway in mammals is presented in Fig. 1B (reviewed in (Denley et al., 2005)). IGF-I, and to a lesser extent IGF-II, bind the extracellular portion of the IGF-1-receptor (IGF-IR) and initiate a phosphorylation cascade that acts through PI3 kinase and AKT to phosphorylate mTOR and FOXO. Phosphorylation of mTOR results in growth, while phosphorylation of FOXO results in reactivation of the cell cycle by removing the block to proliferation by the FOXO target p27. Signaling through insulin via the insulin receptor is very similar. In addition, both insulin and the IGFs can utilize hybrid receptors consisting of INS-R/IGF-1R heterodimers. A third receptor, the IGF-2 receptor, binds ligand but does not transmit the signal intracellularly and thus acts to turn off signaling from all three ligands.
INS/IGF signaling is regulated at several steps, both before and after receptor activation, and is controlled primarily by the quantity of available ligand and the level of the receptor(s). In mammals, IGF signaling is also controlled by six IGF-binding proteins (IGFBPs), which interact with the IGF ligands to increase their stability during circulation (Guler et al., 1989), facilitate the delivery of IGFs to tissues (Stewart et al., 1993), and control accessibility of IGFs for binding the IGF-1R (Angervo et al., 1991; Rutanen et al., 1988). Following receptor activation, INS/IGF signaling is subject to control by phosphatases, such as PTEN, which block phosphorylation of downstream targets and halt the phosphorylation cascade necessary for INS/IGF signal transduction (Maehama and Dixon, 1998).
The INS/IGF signaling proteins are well conserved between invertebrates and mammals, but have a few notable differences. Both C. elegans and Drosophila have a single INS/IGF-receptor but have multiple INS/IGF ligand varieties, 39 insulin-like (ins) genes identified so far in C. elegans (Braeckman and Vanfleteren, 2007) and 7 insulin-like peptides (dILPs) identified in Drosophila (Broeck, 2001; Brogiolo et al., 2001; Cao and Brown, 2001). By contrast, mammals have just three INS/IGF ligands (IGF-I, IGF-II, and insulin) but have four known INS/IGF receptors and multiple IRS isoforms (Navarro et al., 1999).
In humans and mice, the IGF-1 gene consists of 6 exons, separated by 5 introns (Fig. 2) (Adamo et al., 1993). Transcription is controlled by alternate use of two upstream promoters and starts at several transcription start sites located in exons 1 and 2 (Mittanck et al., 1997). Together, alternate promoter usage and alternative splicing at the 5′ and 3′ ends of the gene generate several distinct mRNAs depending on their exon sequences, which code for three isoforms of precursor IGF-1 (Fig. 2) (Daughaday and Rotwein, 1989; Shavlakadze et al., 2005; Stewart and Rotwein, 1996). These isoforms have characteristic N-terminal signal peptide sequences and C-terminal extension (E) peptide domains. Exons 1 and 2 and part of exon 3 encode the signal peptides. The remainder of exon 3 and exon 4 encode the mature IGF-1 peptide and the proximal part of the E peptide, which are shared by all isoforms. Splicing of exon 4 to exon 6 generates the predominant transcript IGF-1Ea. Splicing of exon 4 to exon 5 generates IGF-1Eb, which encodes an isoform with 47 distinct amino acids in the E domain. When part of exon 5 is spliced to exon 6, the IGF-1Ec (IGF-1Eb in mouse) variant is generated. In this case, a frame-shift occurs in exon 5 followed by premature transcription stop in exon 6 that results in a stretch of 25 amino acids unique to this variant (reviewed in (Winn et al., 2002)). All three variants generate the mature 7.6-kDa IGF-1 bioactive peptide upon release of the E peptide domain, but their individual functions are not entirely known.
Fig. 2. Mammalian IGF-1 gene and its products.
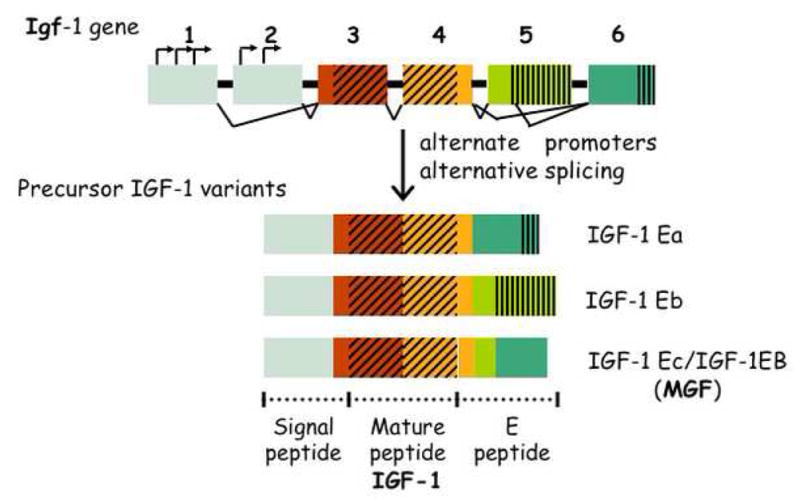
The IGF-1 gene in humans and mice has 6 exons (boxes) and 5 introns and utilizes several promoters (bent arrows) and alternative splicing (connectors) to generate 3 proteins (IGF-1 Ea, Eb, and Ec). Mechano growth factor (MGF) is the name for IGF-1Ec (IGF-1EB in mouse). The mature peptide (IGF-1; hatched area) is generated by cleavage of the signal peptide (gray area) and E peptide (green areas). See text for a description of the IGF-1 isoforms and their individual activities.
The IGF-1Ec/Eb prepeptide is known as mechano growth factor, or MGF. MGF is a non-secreted form of IGF-1 that can be also found in the nucleus of cells in culture (Tan et al., 2002) or perinuclear in hippocampal cells after ischemia (Dluzniewska et al., 2005). Together with IGF-1, MGF functions to regenerate muscle (Hill and Goldspink, 2003) in response to activation of mechano receptors by trauma, stress, or strenuous exercise (McKoy et al., 1999; Yang et al., 1996). MGF stimulates muscle stem cells (satellite cells) to reenter the cell cycle and proliferate (Adams, 1998; Yang and Goldspink, 2002), whereas IGF-1 is necessary for the differentiation of newly generated muscle precursor cells into myoblasts and myofibers..
