

Abstract
In this study, we estimate the statistical significance of structure prediction by threading. We introduce a single parameter ɛ that serves as a universal measure determining the probability that the best alignment is indeed a native-like analog. Parameter ɛ takes into account both length and composition of the query sequence and the number of decoys in threading simulation. It can be computed directly from the query sequence and potential of interactions, eliminating the need for sequence reshuffling and realignment. Although our theoretical analysis is general, here we compare its predictions with the results of gapless threading. Finally we estimate the number of decoys from which the native structure can be found by existing potentials of interactions. We discuss how this analysis can be extended to determine the optimal gap penalties for any sequence-structure alignment (threading) method, thus optimizing it to maximum possible performance.
Protein structure prediction is a complex problem that requires significant approximations and simplifications both in models involved and in search strategy. Currently a popular and reasonably successful method is threading. In threading, a new sequence is mounted on a series of known folds with the goal of finding a fold (a sequence-structure alignment) that provides the best score (lowest energy). A standard quasienergetic scoring scheme assigns energy Es to an alignment s in the hope that the lowest energy alignment bears structural similarity to the native fold of the query sequence. In this regard, threading is similar in spirit to sequence alignment (1). An essential part of all sequence alignment procedures is the evaluation of the statistical significance of obtained scores (2, 3). More recently, the problem of the statistical significance of structural alignments was addressed (3).
Despite the development of numerous approaches and applications (4–10), the statistical significance of predictions from threading calculations has not been systematically analyzed. An empirical approach was proposed by Bryant and coworkers (5, 6), who compared the best threading alignment with the threading of reshuffled random sequences. Bryant and coworkers assumed that scores (energies) of realigned random sequences are normally distributed. This approach is computationally demanding, as it requires realignment of all reshuffled random sequences with all target proteins in the database. Furthermore, the assumption of Gaussian distribution of threading scores of realigned random sequences was not justified in refs. 5 and 6.
In this study, we show that, in contrast to earlier assumptions (5, 6), the probability of successful prediction in threading calculations follows an extreme value distribution (EVD) (11). Furthermore, our analysis identifies a simple parameter that provides a fast and computationally inexpensive clue as to whether the actual threading calculation resulted in a reliable prediction or in a false positive.
Theory Development
As a theoretical background, we use the random energy model (REM). The REM was originally introduced by Derrida (12) to describe a class of spin-glass models. Later Bryngelson and Wolynes postulated (13) and Shakhnovich and Gutin showed [using replica mean-field theory (14)] that the REM provides an adequate description of equilibrium properties of random heteropolymers. Subsequently the REM was successfully applied to various aspects of the protein-folding problem (15–18) as well as to analysis of the protein-structure prediction problem (19, 20). The general applicability and limitations of the REM in describing the energy landscape of heteropolymers have been addressed in refs. 14, 21, and 22.
Here we will formulate the REM and its underlying assumptions by using the language of threading calculations.
The energy of each threading alignment is usually taken as a sum energy of all pairwise contacts:
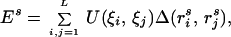 |
1 |
where L is the length of a query sequence, s denotes alignment, and ris is a coordinate of the ith group (usually the Cα or Cβ atom) in this alignment. Δ denotes the cutoff distance for contact potential that determines which groups are interacting (usually taken 7.5–9 Å between the Cα or Cβ atoms). ξi denotes the type of amino acid at position i of the query sequence. U is a 20 × 20 matrix of interaction energy parameters between all types of amino acids. Summations here and below are taken over all residues that are farther apart than two units along the sequence, i.e., j > i + 2.
The REM formulation for threading is as follows:
The set of alignments consists of the “native” alignment having energy EN and a set of M decoys.
The energies of decoys take statistically independent random values.
