

Abstract
Background:
Recent studies raised questions about the severity of cognitive impairment associated with dementia with Lewy bodies (DLB). However, there have been few analyses of large, multicenter data registries for clinical–pathologic correlation.
Methods:
We evaluated data from the National Alzheimer's Coordinating Center registry (n = 5,813 cases meeting initial inclusion criteria) and the University of Kentucky Alzheimer's Disease Center autopsy series (n = 527) to compare quantitatively the severity of cognitive impairment associated with DLB pathology vs Alzheimer disease (AD) and AD+DLB pathologies.
Results:
Mini-Mental State Examination (MMSE) scores showed that persons with pure DLB had cognitive impairment of relatively moderate severity (final MMSE score 15.6 ± 8.7) compared to patients with pure AD and AD+DLB (final MMSE score 10.7 ± 8.6 and 10.6 ± 8.6). Persons with pure DLB pathology from both data sets had more years of formal education and were more likely to be male. Differences in final MMSE scores were significant (p < 0.01) between pure DLB and both AD+DLB and pure AD even after correction for education level, gender, and MMSE–death interval. Even in cases with extensive neocortical LBs, the degree of cognitive impairment was most strongly related to the amount of concomitant AD-type neurofibrillary pathology.
Conclusions:
Dementia with Lewy bodies can constitute a debilitating disease with associated psychiatric, motoric, and autonomic dysfunction. However, neocortical Lewy bodies are not a substrate for severe global cognitive impairment as assessed by the Mini-Mental State Examination. Instead, neocortical Lewy bodies appear to constitute or reflect an additive disease process, requiring Alzheimer disease or other concomitant brain diseases to induce severe global cognitive deterioration.
GLOSSARY
- AD
= Alzheimer disease;
- ADC
= AD Center;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- DLB
= dementia with Lewy bodies;
- LBs
= Lewy bodies;
- MMSE
= Mini-Mental State Examination;
- NACC
= National Alzheimer's Coordinating Center;
- NF
= neurofibrillary;
- NIA-RI
= National Institute of Aging-Reagan Institute;
- UK ADC
= University of Kentucky Alzheimer's Disease Center.
Dementia with Lewy bodies (DLB) is a neurodegenerative disease, defined pathologically by the presence of widespread neocortical Lewy bodies (LBs) and Lewy neurites.1,2 LBs are fibrillar intraneuronal aggregates (“inclusion bodies”) of α-synuclein protein that are thought to induce brain dysfunction.3,4 The hypothesis of direct LB neurotoxicity was supported by the discovery of mutations of α-synuclein that are linked to the DLB phenotype.4,5 In its pure form, DLB lacks any other known substrates for brain dysfunction.
Although autopsy series found that neocortical LBs are present in ∼10%–25% of older adults, far fewer cases are distinguished by the presence of neocortical LBs in cognitively impaired persons without cerebral infarctions or neocortical Alzheimer disease (AD) type neurofibrillary tangles.6–9 Yet many cases of AD have concomitant neocortical LBs,10,11 and a relatively high proportion of nondemented cases have neocortical LBs.12,13 Thus, neocortical LBs are not rare, but only seldom do they seem to be the sole pathologic substrate of clinical dementia. Further, the signs and symptoms of AD+DLB are challenging to distinguish.14 These observations have prompted speculation as to whether the present clinical-pathologic classification system for DLB is appropriate.7,13,15 As we enter an era with ever-greater hopes for tailored neurotherapeutics, we need to learn what the expected degree of cognitive impairment is for pure DLB and how it may add to cognitive impairment severity when other neuropathologies are present.
METHODS
National Alzheimer's Coordinating Center Registry data.