Although the main source of IGF-1 is the liver, where growth hormone (GH) and its associated signaling pathway induce transcription of the IGF-1 gene, the mechanism by which MGF is preferentially made is less clear. From studies in mice and humans, it is clear that GH induces expression of both the endocrine form of IGF-1 and MGF (Hameed et al., 2004; Iida et al., 2004). However, mice deficient in GH (lit/lit mice) respond to administration of GH with an acute increase in MGF, but not IGF-1, in skeletal muscle, but an increase in both MGF and IGF-1 in liver (Iida et al., 2004). The mechanism by which GH has a differential effect on IGF-1 and MGF expression is completely unknown.
IGF-1 isoforms differ in the signaling pathways they activate. By over-expressing IGF-1Ea and MGF in muscle, it has been shown that both isoforms can activate IGF-1R and Akt phosphorylation (Barton, 2006), although the biological relevance of this for MGF, which is not normally secreted, is unclear. In addition, MGF was shown to induce phosphorylation of ERK, a property not shared with IGF-1Ea (Barton, 2006). The potential importance of this distinction in terms of stem cell proliferation will be described in a following section.
The beneficial effects of reduced p53 activity in neurons
The tumor suppressor p53 exerts control over INS/IGF signaling at several key points. First and foremost, p53 controls the level of the IGF-1 receptor (Werner et al., 1996). p53 suppresses Igf-1R transcription by sequestering TBP, the TATA-box binding subunit of the general initiation factor TFIID. The interaction of p53 with TBP blocks the formation of transcription factor complexes on the Igf-1R promoter, thus repressing IGF-1R transcription (Werner et al., 1996). In the absence of p53, TBP is free to initiate IGF-1R expression. p53 also suppresses IGF-1R activity by reducing endogenous levels of IGF-1R mRNA (Werner et al., 1996) and by stimulating the transcription of IGFBP-3, which inhibits ligand-induced activation of the IGF-1R (Buckbinder et al., 1995). Finally, p53 can regulate INS/IGF activity by controlling downstream members of the INS/IGF signaling cascade. p53 controls the level and activity of the dual lipid-protein-phosphatase, PTEN (Stambolic et al., 2001). This is important because PTEN suppresses transduction of the IGF signal to Akt by antagonizing the generation of phosphatidyl-inositol triphosphate (PIP3; indicated as P-PIP2 in Fig. 1B). PIP3 activates PI3kinase, which in turn phosphorylates and activates Akt (P-Akt, Fig. 1B). p53 also counteracts INS/IGF signaling by regulating the level of the GTPase TSC2 (Feng et al., 2007; Feng et al., 2005), a negative regulator of mTOR activity (Inoki et al., 2002). p53 transactivates the tsc2 promoter, resulting in increased TSC2 production and reduced mTOR activity. From the preceding, it is clear that p53 is a critical regulator of insulin/IGF-1 signaling.
We have been examining mice in which the function of p53 is altered in such a way that the IGF-1 signaling pathway is hyperactivated (p44Tg mice) (Maier et al., 2004). In these p44Tg mice, the level of the IGF-1 receptor is increased and downstream effectors such as AKT, FOXO, and mTOR are constitutively phosphorylated. p44Tg mice have a lifespan that is significantly shorter than normal mice. This is consistent with experiments demonstrating that loss-of-function mutations in genes that code for INS/IGF signaling proteins in C. elegans, such as the INS/IGF-receptor (daf-2), PI3K (age-1), and Akt/PKB (akt) (reviewed in (Guarente, 2000)), significantly prolong life-span. Hypomorphic mutations in the INS/IGF-receptor gene (inr) and its substrate (chico) in Drosophila (Clancy et al., 2001; Tatar et al., 2001) resulted in similar extension of lifespan. In mice, two different GH-deficient models (Ames and Snell) mice have been found to have significantly longer life-spans compared to wild-type mice (Flurkey et al., 2001), and reduced levels of the Igf-1 and Ins-receptor genes significantly increase life-span (Bluher et al., 2003; Holzenberger et al., 2003). How does lifespan extension by decreased insulin/IGF signaling relate to the activity of p53? We think that experiments in C. elegans and Drosophila provide the clearest explanation.
The orthologue of p53 in C. elegans is encoded by the cep-1 gene (Derry et al., 2001). To date, two mutant alleles of cep-1 have been described. The gk138 allele is a null allele of cep-1. Loss of Cep-1 function in gk138 homozygotes or in worms fed cep-1 RNAi confers increased longevity (Arum and Johnson, 2007). The w40 allele is a mutant allele that codes for a truncated Cep-1 protein missing the N-terminal region up to and including the DNA binding domain (Derry et al., 2001). Worms with an ectopic copy of cep-1(w40) have a wild-type lifespan under normal growing conditions (Derry et al., 2001). We will return to this difference in lifespan in worms with two different cep-1 alleles in a later section.
In Drosophila, global loss of p53 results in decreased rather than increased lifespan (Bauer et al., 2005). However, targeted reduction of p53 in neurons, results in mean lifespan extension of 20% (Bauer et al., 2005). Flies expressing identical dominant negative forms of p53 in the muscle or fat body had a normal lifespan. To identify the neurons responsible for lifespan extension, p53 reductions were targeted to specific subsets of neurons. Lifespan was extended only by targeted reduction of p53 in the group of 14 insulin-producing neurons (Bauer et al., 2007). These neurons are responsible for secreting three insulin-like peptides (dILPs), dILP2, 3, and 5 19, that hormonally regulate energy utilization by communicating with metabolic tissues such as fat bodies. The mRNA level of one of these, dILP2, was reduced by about 60% in neurons deficient in p53 (Bauer et al., 2007). Targeting p53 deficiency to insulin producing neurons was found to lower insulin/IGF signaling in the fat bodies of larval and adult flies and resulted in nuclear localization of FOXO (Bauer et al., 2007). The lifespan enhancing effects of p53 deficiencies required FOXO, as did lifespan extension by p53 in C. elegans (Arum and Johnson, 2007). Thus, the global effects of p53 loss masked an underlying beneficial effect of reduced p53 in neurons and this in turn has a beneficial effect on longevity. Reduced expression of p53 in neurons resulted in both reduced production of an insulin-like ligand and reduced signaling in the major insulin-responsive organ in the fly (Fig. 3A). These results verify the findings of experiments in C. elegans showing that neurons mediate the lifespan extension effects of reduced INS/IGF signaling. In C. elegans, tissue-specific replacement of the wild-type daf-2 gene in neurons of daf-2 mutant worms restores normal lifespan, whereas replacement of daf-2 in intestine and muscle had no effect on lifespan (Wolkow et al., 2000). Identical results were also reported for tissue-specific replacement of the age-1 gene in C. elegans. In Drosophila, ablation of the insulin-producing neurons (mNSC) was found to extend lifespan (Broughton et al., 2005), and in mice, brain-specific deletion of the IGF-IR adaptor molecule (IRS-2) also significantly increased mean lifespan (Taguchi et al., 2007). The lifespan extending effects of neuronal p53 deficiency in Drosophila, therefore, can be attributed directly to the common pathway that modulates longevity in organisms as diverse as flies, worms, and mammals.