- The most common form for the probability density of energies of decoys is Gaussian:
Eav is the average energy, and Σ is the standard deviation of energies of all decoys. The assumption that energies of decoys are independent random values is validated if contacts in the alternative conformations (decoys) are distributed independently and uniformly. Physically this means that polymer connectivity that may cause correlation between contacts plays a relatively minor role, i.e., long-range (along the sequence) contacts provide dominant contribution to the energy of an alignment. This was shown to be generally true for three-dimensional compact polymers (23, 24).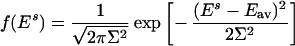
2 - Assuming independence and identical distribution of contacts in the decoys, we can make a simple estimate for the average energy of alignments Eav and standard deviation of alignment energies Σ. These estimates were made in our earlier publication (equations 8–14 of ref. 25); here we provide the results
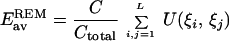
3
where C is the number of contacts in the native conformation (we assume here that all decoys have the same number of contacts) and Ctotal is the total number of possible pairwise interactions between residues (i.e., Ctotal = ∑i,j=1L 1≈L(L−3)/2).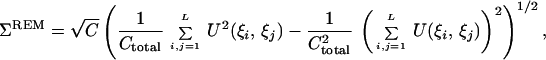
4
The estimates (Eqs. 3 and 4) are potentially very useful because they permit evaluation of the average energy of decoys and their standard variation directly from interaction matrix U and query sequence ξ. No explicit decoys are necessary. Importantly, the Z-score that is used as a common criterion of the quality of discrimination of the native state can be estimated:
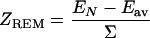 |
5 |
Because Eav and Σ depend only on the composition of the query sequence (and U), one can think of ZREM as a “correction” for sequence composition.
However, the REM estimates for Eav and Σ are obtained under certain assumptions, and the validity of the Z-score must be assessed by comparison with threading calculations (see below).
The density of states (i.e., the number of decoys found in an energy range from E to E + dE) is w(E)dE = Mf(E)dE. We also define a very important threshold energy Ec as
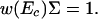 |
6 |
This threshold energy Ec corresponds to the bottom of the continuous part of the energy spectrum when the system “runs out” of decoys. This threshold energy determines qualitatively the features of the density of states:
at E > Ec, the density of states w(E) is very high;
at E < Ec, the density of states w(E) is very low; and
the commonly accepted estimate for Ec is given in refs. 14 and 18,
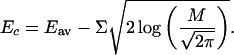 |
7 |
In other words, according to the REM, at E > Ec, there are many decoys in any energy interval Σ whereas at Ec the system runs out of states and the spectrum becomes discrete: one can find only occasional and rare decoys with E < Ec.
In threading calculations, native (or near-native) alignment is obtained with energy EN. It is clear that EN needs to be below Ec to make a successful prediction with native-state ranking first. More specifically, we can determine within the REM approximation the probability that native alignment ranks first. To this end, we require that all M decoys have an energy higher than EN. The probability of this event is
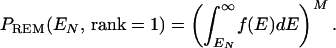 |
8 |
The analysis of Eq. 8 simplifies when the number of decoys M is large (≫1), which is always the case in threading calculations. Straightforward calculations result in
 |
9 |
(see ref. 26 for more details), where
 |
10 |
is the deviation of EN from Ec, and u and α are unitless “center” and “width” of the distribution:
 |
11 |
 |
12 |
The parameter ɛN is also related to the predicted Z-score ZREM via a simple relation:
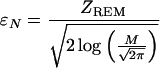 |
13 |
The result in Eq. 9 represents an EVD (11), which is valid for a broad range of distributions f(E) that can be converted to exponential distribution by linearization of the logf(E) at large deviations from the mean (11). However, the specific expressions for parameters u and α given by Eqs. 11 and 12 are valid for Gaussian distribution f(E) (Eq. 2) and large M.
Because for any sequence-structure pair M depends only on its length, one can consider transformation from ZREM to ɛ as a “correction” for the length of the template and the query sequence.
It is very instructive to examine the qualitative features of the probability distribution (Eq. 9). When the number of decoys is very large, u→1 and α→∞, which means that ɛ serves as a very good predictor of success in the threading simulations: when ɛ > 1, the native fold ranks first with a very high degree of certainty, whereas at ɛ < 1, the native fold will surely not rank first. The results become less clearcut when the number of decoys is small. It is crucial to note that one does not need to know the native fold to evaluate Ec. Eq. 7 suggests that the number of decoys M and the standard variance define Ec completely. Both M and Ec can either be estimated (by using Eq. 4) or obtained directly from threading calculations (see below). Importantly, the computation of Ec does not require costly runs of threading with “shuffled” sequences, a method widely used to estimate the statistical significance of threading (27). Given a query sequence and a potential, one can compute Ec and use it as a cutoff for assessing the significance of structure prediction.