The National Alzheimer's Coordinating Center (NACC) Registry represents detailed data obtained from 31 AD Centers (ADCs) across the United States.16 NACC data were obtained by request according to the regular application methods. Exclusion criteria (as described in detail in table e-1 on the Neurology® Web site at www.neurology.org) were applied a priori to enable clinical-pathologic correlations specific to the diagnoses of cortical LB and AD-type pathology. Briefly, cases were excluded for the following reasons: no Mini-Mental State Examination (MMSE) score; no Braak neurofibrillary (NF) stage17 data; no Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuritic plaques18 data; clinical history of stroke; or clinical history of prion disease, triplet-repeat disease, or any other disease that would by itself explain the clinical syndrome. Patients with autopsies between 1995 and 2008 were included to assess diagnostic accuracy both before and subsequent to implementation of the National Institute of Aging-Reagan Institute (NIA-RI)19 and DLB2 consortia/consensus criteria. Most cases meeting final inclusion criteria were derived from 27 ADCs. The average number of cases per center was 142, the median was 108 (range 37–367).
University of Kentucky Alzheimer's Disease Center data.
Details of University of Kentucky Alzheimer's Disease Center (UK ADC) Internal Review Board protocols, patient recruitment, and longitudinal follow-up have been described previously.11,20 Briefly, patients who came to autopsy from the UK ADC cohort (total n = 527) were studied. The autopsy rate for the study group is >90%. Mental status testing has been described.11,20 Clinical diagnoses were documented by the clinicians at the UK ADC Consensus Conference, which included neurologists, neuropsychologists, and social workers. Detailed neuropathological studies were performed at the UK ADC as described previously.10,11,21 For direct comparison with data from other ADCs represented in NACC Registry, UK ADC data were pulled from the actual NACC results to ensure all the relevant inclusion and exclusion criteria were identical. Serial MMSE scores were the basis for studying the rate of cognitive deterioration of patients from various neuropathologic diagnostic groups as described below. To be included in this longitudinal analysis, there had to be at least 4 MMSE scores and autopsy confirmation. Cases with cerebrovascular disease or hippocampal sclerosis were excluded as described,22 leaving 143 cases in these analyses: 107 pure AD cases, 27 AD+DLB cases, and 9 pure DLB cases.
Pathologic criteria.
The presence or absence of neocortical LBs was inferred, in both data sets, by indication of “diffuse neocortical type LB” disease in the data registry. The presence of AD-type pathology was determined using the NIA-RI criteria, where Braak Stages V/VI with CERAD “moderate” or “frequent” indicate the disease has pathologic impact (NIA-RI High Likelihood). In the presence of sufficient neocortical LBs and Lewy neurites to merit the diagnosis of DLB, AD+DLB was determined to be present with Braak neurofibrillary stages IV, V, or VI (with CERAD “moderate” or “frequent”), since in the presence of neocortical LBs, the neurofibrillary pathology tends to be lower because of the additive clinical impact of these pathologies.11
Statistical methods.
Mean response (MMSE) was compared among 3 pathologic groups from the NACC Registry by constructing an analysis of variance procedure for a one-way layout with post hoc comparisons of means based on Fisher protected least significant differences procedure (table 1). Final MMSE scores refer to scores derived from final minimum data set data. Percent female was compared among groups using a χ2 test. To compare MMSE scores across a range of AD-type pathology, generalized linear model for a 2-factor design was constructed with post hoc comparisons to check the differences in final MMSE scores between the presence and absence of neocortical LBs at each AD-type Braak stage (0–I, II/III, IV/V, and VI). Post hoc comparisons of the means were based on t statistics. Statistical significance was determined at the 0.05 level. For assessment of longitudinal UK ADC data, a linear mixed model was fitted to the last 4 MMSE scores before death. The purpose of the analysis was to determine how the average intercept and slope varied among the 3 neuropathologic diagnostic groups. In this model it was assumed that each subject had a linear decline in their MMSE scores with each person having their own intercept and slope for this decline (random intercept and slope). This latter analysis was adjusted for years of education. Statistical significance was set at the 0.05 level.