Fig. 3. Control of INS/IGF activity by p53.
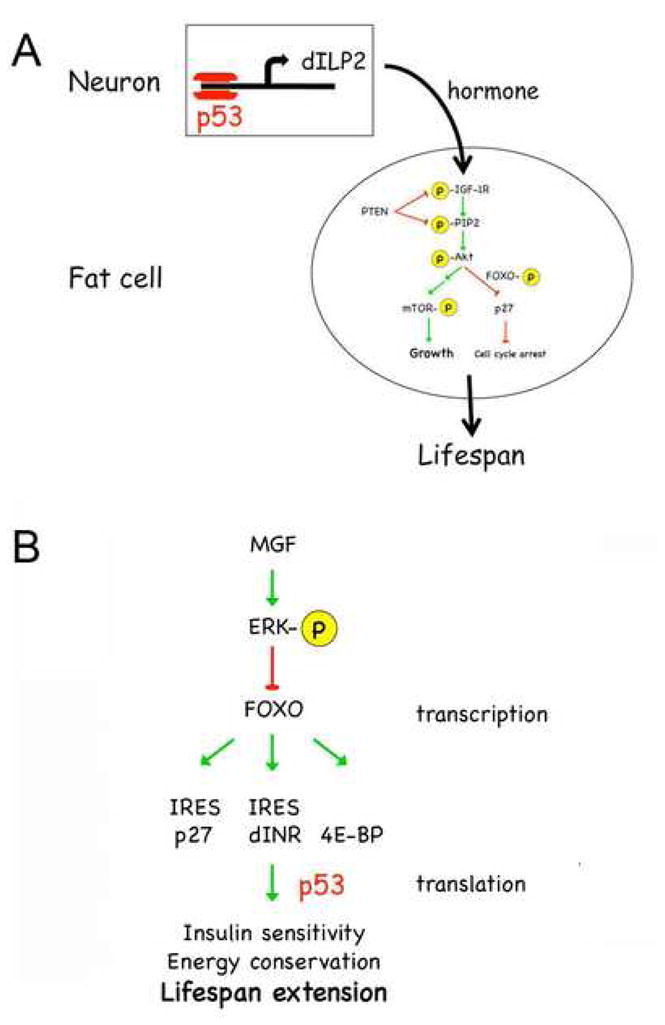
A. In Drosophila, p53 controls the level of dILP2, one of 5 insulin-like peptides produced and secreted by 14 insulin-producing neurons in the fly brain. dILPs activate INS/IGF signaling in peripheral tissues, such as fat bodies.
B. Under conditions of severe nutrient deprivation, FOXO transcribes genes such as the insulin receptor (dINR), p27, and 4E-BP, which increase insulin sensitivity and decrease energy consumption and allow cells to survive. We postulate that p53 binds to complex structures in the 5′ UTRs of these FOXO targets and facilitates their translation.
From worms and flies to mice
In mammals, of course, insulin is not produced by neurons but by the pancreatic islet cells; and, the insulin-like growth factors are produced mainly in extra-neural sources as well, such as the liver. The production of these hormones, furthermore, is not known to be under p53 control in mammals. What then is the relevance of the model represented in Fig. 3A to organisms that have neural mechanisms for controlling insulin/IGF production, secretion, and activity, which act at a higher level than that of the cells in which these hormones are made? In this regard, it is pertinent to recall that earlier experiments suggest that there is a rudimentary neural axis regulating insulin/IGF control of longevity even in lower organisms such as C. elegans. Laser ablation of specific sensory neurons has revealed that a pair of neurons regulates daf-2 activity in the periphery through a signal transduction pathway that requires daf-16 (Alcedo and Kenyon, 2004). One neuron (ASI or ASG) suppresses a second neuron (ASJ or ASK) that controls the level of DAF-2 activity. Loss of the suppressor neuron increases DAF-2 signaling and decreases lifespan. On the other hand, loss of the sensory neuron that activates DAF-2 results in decreased DAF-2 activity and increased lifespan. These simple systems, in which relatively few neurons communicate with and control the activity of visceral organs by electrically or hormonally transmitted signals, foreshadow the more complex mammalian systems that have evolved to maintain homeostasis and regulate longevity in larger organisms with longer life-spans. For example, the sensory neuron that activates DAF-2 activity could be analogous to the dILP-producing neurons in Drosophila or to the non-neuronal insulin or IGF producing cells in mammals that produce and secrete insulin-like hormones (Apfeld and Kenyon, 1999). The suppressor neuron, then, would be analogous to neurons that control hormone release, such as those in the hypothalamus of the mammalian brain.