Comparison with Gapless Threading
Now we compare the predictions from the REM model with the results of “gapless threading.” In gapless threading, one takes an amino acid sequence and mounts it in every possible way (without gaps) onto known protein structures of greater length. If the sequence has a length L and the structure it is mounted onto has a length Lstr, the total number of decoys generated by gapless threading is Lstr − L + 1. Hence a database of about 103 protein structures allows generation of about 105. . . 106 decoys. Decoys obtained in this way are sorted by energy. The goal is to recognize the native structure, i.e., to have it rank first in the sorted list. Importantly, gapless threading is inappropriate for real structure prediction because native structure is not present among the set of decoys, and structures similar to the native (analogues) cannot be recognized in gapless threading for the vast majority of proteins (L.A.M. and E.I.S., unpublished results). However, gapless threading is a useful tool for generating decoys and for testing the recognition abilities of the energy function.
To construct a database, we selected a representative set of nonhomologous proteins from the fssp database (28). From this set, all structures that have no coordinates for side chains and those that are longer than 500 residues were removed. The final database contained 1,011 nonhomologous proteins. We performed all-against-all recognition by gapless threading of every sequence through all proteins of greater length. Energy was computed by using an optimized potential of interactions U(σ,η) taken from ref. 25. Of 1,011 sequences, the native structure was recognized as having rank 1 for 763 (75%) proteins. The native structure had rank = 2 for 30 other proteins. For 13 sequences, a structure similar to the native (root-mean-square deviation < 5 Å) ranked first. A small fraction of proteins in the dataset are not stable by themselves but are stabilized only in larger complexes (e.g., protein rop is tetrameric) or by metal ions, large numbers of disulfides, etc. For those proteins, gapless threading is not expected to recognize their native structure correctly based on interactions within a monomer, and it does not (see below).
First, we test the main REM assumption that energies of all decoys are normally distributed. In Fig. 1, we plotted the energy distributions of decoys for 10 proteins and fitted each of them into Gaussian distribution. Clearly, the fit is very good throughout the whole range of energies except the lowest energies, which show a deviation from Gaussian distribution in the form of a characteristic “shoulder.” Such a “shoulder” is a typical feature of the density of states of nonrandom protein-like sequences that fold into its native conformation with low (compared with a random sequence) energy (see also figure 3 of ref. 29). The existence of this “shoulder” suggests that potentials are good enough to distinguish the native structure from the set of decoys generated in gapless threading. Furthermore, Fig. 1B confirms the condition ɛ = 1 for the boundary of the density of states for decoys that are structurally unrelated to the native state. Decoys with ɛ > 1.2 are likely to be structurally similar to the native state (see below and Fig. 4 Inset).
Figure 1.

(A) Normalized distribution of energies f(E) = w(E)/M for decoys obtained by gapless threading (red lines). Ten red lines correspond to the distributions obtained for 10 different proteins (153 l, 1aa0, 1aa8, 1aac, 1aaf, 1aay, 1ab3, 1ab8, 1ab9, and 1aba), each threaded against the full database. Blue broken line shows standard normal distribution. Notice deviation from normal distribution at the low energy tail. (B) Same data shown not normalized [w(E)] as a function of −ɛ (red lines). Notice that Gaussian approximations (blue lines) all cross w(E) = 1 at −ɛ = −1, where the actual number of decoys at this point (red lines) is of the order of 10.
Figure 4.