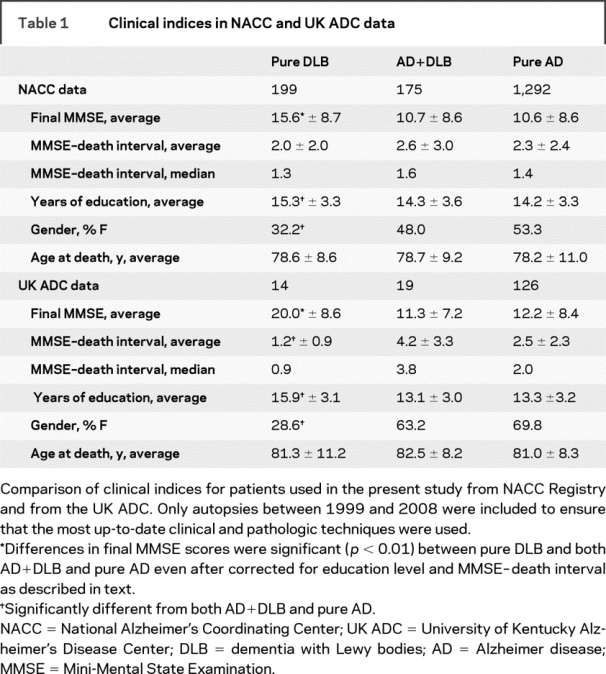
Table 1 Clinical indices in NACC and UK ADC data
RESULTS
An initial analysis of the UK ADC and NACC data indicated that some clinical indices differ between groups with pure DLB, pure AD, or AD+DLB (tables 1 and 2). UK ADC data were analyzed separately from NACC data because further studies were performed on the UK ADC cohort. In both data sets, patients with pure DLB tend to die with higher final MMSE scores in comparison to the other dementia groups (NACC data—pure DLB vs pure AD: p < 0.0001; pure DLB vs AD+DLB: p < 0.0001. UK ADC data—pure DLB vs pure AD: p = 0.001; pure DLB vs AD+DLB: p = 0.0033). Note that persons with pure DLB pathology also tended to have more years of formal education (NACC data—pure DLB vs pure AD: p < 0.0001; pure DLB vs AD+DLB: p = 0.0039. UK ADC data—pure DLB vs pure AD: p = 0.004; pure DLB vs AD+DLB: p = 0.0105) and were more likely to be male (NACC data—pure DLB vs pure AD: p < 0.0001; pure DLB vs AD+DLB: p = 0.0018. UK ADC data—pure DLB vs pure AD: p = 0.002; pure DLB vs AD+DLB: p = 0.0494).
Table 2 Evaluation of different data sets
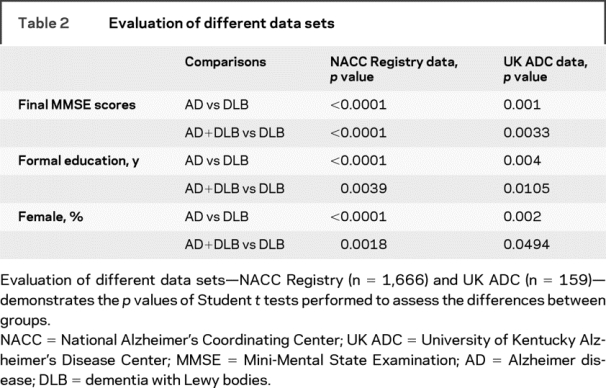
Differences between groups in time periods between final MMSE evaluations and death and differences in formal education levels between groups were possible sources of bias. We thus performed separate analyses exclusively on patients who had been tested within a year of death, adjusted for education. In the UK group, this analysis (n = 58 total) still had large differences in final MMSE scores (pure DLB vs AD+DLB: p = 0.008; pure DLB vs pure AD: p = 0.008). This difference in final MMSE scores also held true for the NACC cohort (n = 674 total; pure DLB vs AD+DLB: p < 0.001; pure DLB vs pure AD: p < 0.0001).