These experiments in C. elegans demonstrate that neural circuits that involve certain sensory neurons can control longevity by altering signaling through the insulin/IGF receptor in peripheral (non-neural) sites. Loss of neurons impairs these circuits and alters lifespan. In mammals, where neurons are maintained throughout life by ongoing neurogenesis, factors that impair adult neurogenesis could alter longevity. We have recently demonstrated that neurogenesis is under the control of p53, which is responsible for maintaining the regenerative potential of stem cells that supply the adult brain with replacement neurons that subserve at least two known functions, olfaction and cognition (Medrano et al., 2007). We propose that this hierarchy of control over the insulin/IGF axis in mice is structurally analogous to direct control of insulin production by p53 in Drosophila neurons. In both cases, loss of p53 alters endocrine control of metabolic pathways that enhance or impair longevity.
p53 is expressed postnatally in neurogenic areas of the mammalian brain (van Lookeren Campagne and Gill, 1998). In mammals, neurogenesis continues throughout life in the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the hippocampus (Altman and Bayer, 1990; Corotto et al., 1993). The integration of newly generated neurons into existing circuits in the olfactory bulb and the dentate gyrus is important for odor discrimination (Gheusi et al., 2000), and for learning and memory (Brown, 2003; Gould et al., 1999; Shors et al., 2001; Shors et al., 2002), respectively. Recently, the hypothalamus has been shown to support adult neurogenesis in mice. Upon growth factor stimulation, stem/progenitor cells in the periventricular region generate new hypothalamic neurons necessary to maintain energy balance function (Kokoeva et al., 2005).
Mice deficient in p53 exhibit increased proliferation and self-renewal of neural stem cells (Gil-Perotin et al., 2006; Meletis et al., 2006). In contrast, mice with increased p53 (p44Tg mice) exhibit decreased proliferation resulting in fewer new neurons in the olfactory bulb, the target of SVZ neurogenesis, and impaired olfactory acuity (Medrano, 2007). Reduced neurogenesis in the SVZ of p44Tg mice was due to factors intrinsic to the stem/progenitor cells. Studies in isolated SVZ cells grown in culture were indicative of reduced stem/progenitor cell number and proliferation. The self-renewal and amplification capacities were limited, and the cells were more likely to remain in G0 compared to cells from normal mice. These results demonstrate that p53 plays a crucial role in maintaining brain function by regulating the cell cycle in stem cells and that increased p53 activity limits stem cell proliferation.
The mechanism by which increased activity of p53 impairs stem cell proliferation and neurogenesis in the adult brain can be explained on the basis of increased activity of the insulin/IGF axis, as described above. In mice in which this axis is less active, for example in Ames dwarf mice, the capacity for adult neurogenesis is preserved even in very old animals (Sun and Bartke, 2007; Sun et al., 2005). These mice have a loss-of-function mutation in the prop-1 gene, resulting in the complete lack of growth hormone (GH)-producing cells in the pituitary (reviewed in (Bartke et al., 2003)). As a result, systemic levels of IGF-1 are very low and the animals have a significantly longer lifespan and appear to age more slowly than normal mice (Brown-Borg et al., 1996). Compared to their normal siblings, Ames dwarfs can generate hippocampal neurons at a significantly higher level, even when they are old (Sun and Bartke, 2007; Sun et al., 2005). In mice in which the IGF axis is hyperactivated, such as p44Tg mice, the capacity for adult neurogenesis is impaired and replacement neurons are generated at a lower level than in age-matched normal controls (Medrano et al., 2007).
These effects of altered IGF signaling appear to conflict with evidence that IGF-1 plays a role in ameliorating the effects of age on adult neurogenesis in the mouse. IGF-I increases the number of neural cells (neurons, astrocytes, and oligodendrocytes) in vivo and in vitro by increasing the proliferation of precursors (see references in (Ye and D’Ercole, 2006)) and the differentiation of proliferating cells into neurons and glia (reviewed in (D’Ercole et al., 1996; Mason et al., 2003; Otaegi et al., 2006). Addition of IGF-1 to SVZ NSCs cultures enhanced the number of neuron-containing clones and the number of neurons per clone, an effect that could be blocked by anti-IGF-1 antibodies (Brooker et al., 2000). Mice with a deficiency in the IGF-1 gene have smaller than normal brains (Liu et al., 1993) with reductions in oligodendrocytes (Otaegi et al., 2006), white matter regions, and hippocampal pyramidal cells and granule cells of the dentate gyrus and striatum (Beck et al., 1995).
The differential effects of IGF signaling and IGF-1 on neurogenesis clearly demonstrate that while altered IGF-1 activity has a major impact on the ability of neurogenic cells to proliferate, IGF-1 signaling cannot explain all of this IGF effect. The idea that there is an IGF-1 receptor-independent activity of IGF-1 in neurons is supported by the recent report that the neuroprotective effect of a synthetic bioactive MGF peptide on cultured hippocampal neurons subjected to oxidative or excitotoxic stress was not blocked by inactivation of the IGF-1 receptor (Dluzniewska et al., 2005). In vivo, furthermore, injection of this peptide into the carotid artery protected hippocampal neurons from global ischemic damage and induced endogenous MGF expression (Dluzniewska et al., 2005). MGF was also more effective than IGF-1 at protecting motor neurons after crush injury (Aperghis et al., 2004). Furthermore, Ames dwarf mice have demonstrable levels of IGF-1 that is not affected by the loss of prop-1 function, including in the brain (Sun et al., 2005). This IGF-1, which presumably derives from an autocrine or paracrine mechanism, is most likely MGF. In addition to a potential role in ongoing neurogenesis in the adult, we think that MGF may play a critical role under circumstances that put brain function at risk, as described in the next section. In times of severe nutrient stress, cells that express MGF would be exempt from regulatory mechanisms that would prevent them from proliferating, thus allowing preservation of function in brain regions that control energy usage, health, and lifespan.
The detrimental effects of reduced p53 in metabolic tissues: a model
As mentioned previously, cep-1(w40) has no effect on longevity in the worm. This mutant form of C. elegans p53 codes for a truncated protein missing the N-terminal domain (Derry et al., 2001), which is known to interact with other transcription factors and the basal transcription machinery to control gene expression. A similar mutation in the murine p53 gene (p53m), however, causes accelerated aging and decreased lifespan in a mammalian species (Tyner et al., 2002). In contrast, a null allele of cep-1 (gk138) actually extends lifespan in the worm. Why the differences in phenotype?