Probability P(e > ɛ, rank = 1) of having Rank = 1 for a structure with normalized energy above ɛ. Black lines correspond to the probability expected for a Gaussian distribution (Eqs. 8, 12, and 11). Because the number of decoys M is different for different proteins, each of 1,011 proteins has its own line (i.e., different u and α). Black squares show the same probability as obtained from gapless threading. (P(e > ɛ, rank = 1) = 1 − Pdecoy(e > ɛ), where Pdecoy(e > ɛ) = ∫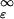 fdecoy(e) de was obtained by threading). The green line is the best fit of the observed probability by Eq. 9. The best fit is achieved at ufit = 1.01,αfit = 8.03. Corresponding values for a Gaussian distribution are uG = 0.92 and αG = 18.8 (see text for details). (Inset) Distribution of energies of the lowest-energy decoys fdecoy(ɛ). Notice the typical EVD shape of the distribution. Red and blue histograms correspond to the lowest-energy decoys that are native like (Q ≥ 0.7) and nonnative (Q < 0.7), accordingly. Histogram of native-like decoys stretches till ɛ ≈ 2.5, whereas nonnative decoys are not observed above ɛ = 1.8.
fdecoy(e) de was obtained by threading). The green line is the best fit of the observed probability by Eq. 9. The best fit is achieved at ufit = 1.01,αfit = 8.03. Corresponding values for a Gaussian distribution are uG = 0.92 and αG = 18.8 (see text for details). (Inset) Distribution of energies of the lowest-energy decoys fdecoy(ɛ). Notice the typical EVD shape of the distribution. Red and blue histograms correspond to the lowest-energy decoys that are native like (Q ≥ 0.7) and nonnative (Q < 0.7), accordingly. Histogram of native-like decoys stretches till ɛ ≈ 2.5, whereas nonnative decoys are not observed above ɛ = 1.8.
We then test another important REM assumption of the independence of contacts. To this end, we compare the ZREM estimated in Eq. 5 based on this assumption with the Z-score obtained directly from threading:
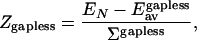 |
14 |
where Eavgapless and Σgapless are obtained for each protein directly from the threading calculation as average energy and its standard deviation over all decoys. Comparison of Zgapless with ZREM is the first test of the REM applied to fold recognition. Fig. 2 compares Zgapless with ZREM for every protein in the database. The correlation between the two measures is 0.95. Notice there is no bias or nonlinearity in the plot. This comparison indicates that ZREM is a very good estimate of Zgapless. Note that the major assumption in estimating ZREM is the statistical independence of frequencies of interresidue contacts, which is fundamental for the whole REM analysis. The good correlation shown in Fig. 2 supports the applicability of the REM to threading.
Figure 2.

Comparison of Z-scores computed by Eq. 5 ZREM and obtained by gapless threading (Zgapless). the correlation coefficient is 0.95. Solid line: best linear fit by 0.87ZREM − 0.30.
The next question we address is how well the rank of the native structure (which is the measure of success in fold recognition) can be predicted by the value of ɛ [defined in Eq. 10 and computed as ɛ = Zgapless/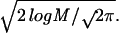
Fig. 3 presents a number of important results. It shows that ɛ is a very good predictor of success in fold recognition, particularly:
Two distinct regions can be seen: ɛ > 1 and ɛ < 1. As expected from the REM, when ɛ < 1, the rank of the native structure is >1 in 95% of the cases, i.e., the native structure is not recognized.
When ɛ > 1, the native structure ranks first for 94% of the sequences but not for all of them. However, even when the native structure is not recognized (rank > 1) for ɛ > 1, in the vast majority of cases (41 of 43≈95%), it is located among the 10 top-scoring decoys (i.e., rank ≤ 10). According to the REM, this happens when the native structure is below the bottom of the continuous spectrum (EN < Ec) but is intermixed with rare low-energy decoys that also have E < Ec. However, there are only few such decoys, and the rank of the native structure stays low.
When ɛ > 1.5, the native structure has the first rank in 99% of the cases. In 3 of 490 cases, the native structure has rank = 2, and in 1 case, it has rank = 3. Most importantly in these four cases, the top-scoring decoy has a structure similar to the native one (root-mean-square deviation < 5 Å).