To assess with higher resolution how AD-type and DLB-type pathologies were associated with antemortem cognitive impairment, we evaluated the association of MMSE scores with LB pathology across a range of severity of AD-type pathology (figure 1). The severity of AD pathology was stratified by Braak stages, which by convention are scaled using Roman numerals (0–VI).17 Across a range of Braak stages, in both the NACC Registry data and the UK ADC data, the neocortical LB pathology was associated with an incremental amount (∼5–10 final MMSE score points) of cognitive impairment. Note that for this assessment, the UK ADC data had been removed from the NACC Registry data to evaluate them independently. For NACC Registry data (total subjects n = 2,667), the mean MMSE scores vary significantly at the first 3 Braak stages 0–I, II/III, and IV/V (the means with presence of neocortical LBs are lower than the means with absence of LBs; p = 0.0024, p < 0.0001, and p = 0.0353) but not the most severe Braak NF stage (p = 0.3990). For UK ADC data (total subjects n = 259), only 2 of the 4 Braak NF stages vary in means (II/III: p = 0.0005 and IV/V: p = 0.0002).

Figure 1 Evaluation of average final antemortem MMSE scores for patients with and without appreciable neocortical LBs present according to Braak neurofibrillary stages on neuropathology reports
Note that the presence of neocortical Lewy bodies (LBs) is associated with a relatively modest decrease in final Mini-Mental State Examination (MMSE) scores at some Braak stages. The University of Kentucky Alzheimer's Disease Center (UK ADC) data are shown separately from the National Alzheimer's Coordinating Center (NACC) Registry data because each study group has unique characteristics. For these figures, only cases with autopsies from 1999 to 2008 were used to ensure most up-to-date pathologic techniques. Statistically, the mean final MMSE scores were not different in Braak stage VI patients in the presence vs absence of neocortical LBs (using NACC Registry, UK ADC, or combining data). Error bars = standard deviation. MMSE scores vary by Alzheimer disease–type Braak staging and presence or absence of neocortical LBs.
In the UK ADC data, there was no difference in the mean final MMSE scores, with or without LBs, for patients at NF stages 0–I or VI (Braak NF stage 0–I: p = 0.41, perhaps due to small n = 4 for patients with neocortical LBs in this category; Braak NF stage VI: 0.69). When the NACC Registry data and UK ADC data were combined (total subjects n = 2,926), mean MMSE scores vary at the first 3 groups (Braak NF stages 0–I: p = 0.0012; II/III: p < 0.0001; IV/V: p = 0.0055), but not Braak stage VI (p = 0.5302). Thus, in all the analyses the mean MMSE scores between cases with or without neocortical LBs were not significantly different for patients with Braak stage VI NF pathology.
To assess a survivor bias, we also analyzed the duration of the disease, i.e., the interval between onset of dementia symptoms and death, in a subset of cases from UK ADC where those data were available (AD n = 85; AD+DLB n = 23; pure DLB n = 6). Means of duration of disease were compared using t tests. There were no differences in duration of disease among the groups (p > 0.15 for all comparisons). The “snapshot” of cognitive impairment severity as determined by final MMSE scores does not necessarily indicate how rapidly the patients declined in the prior years. To address this issue, we used data from the UK ADC because we had multiple annual MMSE scores for cases with autopsy confirmation of pathology. MMSE scores were charted in relationship to time for 9 patients who had pure DLB with autopsy confirmation of pathology as shown in figure 2.