“p53” is a family of protein isoforms, currently nine in number, that include full-length p53 and eight variants with different N- and C-terminal regions that control transactivation and tetramerization (Bourdon et al., 2005). At least one of these isoforms, an N-terminally truncated variant designated as Δ40p53, is capable of binding to and altering the activity of full-length p53 (Courtois et al., 2002; Yin et al., 2002). Conservation of gene structure in Drosophila has demonstrated the existence of at least two p53 isoforms in the fly (Bourdon et al., 2005). In C. elegans, the Cep-1 tetramerization domain shows poor homology with vertebrate forms of p53, yet structural analysis and gel filtration studies reveal that Cep-1 is capable of forming dimers (Ou et al., 2007). The cep-1(w40) mutation utilized by Derry and colleagues (Derry et al., 2001) codes for a form of p53 that resembles the naturally occurring Δ133 isoform of mammalian p53, but not the full length protein. The phenotype of cep-1(w40) worms, therefore, reflects p53 activity that is altered due to an imbalance in the normal ratio of isoforms rather than simply lost, as it is the case in cep-1(gk138) worms. This could help account for the phenotypic differences between the two cep-1 mutant strains. However, it leaves unresolved the issue of why a similar imbalance in isoforms in both p53+/m and p44Tg mice decreases longevity.
Worms with an ectopic cep-1(w40) gene (in addition to the normal cep-1 gene) have a wild-type lifespan under normal growing conditions, but show increased mortality (decreased lifespan) following a period of severe nutrient deprivation during development (Derry et al., 2001). This suggests that survival under starvation conditions requires p53. Recent experiments in Drosophila have revealed how the fly can survive under conditions of severe nutrient deprivation (Marr et al., 2007), reviewed in (Lasko and Sonenberg, 2007). Low insulin or nutrient levels activate a coupled program of transcription and translation that results in increased insulin sensitivity and reduced energy consumption (Fig. 3B). The mechanism is based on enhanced translation of the insulin receptor (dINR) and 4E-BP, which are transcribed by FOXO (Marr et al., 2007). The dINR has a complicated secondary structure in the 5′ UTR of its transcript that might function as an internal ribosome entry sequence, or IRES (Marr et al., 2007; these sequences are sometimes referred to as “internal regulatory expression sequences” instead to account for their disputed roles as true ribosome entry sequences; see Kozak, 2003). When insulin levels are low and insulin signaling is inactive, both FOXO and 4E-BP would be unphosphorylated. As a result, FOXO would be in the nucleus and active and 4E-BP would bind to eIF4E and block cap-dependent translation. Blocking cap-dependent translation would lower metabolic activity and increasing the level of the insulin receptor would increase insulin sensitivity, both of which are associated with the lifespan extending effects of calorie restriction. The cell cycle inhibitor p27 can also be translated from a putative IRES element in its mRNA (Kullmann et al., 2002) and increased p27 in times of nutrient deprivation would keep cells from proliferating.
In addition to its DNA binding capabilities, p53 is an RNA binding protein that binds to stem-loop elements in mRNA leader sequences and can activate or suppress their translation (Gonzalez-Herrera et al., 2006; Scrable et al., 2005). One consequence of reducing p53, therefore, could be changes in IRES-mediated translation and response to nutrient stress, which would make the flies more sensitive to nutrient restriction. In flies in which p53 deficiency was restricted to neurons, for example, lifespan extension was improved when the animals were fed a high calorie diet compared to a standard diet (Bauer et al., 2007). This indicates that p53 may play an important role in the nutrient stress response mediated by FOXO-dependent transcription and IRES-mediated translation.
One of the mammalian genes potentially regulated by p53 in this manner is the IGF-1 receptor (Meng et al., 2005). As Tjian has pointed out, small increases in transcription are amplified by large increases in translation when IRES-dependent mechanisms are activated (Marr et al., 2007), as would occur when cap-dependent translation is blocked. In fact, we have observed much greater differences in IGF-1R protein levels than RNA levels in cells from p44Tg mice (Maier et al., 2004), suggesting that one consequence of isoform imbalance in mammals is loss of p53 suppression of IRES-dependent translation. If we assume that w40 acts like Δ133p53 or p44 in mice, then increased sensitivity to metabolic stress could reflect the ability of w40 to interfere with p53-mediated suppression of IRES-dependent translation. As described above, when nutrients are low in Drosophila, FOXO transcribes genes whose transcripts include IRES elements, such as the insulin/IGF receptor, and 4E-BP. This increases insulin sensitivity and blocks cap-dependent translation allowing cells to conserve energy. In p44Tg mice, growth is impaired despite having higher than normal levels of the IGF-1 receptor (Maier et al., 2004). Loss of p53 suppression of 4E-BP translation could account for this. This in turn would compromise viability under conditions of severe nutrient deprivation such as starvation.
In the cep-1(gk138) mutant strain in which p53 activity is lost, lifespan extension was dramatically enhanced when the worms were fed heat-killed rather than live bacteria. Because wild-type worms fed bacteria that are dead, UV-irradiated, or grown in the presence of bacteriostatic drugs such as carbenicillin (Garigan et al., 2002) also live longer than worms grown on living bacteria, the mechanism underlying this phenomenon cannot be cep-1 dependent. Kenyon proposed that the difference in lifespan was due to a propensity for live bacteria to buildup in the posterior intestine (“constipation”) and suggested that increased pathogen load contributed to mortality (Garigan et al., 2002). If this is true, then long-lived cep-1(gk138) worms have decreased resistance to bacterial pathogens. However, worms with hypomorphic alleles of daf-2, which are also long-lived, demonstrate increased resistance to bacterial pathogens (Bolm et al., 2004). These results collectively indicate that resistance to bacterial pathogens is not necessary for lifespan extension. An alternative explanation for the difference in lifespan is that lifespan improves when the food supply is limited (dead bacteria) and decreases when it is overly abundant (proliferating bacteria), and that this is true for cep-1(gk138) as well as wild type worms. In worms in which daf-2 is expressed at very low levels, reduced signaling, in addition to enhancing nuclear daf-16 activity, would downregulate TOR and lower metabolic activity, thus ameliorating the effects of overfeeding, including its negative effects on lifespan. Would reduced p53 lead to similar effects? Loss of p53 would result in increased levels of proteins translated by an IRES-dependent mechanism, which includes both the insulin and IGF-1 receptors (Giraud et al., 2007; Marr et al., 2007). As described for the w40 allele, this would result in increased insulin sensitivity and energy conservation due to 4E-BP blockage of cap-dependent translation downstream of TOR. Thus, in the case of over-nutrition, loss of p53 would be beneficial, while in the case of under-nutrition, it would be detrimental, exactly what was observed with the two cep-1 mutant strains.