- The color code of squares in Fig. 3 indicates the degree of similarity between the native structure rN and the structure that ranks first r1. This quantity is defined as
the overlap between contact maps of the two structures. Clearly, when the native structure has rank = 1, then Q = 1, and the square on Fig. 3 is colored red. More important are the cases when the native structure is not recognized but a similar structure (analogue) comes with rank = 1. One would expect an analogue to have energy similar to the native one. Hence, when the native structure is intermixed with low-energy decoys (i.e., has a low, but not the first, rank), an analogue can rank first instead. In agreement with this expectation, we observe the following: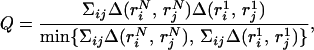
15
When the native structure is not recognized but is not very high in rank (≤10) (i.e., still below the continuous part of the spectrum), then in about 10% of the cases a native-like structure (Q ≥ 0.7) ranks first.
In the opposite case, when the rank of the native structure is high (>10) in only 1 case of 173, the decoy with the first rank has Q ≥ 0.7.
Figure 3.

Rank of the native structure in gapless threading against ɛ of this structures. The color of each point shows the similarity Q between the structure ranking first and the native structure. The high value of Q indicates structural similarity. To emphasize points with higher Q, we plot them on the front; those with lower Q (which dominate at low ɛ) are on the background.
These results bring us to the conclusion that when ɛ < 1, the native structure is not recognized, and the top-scoring decoy is very unlikely to have a native-like structure.
We also notice that when ɛ > 1, the energy of the native state belongs to the discrete spectrum (see Fig. 1). However, that does not guarantee the recognition of the native structure, because an occasional low-energy decoy belonging to the discrete spectrum can still rank first. However, the probability of finding a random decoy with energy E well below Ec is small (see Fig. 3 Inset).
Now we come to the central point of our study: estimating the probability of the native fold (with energy EN) ranking first among its decoys.
The result is shown in Fig. 4. It can be seen that the EVD (Eq. 9) provides the perfect functional form for the observed probability P(EN, rank = 1). However, there is some quantitative disagreement between the parameters of the EVD predicted by Eqs. 11 and 12 [black lines in Fig. 4 and parameters ufit and αfit obtained by fitting the data into the EVD (9) (green line in Fig. 4)]. Although the predicted u and fitted ufit are close to each other, the discrepancy in parameter α is more substantial, close to a factor of 2. This discrepancy indicates that the actual distribution fdecoys(E) for the energy of the decoys deviates from the Gaussian on the tails. In fact, fdecoys(E) have a Gaussian form for E close to Eav (E − Eav < 3Σ) but decays exponentially at larger deviations (see Fig. 1).
However, the form of the distribution (Eq. 9) does not depend on the Gaussian form of f(E) and thus is more generally applicable. Parameter α then can be viewed as an empirical parameter or can be obtained directly from the form of the distribution of energies of decoys.
The results presented in Figs. 3 and 4 address the crucial issue of “false positives” in protein threading: a comparison of the lowest-energy predicted alignment with Ec makes it possible to assess with a high degree of certainty whether the threading calculation returned a native-like structure or a “false-positive” misfold.
It is clear from Fig. 4 that ɛ > 1.2 almost guarantees that the native structure ranks first in threading calculations. As can be seen from Fig. 4 Inset, most of the decoys at ɛ > 1.2 are native like. Parameter ɛ depends on two factors: (i) the quality of the model and the potential U (good model and precise potential provide low EN); and (ii) the number of decoys M that affect ɛ via Ec. Having established ɛ as a powerful criterion of success in threading prediction, we can now address the next question: among how many decoys can existing models and potential functions select the native state as first ranking (30). To this end, we solve the inequality ɛ > 1.2 vis à vis the number of decoys by using the definitions of ɛ (10) and Ec from Eq. 7.
Fig. 5 shows the maximal number of decoys Mmax that can be distinguished from the native state for the protein model we use. Different data points correspond to different proteins. Each protein has its own value of EN and ɛ, and therefore the criterion ɛ > 1.2 determines a different limiting number of decoys for different proteins. Nevertheless, we come to an estimate that present models and potential functions can select the native state out of about 1012 decoys for a protein of 150… 250 amino acids.
Figure 5.