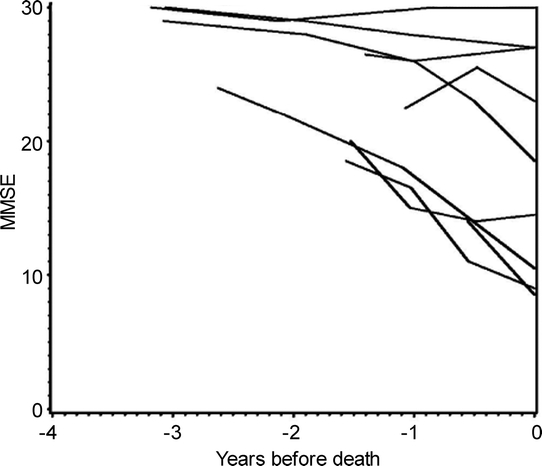
Figure 2 Cognitive impairment is seen in some but not all patients with pure dementia with Lewy bodies confirmed at autopsy
The rate of cognitive impairment for 9 individuals with pure dementia with Lewy bodies from the University of Kentucky Alzheimer's Disease Center database with 3 or more serial Mini-Mental State Examination (MMSE) scores. The abscissa refers to years before death.
MMSE score changes over time (UK ADC data) were then used to compare the rate of cognitive deterioration for patients with autopsy-confirmed pure DLB, pure AD, or AD+DLB (figure 3). The slopes (p = 0.049) and intercepts (p < 0.005) varied among the groups. Post hoc comparisons of the slopes showed that the pure AD group had a less steep slope (−2.37) than the AD+DLB group (−3.37, p = 0.05) while the pure DLB group had a slope in between (−2.84, p = 0 0.5 when compared to either of the 2 other groups). Post hoc comparison of the intercepts showed that the pure DLB group had a higher intercept (19.8) than the mixed pathology group (11.12, p = 0.016) while the pure AD group had an intercept in between (14.42, p = 0.10 when compared to either of the other 2 groups). Hence, although it was a small group (n = 9 subjects) the pure DLB group had higher MMSE scores 4 visits before death than the other 2 groups and declined less than the AD+DLB group.

Figure 3 Rate of cognitive deterioration in patients with autopsy-confirmed pure DLB, pure AD, or AD+DLB
Analysis of last 4 Mini-Mental State Examination (MMSE) scores before death of persons from the University of Kentucky Alzheimer's Disease Center autopsy series with pathologically confirmed pure dementia with Lewy bodies (DLB; n = 9), pure Alzheimer disease (AD; n = 107), or AD+DLB (n = 27) was performed to determine how the average intercept and slope varied among the 3 neuropathologic diagnostic groups after adjusting for years of education. With the caveat that it is a small group, the pure DLB group had higher MMSE scores 4 visits before death compared to the other groups and declined less perceptibly than the AD+DLB group (see text for details).
DISCUSSION
We analyzed large data sets of longitudinally assessed patients followed to autopsy, across a broad clinical spectrum, showing that MMSE score loss in pure DLB is less severe when compared to cases with significant AD pathology. These results are concordant with recent studies that have called into question the impact of neocortical LBs on global cognition and suggested that LBs are an additive pathology.7,11,13,21,23,24 However, the current study involves a larger sample encompassing 31 different academic medical centers, with explicit distinction made between AD, DLB, and AD+DLB cases. Collectively these data suggest that pure DLB pathology, without AD pathology, is associated with a relatively moderate impact on global cognition.
Older studies may report different outcomes partly because the rates of evaluation for neocortical LBs have changed significantly over time. The DLB Consortium published the first clinical and pathologic guidelines for diagnosis of the disease in 1996.2 Further consensus revisions of the diagnostic guidelines for DLB were even more recent.1,25 Thus, the clinical-biologic understanding of DLB is still evolving. Retrospective analyses should take these considerations into account.
Clinically, patients with DLB may manifest fluctuating clinical course, early well-formed hallucinations, delusions, paranoia, parkinsonism, and REM sleep disorders.1,26 The contradistinctions between DLB and Parkinson disease dementia are somewhat vague—clinical and pathologic features are virtually identical but DLB is distinguished by the cognitive symptoms presenting before parkinsonism.1,2 A number of studies assumed that the clinical signs of DLB are strongly predictive for the relevant pathology. However, studies that have evaluated the sensitivity and specificity of those clinical signs/symptoms in predicting neocortical LB pathology in a prospective manner have had varying results.1,27–29 Thus, specific clinical features may be imperfect at indicating exactly which cases will show pure DLB at autopsy. This is why pathologic diagnoses were used as the gold standard in the present study.