In higher organisms in which adult stem cells repair age- or injury-damaged tissue, a response to nutrient deprivation that could include a block to cell proliferation, such as that illustrated in Fig. 3B, would limit regeneration and presumably also lifespan. Stem cells, therefore, should have a way to escape this damage response. One possibility is that MGF, the proliferation-inducing form of IGF-1, can block this pathway upstream of FOXO. MGF was shown to be induced in muscle stem cells after muscle damage (Adams, 1998; Yang and Goldspink, 2002), and we know from preliminary results from our lab that it is expressed in neurogenic regions of the adult brain. Expression of MGF results in ERK phosphorylation and activation (Barton, 2006), which blocks FOXO activity in the nucleus (Asada et al., 2007). Because MGF is not secreted and does not act through the IGF-1 receptor, its activity would be insensitive to the suppressive effects of nutrient deprivation that drives gene expression into cap-independent translation. Furthermore, in those cells that express MGF, such as stem cells, selective inactivation of the coupled program of FOXO-mediated transcription and IRES-mediated translation that shuts down energy consuming processes in other cells would permit these cells to proliferate even when others are inhibited. Regeneration of neurons that control the insulin/IGF axis by proliferation of neural stem cells utilizing MGF would facilitate recovery and improve chances for continued survival when the nutritional state improves.
Summary
In good times, when nutrients are plentiful, reduced p53 can be beneficial by limiting the supply of INS/IGF ligands and reducing the activity of INS/IGF signaling pathways. In times of nutrient stress, however, when these signaling pathways are inactive, p53 could be beneficial by facilitating alternate modes of protein translation that increase insulin sensitivity and maximize energy conservation. Thus, p53 plays a dual role in homeostatic mechanisms that maintain longevity, first by regulating cell proliferation and the replacement of worn-out, dead, or damaged cells, and second by supporting cell survival during fluxes in nutrient availability.
Acknowledgments
We thank the members of the Scrable Lab, past and present, for many hours of interesting and enlightening discussions. We are also grateful to our many collaborators and colleagues who have shared ideas and unpublished results and have helped us understand and interpret the results of experiments we have done ourselves. These experiments were supported by grants from the National Institute on Aging, NIH, and the Ellison Medical Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamo ML, Neuenschwander S, LeRoith D, Roberts CT., Jr Structure, expression, and regulation of the IGF-I gene. Adv Exp Med Biol. 1993;343:1–11. doi: 10.1007/978-1-4615-2988-0_1. [DOI] [PubMed] [Google Scholar]
- Adams GR. Role of insulin-like growth factor-I in the regulation of skeletal muscle adaptation to increased loading. Exerc Sport Sci Rev. 1998;26:31–60. [PubMed] [Google Scholar]
- Alcedo J, Kenyon C. Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron. 2004;41:45–55. doi: 10.1016/s0896-6273(03)00816-x. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol. 1990;301:365–81. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- Angervo M, Koistinen R, Suikkari AM, Seppala M. Insulin-like growth factor binding protein-1 inhibits the DNA amplification induced by insulin-like growth factor I in human granulosa-luteal cells. Hum Reprod. 1991;6:770–3. doi: 10.1093/oxfordjournals.humrep.a137426. [DOI] [PubMed] [Google Scholar]
- Aperghis M, Johnson IP, Cannon J, Yang SY, Goldspink G. Different levels of neuroprotection by two insulin-like growth factor-I splice variants. Brain Res. 2004;1009:213–8. doi: 10.1016/j.brainres.2004.02.049. [DOI] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature. 1999;402:804–9. doi: 10.1038/45544. [DOI] [PubMed] [Google Scholar]
- Arum O, Johnson TE. Reduced expression of the Caenorhabditis elegans p53 ortholog cep-1 results in increased longevity. J Gerontol A Biol Sci Med Sci. 2007;62:951–9. doi: 10.1093/gerona/62.9.951. [DOI] [PubMed] [Google Scholar]
- Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, Iwashita S, Kako K, Kishi T, Kasuya Y, Fukamizu A. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–27. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Bartke A, Chandrashekar V, Dominici F, Turyn D, Kinney B, Steger R, Kopchick JJ. Insulin-like growth factor 1 (IGF-1) and aging: controversies and new insights. Biogerontology. 2003;4:1–8. doi: 10.1023/a:1022448532248. [DOI] [PubMed] [Google Scholar]
- Barton ER. Viral expression of insulin-like growth factor-I isoforms promotes different responses in skeletal muscle. J Appl Physiol. 2006;100:1778–84. doi: 10.1152/japplphysiol.01405.2005. [DOI] [PubMed] [Google Scholar]
- Bauer JH, Chang C, Morris SN, Hozier S, Andersen S, Waitzman JS, Helfand SL. Expression of dominant-negative Dmp53 in the adult fly brain inhibits insulin signaling. Proc Natl Acad Sci U S A. 2007;104:13355–60. doi: 10.1073/pnas.0706121104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer JH, Poon PC, Glatt-Deeley H, Abrams JM, Helfand SL. Neuronal expression of p53 dominant-negative proteins in adult Drosophila melanogaster extends life span. Curr Biol. 2005;15:2063–8. doi: 10.1016/j.cub.2005.10.051. [DOI] [PubMed] [Google Scholar]
- Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–30. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–4. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Bolm M, Chhatwal GS, Jansen WT. Bacterial resistance of daf-2 mutants. Science. 2004;303:1976. doi: 10.1126/science.303.5666.1976a. author reply 1976. [DOI] [PubMed] [Google Scholar]
- Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–37. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckman BP, Vanfleteren JR. Genetic control of longevity in C. elegans. Exp Gerontol. 2007;42:90–8. doi: 10.1016/j.exger.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Broeck JV. Insect G protein-coupled receptors and signal transduction. Arch Insect Biochem Physiol. 2001;48:1–12. doi: 10.1002/arch.1054. [DOI] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol. 2001;11:213–21. doi: 10.1016/s0960-9822(01)00068-9. [DOI] [PubMed] [Google Scholar]
- Brooker GJ, Kalloniatis M, Russo VC, Murphy M, Werther GA, Bartlett PF. Endogenous IGF-1 regulates the neuronal differentiation of adult stem cells. J Neurosci Res. 2000;59:332–41. doi: 10.1002/(sici)1097-4547(20000201)59:3<332::aid-jnr6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Broughton SJ, Piper MD, Ikeya T, Bass TM, Jacobson J, Driege Y, Martinez P, Hafen E, Withers DJ, Leevers SJ, Partridge L. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci U S A. 2005;102:3105–10. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Brown K. Neuroscience. New attention to ADHD genes. Science. 2003;301:160–1. doi: 10.1126/science.301.5630.160. [DOI] [PubMed] [Google Scholar]
- Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, Kley N. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature. 1995;377:646–9. doi: 10.1038/377646a0. [DOI] [PubMed] [Google Scholar]
- Cao C, Brown MR. Localization of an insulin-like peptide in brains of two flies. Cell Tissue Res. 2001;304:317–21. doi: 10.1007/s004410100367. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–6. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Corotto FS, Henegar JA, Maruniak JA. Neurogenesis persists in the subependymal layer of the adult mouse brain. Neurosci Lett. 1993;149:111–4. doi: 10.1016/0304-3940(93)90748-a. [DOI] [PubMed] [Google Scholar]
- Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–8. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–55. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10:68–91. doi: 10.1210/edrv-10-1-68. [DOI] [PubMed] [Google Scholar]
- Denley A, Cosgrove LJ, Booker GW, Wallace JC, Forbes BE. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005;16:421–39. doi: 10.1016/j.cytogfr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science. 2001;294:591–5. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- Dluzniewska J, Sarnowska A, Beresewicz M, Johnson I, Srai SK, Ramesh B, Goldspink G, Gorecki DC, Zablocka B. A strong neuroprotective effect of the autonomous C-terminal peptide of IGF-1 Ec (MGF) in brain ischemia. Faseb J. 2005;19:1896–8. doi: 10.1096/fj.05-3786fje. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98:6736–41. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161:1101–12. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheusi G, Cremer H, McLean H, Chazal G, Vincent JD, Lledo PM. Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc Natl Acad Sci U S A. 2000;97:1823–8. doi: 10.1073/pnas.97.4.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–16. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Herrera IG, Prado-Lourenco L, Teshima-Kondo S, Kondo K, Cabon F, Arnal JF, Bayard F, Prats AC. IRES-dependent regulation of FGF-2 mRNA translation in pathophysiological conditions in the mouse. Biochem Soc Trans. 2006;34:17–21. doi: 10.1042/BST20060017. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–5. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–6. [PubMed] [Google Scholar]
- Guler HP, Zapf J, Schmid C, Froesch ER. Insulin-like growth factors I and II in healthy man. Estimations of half-lives and production rates. Acta Endocrinol (Copenh) 1989;121:753–8. doi: 10.1530/acta.0.1210753. [DOI] [PubMed] [Google Scholar]
- Hameed M, Lange KH, Andersen JL, Schjerling P, Kjaer M, Harridge SD, Goldspink G. The effect of recombinant human growth hormone and resistance training on IGF-I mRNA expression in the muscles of elderly men. J Physiol. 2004;555:231–40. doi: 10.1113/jphysiol.2003.051722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M, Goldspink G. Expression and splicing of the insulin-like growth factor gene in rodent muscle is associated with muscle satellite (stem) cell activation following local tissue damage. J Physiol. 2003;549:409–18. doi: 10.1113/jphysiol.2002.035832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–7. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Iida K, Itoh E, Kim DS, del Rincon JP, Coschigano KT, Kopchick JJ, Thorner MO. Muscle mechano growth factor is preferentially induced by growth hormone in growth hormone-deficient lit/lit mice. J Physiol. 2004;560:341–9. doi: 10.1113/jphysiol.2004.069500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 2005;310:679–83. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]
- Kozak M. Alternative ways to think about mRNA sequences and proteins that appear to promote internal initiation of translation. Gene. 2003;318:1–23. doi: 10.1016/s0378-1119(03)00774-1. [DOI] [PubMed] [Google Scholar]
- Kullmann M, Gopfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 2002;16:3087–99. doi: 10.1101/gad.248902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Girnita A, Girnita L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br J Cancer. 2005;92:2097–101. doi: 10.1038/sj.bjc.6602627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko P, Sonenberg N. Coordinated transcriptional and translational control in metabolic homeostasis in flies. Genes Dev. 2007;21:235–7. doi: 10.1101/gad.1524707. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–19. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr MT, 2nd, D’Alessio JA, Puig O, Tjian R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. 2007;21:175–83. doi: 10.1101/gad.1506407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Xuan S, Dragatsis I, Efstratiadis A, Goldman JE. Insulin-like growth factor (IGF) signaling through type 1 IGF receptor plays an important role in remyelination. J Neurosci. 2003;23:7710–8. doi: 10.1523/JNEUROSCI.23-20-07710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKoy G, Ashley W, Mander J, Yang SY, Williams N, Russell B, Goldspink G. Expression of insulin growth factor-1 splice variants and structural genes in rabbit skeletal muscle induced by stretch and stimulation. J Physiol. 1999;516 (Pt 2):583–92. doi: 10.1111/j.1469-7793.1999.0583v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano S, Burns-Cusato M, Atienza MB, Rahimi D, Scrable H. Regenerative capacity of neural precursors in the adult mammalian brain is under the control of p53. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meletis K, Wirta V, Hede SM, Nister M, Lundeberg J, Frisen J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–9. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- Meng Z, King PH, Nabors LB, Jackson NL, Chen CY, Emanuel PD, Blume SW. The ELAV RNA-stability factor HuR binds the 5′-untranslated region of the human IGF-IR transcript and differentially represses cap-dependent and IRES-mediated translation. Nucleic Acids Res. 2005;33:2962–79. doi: 10.1093/nar/gki603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittanck DW, Kim SW, Rotwein P. Essential promoter elements are located within the 5′ untranslated region of human insulin-like growth factor-I exon I. Mol Cell Endocrinol. 1997;126:153–63. doi: 10.1016/s0303-7207(96)03979-2. [DOI] [PubMed] [Google Scholar]