Even using pathologic diagnoses involves potential pitfalls and alternative hypotheses must be considered. It is possible that the differences in MMSE scores between groups with different pathologic diagnoses could represent a survival effect. In this case, patients with pure DLB may die at an earlier age, before more severe cognitive decline could be identified. Possible causes for early mortality in DLB could include adverse medication effects as can be seen with atypical and typical antipsychotic therapies, or autonomic dysfunction related to widespread brainstem and limbic involvement of LBs. However, the age at death for pure DLB was not different from the pure AD or AD+DLB cases in either the NACC or the UK ADC data. Further, neither our results nor other sources30–32 have indicated that DLB duration of disease differs systematically from AD or AD+DLB. Still, the possibility of a survivor bias has not been completely ruled out.
There are other limitations to the present study. MMSE scores proximal to death were used as the sole metric of global cognitive status. Prior studies have shown that overlapping but non-identical cognitive domains are affected in DLB and in AD, and the difference can be a challenge to capture in a single test.33–35 The MMSE evaluates a number of cognitive domains, including orientation, language, attention/calculation, short-term memory/recall, and praxis. MMSE scores offer good sensitivity in predicting some aspects of brain disease, producing universally understood data with known normative standards for multicenter studies.21 The MMSE is also more clinically relevant than longer tests for the simple reason that many non-academic clinicians worldwide actually use it. At the present time, there are no other cognitive assessments that are consistent within and between ADC/NACC data sets that would allow for the comparisons of clinical (cognitive) and pathologic data. The current updated NACC protocol is designed to provide such data in the future but this involves years of longitudinal data collection. For all their merits, MMSE tests are imperfect metrics of “cognition” and do not explicitly assess psychiatric, executive, or autonomic domains that tend to be affected in DLB. Visuospatial dysfunction is prominent in DLB36 and can have profound impact on daily function yet may theoretically only cause a single point lost on the MMSE for copy of the intersecting pentagons. In sum, although patients with DLB have higher final MMSE scores, they may be equally or more adversely affected by their disease in other ways that are not tracked as extensively by the MMSE.
Using data derived from 31 different ADCs necessitates pooling of data that are not exactly analogous—different academic centers follow different protocols in both the clinical and neuropathologic settings. For example, the rate of obtaining autopsies can be a systematic bias in a community setting.37 We compensated partly by comparing the NACC results with those of the UK ADC database, which has followed a relatively consistent approach and has an autopsy rate of over 90%. We found good concordance in the results despite differences in cohort characteristics.
We discussed cases of AD, DLB, and AD+DLB pathologies as though each were a different disease. This is partly because some cases of AD+DLB may represent an entirely different disease subtype as underscored by the discovery of families with congenital, i.e., monoallelic, vulnerability to dementia with AD+DLB pathology.38–40 However, it may well be that in many cases the AD and neocortical LB diseases were pathogenetically independent.
Finally, we are unaware of reasons that pure DLB would be significantly more likely to affect males and persons with more years of formal education, although these are highly intriguing correlative phenomena that hold true over multiple data sets. Thus, additional questions remain at the fascinating nexus of cognitive impairments and their neuropathologic correlates.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by Dr. Kryscio, L.O. Xu, and E.L. Abner.
ACKNOWLEDGMENT
The authors thank all the patients; Ela Patel, Ann Tudor, Paula Thomason, Dr. Huaichen Liu, and Sonya Anderson for technical support; Nancy Stiles, MD, and Allison Caban-Holt, PhD, for clinical evaluations; Daron Davis, MD, for pathologic evaluations; and Leslie E. Phillips, MS, for help with NACC data.