- Navarro I, Leibush B, Moon TW, Plisetskaya EM, Banos N, Mendez E, Planas JV, Gutierrez J. Insulin, insulin-like growth factor-I (IGF-I) and glucagon: the evolution of their receptors. Comp Biochem Physiol B Biochem Mol Biol. 1999;122:137–53. doi: 10.1016/s0305-0491(98)10163-3. [DOI] [PubMed] [Google Scholar]
- Otaegi G, Yusta-Boyo MJ, Vergano-Vera E, Mendez-Gomez HR, Carrera AC, Abad JL, Gonzalez M, de la Rosa EJ, Vicario-Abejon C, de Pablo F. Modulation of the PI 3-kinase-Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J Cell Sci. 2006;119:2739–48. doi: 10.1242/jcs.03012. [DOI] [PubMed] [Google Scholar]
- Ou HD, Lohr F, Vogel V, Mantele W, Dotsch V. Structural evolution of C-terminal domains in the p53 family. Embo J. 2007;26:3463–73. doi: 10.1038/sj.emboj.7601764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper MD, Selman C, McElwee JJ, Partridge L. Separating cause from effect: how does insulin/IGF signalling control lifespan in worms, flies and mice? J Intern Med. 2008;263:179–91. doi: 10.1111/j.1365-2796.2007.01906.x. [DOI] [PubMed] [Google Scholar]
- Rutanen EM, Pekonen F, Makinen T. Soluble 34K binding protein inhibits the binding of insulin-like growth factor I to its cell receptors in human secretory phase endometrium: evidence for autocrine/paracrine regulation of growth factor action. J Clin Endocrinol Metab. 1988;66:173–80. doi: 10.1210/jcem-66-1-173. [DOI] [PubMed] [Google Scholar]
- Scrable H, Sasaki T, Maier B. DeltaNp53 or p44: priming the p53 pump. Int J Biochem Cell Biol. 2005;37:913–9. doi: 10.1016/j.biocel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Shavlakadze T, Winn N, Rosenthal N, Grounds MD. Reconciling data from transgenic mice that overexpress IGF-I specifically in skeletal muscle. Growth Horm IGF Res. 2005;15:4–18. doi: 10.1016/j.ghir.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–6. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–84. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–25. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- Stewart CE, Bates PC, Calder TA, Woodall SM, Pell JM. Potentiation of insulin-like growth factor-I (IGF-I) activity by an antibody: supportive evidence for enhancement of IGF-I bioavailability in vivo by IGF binding proteins. Endocrinology. 1993;133:1462–5. doi: 10.1210/endo.133.3.7689959. [DOI] [PubMed] [Google Scholar]
- Stewart CE, Rotwein P. Growth, differentiation, and survival: multiple physiological functions for insulin-like growth factors. Physiol Rev. 1996;76:1005–26. doi: 10.1152/physrev.1996.76.4.1005. [DOI] [PubMed] [Google Scholar]
- Sun LY, Bartke A. Adult neurogenesis in the hippocampus of long-lived mice during aging. J Gerontol A Biol Sci Med Sci. 2007;62:117–25. doi: 10.1093/gerona/62.2.117. [DOI] [PubMed] [Google Scholar]
- Sun LY, Evans MS, Hsieh J, Panici J, Bartke A. Increased neurogenesis in dentate gyrus of long-lived Ames dwarf mice. Endocrinology. 2005;146:1138–44. doi: 10.1210/en.2004-1115. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–72. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–10. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee Park S, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Ungewitter E, Scrable H. Antagonistic pleiotropy and the p53 gene. Mechanisms of Aging & Development. doi: 10.1016/j.mad.2008.06.002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaessen N, Janssen JA, Heutink P, Hofman A, Lamberts SW, Oostra BA, Pols HA, van Duijn CM. Association between genetic variation in the gene for insulin-like growth factor-I and low birthweight. Lancet. 2002;359:1036–7. doi: 10.1016/s0140-6736(02)08067-4. [DOI] [PubMed] [Google Scholar]
- van Heemst D, Beekman M, Mooijaart SP, Heijmans BT, Brandt BW, Zwaan BJ, Slagboom PE, Westendorp RG. Reduced insulin/IGF-1 signalling and human longevity. Aging Cell. 2005;4:79–85. doi: 10.1111/j.1474-9728.2005.00148.x. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene Bax. J Comp Neurol. 1998;397:181–98. doi: 10.1002/(sici)1096-9861(19980727)397:2<181::aid-cne3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Werner H, Karnieli E, Rauscher FJ, LeRoith D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc Natl Acad Sci U S A. 1996;93:8318–23. doi: 10.1073/pnas.93.16.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution. 1957;11:398–411. [Google Scholar]
- Winn N, Paul A, Musaro A, Rosenthal N. Insulin-like growth factor isoforms in skeletal muscle aging, regeneration, and disease. Cold Spring Harb Symp Quant Biol. 2002;67:507–18. doi: 10.1101/sqb.2002.67.507. [DOI] [PubMed] [Google Scholar]
- Wolkow CA, Kimura KD, Lee MS, Ruvkun G. Regulation of C. elegans lifespan by insulinlike signaling in the nervous system. Science. 2000;290:147–50. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- Yang S, Alnaqeeb M, Simpson H, Goldspink G. Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J Muscle Res Cell Motil. 1996;17:487–95. doi: 10.1007/BF00123364. [DOI] [PubMed] [Google Scholar]
- Yang SY, Goldspink G. Different roles of the IGF-I Ec peptide (MGF) and mature IGF-I in myoblast proliferation and differentiation. FEBS Lett. 2002;522:156–60. doi: 10.1016/s0014-5793(02)02918-6. [DOI] [PubMed] [Google Scholar]
- Ye P, D’Ercole AJ. Insulin-like growth factor actions during development of neural stem cells and progenitors in the central nervous system. J Neurosci Res. 2006;83:1–6. doi: 10.1002/jnr.20688. [DOI] [PubMed] [Google Scholar]
- Yin Y, Stephen CW, Luciani MG, Fahraeus R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462–7. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]