DISCLOSURE
Dr. Nelson is supported by NIH 1R01NS061933 (PI), K08 NS050110 (PI), and an Alzheimer's Association NIRG grant (PI). Dr. Kryscio serves as an Associate Editor of Neurology® and receives research support from the NIA [ADC Core grant P30 AG028383 (sponsor)]. Dr. Jicha served as a consultant for Medivation; and receives research support from Pfizer, Inc., Martek Biosciences Corporation, Elan Pharmaceuticals, NIH/NIA [1 P30 AG028383-03 (Co-I), 2R01AG019241-06A2 (Co-I), ADC-0272U01AG010483 (site PI)], and from the Alzheimer's Association [NIRG-07-59967 (PI)]. Ms. Abner reports no disclosures. Dr. Schmitt receives research support from the NIA [1 P30 AG028383-02 (Co-I) and 1 R01 AG1924-05S2 (Co-I)]. Ms. Xu reports no disclosures. Dr. Cooper serves on speakers' bureaus for Pfizer Inc. and Forest Pharmaceuticals; receives research support from Eli Lilly, Eisai Limited, and Myriad Pharmaceuticals; and has provided expert consultation regarding nursing home care of patients with dementia. Dr. Smith receives research support from the NIH [NIA R01 AG025159 (PI), P30 AG028383 (core leader), NIA T32 000242 (faculty), NIA U01 AG024904 (site PI), NIMH R01MH078228 (Co-I), NCRR-Shared Instrumentation 1S10RR024553 (PI), NINDS R01-36660 (PI), and NIMH R01MH063817-04 (Co-I). Dr. Markesbery receives research support from P30 AG028383 (PI), P01 AG05119 (PI), R01 AB1924 (PI), P50 NS39787 (Co-I), the Kleberg Foundation and the Healy Family Foundation.
Supplementary Material
Address correspondence and reprint requests to Dr. Peter T. Nelson, Department of Pathology, Division of Neuropathology, and the Sanders-Brown Center on Aging, Rm 311, Sanders-Brown Center Building, 800 S. Limestone, University of Kentucky, Lexington, KY 40536-0230 pnels2@email.uky.edu.
Supplemental data at www.neurology.org.
Supported by grants R01 NS061933, K08 NS050110, P30 AG028383, and U01 AG016976 from NIH.
Disclosure: Author disclosures are provided at the end of the article.
Received March 13, 2009. Accepted in final form July 16, 2009.
REFERENCES
- 1.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 2.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- 4.Waxman EA, Giasson BI. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta 2009;1792:616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997;276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger KA. Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm 2004;111:1219–1235. [DOI] [PubMed] [Google Scholar]
- 7.Jellinger KA. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 2009;1792:730–740. [DOI] [PubMed] [Google Scholar]
- 8.Jellinger KA, Attems J. Neuropathological evaluation of mixed dementia. J Neurol Sci 2007;257:80–87. [DOI] [PubMed] [Google Scholar]
- 9.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 2007;62:406–413. [DOI] [PubMed] [Google Scholar]
- 10.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer's disease: a complex but coherent relationship. J Neuropathol Exp Neurol 2009;68:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol 2007;66:1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 13.Parkkinen L, Pirttila T, Alafuzoff I. Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol 2008;115:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez OL, Hamilton RL, Becker JT, Wisniewski S, Kaufer DI, DeKosky ST. Severity of cognitive impairment and the clinical diagnosis of AD with Lewy bodies. Neurology 2000;54:1780–1787. [DOI] [PubMed] [Google Scholar]
- 15.Parkkinen L, Soininen H, Alafuzoff I. Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol 2003;62:363–367. [DOI] [PubMed] [Google Scholar]
- 16.Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord 2004;18:270–277. [PubMed] [Google Scholar]
- 17.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol 1993;33:403–408. [DOI] [PubMed] [Google Scholar]
- 18.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: a commentary. Neurobiol Aging 1997;18:S91–94. [DOI] [PubMed] [Google Scholar]
- 19.The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging 1997;18:S1–S2. [PubMed] [Google Scholar]
- 20.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol 2006;63:38–46. [DOI] [PubMed] [Google Scholar]
- 21.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol Epub 2008 Nov 19. [DOI] [PMC free article] [PubMed]
- 22.Nelson PT, Kryscio RJ, Abner EL, et al. Acetylcholinesterase inhibitor treatment is associated with relatively slow cognitive decline in patients with Alzheimer's disease and AD+DLB. J Alzheimers Dis 2009;16:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkkinen L, Soininen H, Laakso M, Alafuzoff I. Alpha-synuclein pathology is highly dependent on the case selection. Neuropathol Appl Neurobiol 2001;27:314–325. [DOI] [PubMed] [Google Scholar]
- 24.Merdes AR, Hansen LA, Jeste DV, et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 2003;60:1586–1590. [DOI] [PubMed] [Google Scholar]
- 25.Leverenz JB, Hamilton R, Tsuang DW, et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 2008;18:220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crystal HA, Dickson DW, Lizardi JE, Davies P, Wolfson LI. Antemortem diagnosis of diffuse Lewy body disease. Neurology 1990;40:1523–1528. [DOI] [PubMed] [Google Scholar]
- 27.Luis CA, Barker WW, Gajaraj K, et al. Sensitivity and specificity of three clinical criteria for dementia with Lewy bodies in an autopsy-verified sample. Int J Geriatr Psychiatry 1999;14:526–533. [PubMed] [Google Scholar]
- 28.McKeith I, Mintzer J, Aarsland D, et al. Dementia with Lewy bodies. Lancet Neurol 2004;3:19–28. [DOI] [PubMed] [Google Scholar]
- 29.Lopez OL, Litvan I, Catt KE, et al. Accuracy of four clinical diagnostic criteria for the diagnosis of neurodegenerative dementias. Neurology 1999;53:1292–1299. [DOI] [PubMed] [Google Scholar]
- 30.Nervi A, Reitz C, Tang MX, et al. Comparison of clinical manifestations in Alzheimer disease and dementia with Lewy bodies. Arch Neurol 2008;65:1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujishiro H, Ferman TJ, Boeve BF, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol 2008;67:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jicha GA, Schmitt FA, Abner E, et al. Prodromal clinical manifestations of neuropathologically confirmed Lewy body disease. Neurobiol Aging Epub 2008 Nov 19. [DOI] [PMC free article] [PubMed]
- 33.Olichney JM, Galasko D, Salmon DP, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology 1998;51:351–357. [DOI] [PubMed] [Google Scholar]
- 34.Hamilton JM, Salmon DP, Galasko D, et al. A comparison of episodic memory deficits in neuropathologically-confirmed Dementia with Lewy bodies and Alzheimer's disease. J Int Neuropsychol Soc 2004;10:689–697. [DOI] [PubMed] [Google Scholar]
- 35.Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol 2006;63:1450–1456. [DOI] [PubMed] [Google Scholar]
- 36.Galasko D, Katzman R, Salmon DP, Hansen L. Clinical and neuropathological findings in Lewy body dementias. Brain Cogn 1996;31:166–175. [DOI] [PubMed] [Google Scholar]
- 37.Tsuang D, Simpson KL, Li G, et al. Evaluation of selection bias in an incident-based dementia autopsy case series. Alzheimer Dis Assoc Disord 2005;19:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg CK, Pericak-Vance MA, Saunders AM, Gilbert JR, Gaskell PC, Hulette CM. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol 2000;100:145–152. [DOI] [PubMed] [Google Scholar]
- 39.Trembath Y, Rosenberg C, Ervin JF, et al. Lewy body pathology is a frequent co-pathology in familial Alzheimer's disease. Acta Neuropathol 2003;105:484–488. [DOI] [PubMed] [Google Scholar]
- 40.Tsuang DW, Riekse RG, Purganan KM, et al. Lewy body pathology in late-onset familial Alzheimer's disease: a clinicopathological case series. J Alzheimers Dis 2006;9:235–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.