The maximal number Mmax = 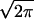 exp((Z/1.2)2/2) of decoys a protein structure calculation can sustain while having the correct native structure recognized (i.e., ɛ > 1.2) vs. the length L of the protein. Blue dots: Mmax vs. L for proteins whose native structure has been recognized in gapless threading. Red crosses: Mmax vs. L for proteins whose native structure has not been recognized. Solid line: the actual number of decoys M obtained by gapless threading as a function of protein length. Notice how well the solid line separates regions of recognized and nonrecognized proteins. This indicates that Mmax is a very good predictor of whether a protein is recognized among a pool of M decoys.
exp((Z/1.2)2/2) of decoys a protein structure calculation can sustain while having the correct native structure recognized (i.e., ɛ > 1.2) vs. the length L of the protein. Blue dots: Mmax vs. L for proteins whose native structure has been recognized in gapless threading. Red crosses: Mmax vs. L for proteins whose native structure has not been recognized. Solid line: the actual number of decoys M obtained by gapless threading as a function of protein length. Notice how well the solid line separates regions of recognized and nonrecognized proteins. This indicates that Mmax is a very good predictor of whether a protein is recognized among a pool of M decoys.
Is this sufficient for a realistic threading calculation? Such a calculation should allow for the possibility of gaps and insertions both in the target structure and in a sequence. This is a key requirement to make threading strategy capable of recognizing analogs. The ability to recognize analogs is crucial because in real life, threading application native structure is not available, and the only hope is that there will be a structure in the database that is not identical but is similar to the native state of the query sequence.
Practical Implications for Protein Structure Prediction
Although gapless threading is used in this paper to illustrate the important points of our analysis, our results are not limited to it. Rather they may be applicable to any threading calculation for which energies of decoys can be considered as independent random values. This assumption is clearly corroborated in the present study for gapless threading (see Fig. 2). An advanced Monte Carlo threading technique that allows gaps and insertions in both sequence and target was reported recently (8). The energy landscape of decoys generated by this threading technique was analyzed with the conclusion that the REM may be adequate to describe it (8).
We showed that parameter ɛ can serve as a reliable and computationally inexpensive predictor of success in threading calculation. This parameter is related to stability gap or “Tf/Tg,” which was shown in protein folding theory to be good a determinant of sequence stability and fast folding (13, 29, 31–33).
The estimate of ɛ for gapless threading is straightforward and does not require sequence reshuffling and realignment, a computationally costly procedure. The main difference in the evaluation of ɛ for realistic gapped threading comes from the fact that the length of an alignment now varies. Σ and Eav depend on alignment length so that the full distribution of alignment scores cannot be described by a single Gaussian. However, the analysis of gapped threading (L.A.M., W. Chen & E.I.S., unpublished work) suggests that the distribution of scores for each alignment length l can be described by its own Gaussian with alignment-length dependent Σ(l) and Eav(l). Then the density of states can be generalized to gapped threading:
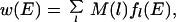 |
16 |
where fl(E) is the Gaussian probability density corresponding to an alignment of length l with its own Eav(l) and Σ(l). M(l) is the number of decoys for alignments of length l. This number can be determined from combinatorics. The average energy of all alignments is determined from w(E), and Ec can also be found from w(E) by using Eq. 6 [where maximal Σ(l) can be used]. Then ɛ for gapped threading can be determined from Eq 10. Note that the evaluation of ɛ does not require sequence reshuffling. This provides a fast way to recognize false positives in realistic threading calculations. In cases when lowest scoring threading alignment features ɛ ≤ 1, such alignment is most likely to be a false positive that is structurally unrelated to the native state. Alignments featuring ɛ > 1.5 are most likely to be correct predictions. A more detailed quantitative estimate of the probability of correct prediction in the range 1 < ɛ < 1.5 requires evaluation of the EVD of ɛ. Parameters α and u can be determined from the fitting of distribution of ɛ for the threading of many reshuffled random sequences of various lengths into the EVD with subsequent tabulation of the results for the range of lengths. (Each reshuffled random sequence “EN ” entering the definition of ɛ in Eq. 10 corresponds to the lowest-scoring alignment.) These procedures will be discussed in detail in subsequent publications.
Although gaps and insertions in realistic threading calculations are crucial, their introduction comes at the cost of a serious increase in the number of decoys, to the point that even the best potentials for the present models are unable to find analogs with lowest energy Eanalog < Ec (or ɛ > 1) (25). Our results suggest a constructive way to address this problem. The allowed length of an alignment and the gap penalty may be chosen in such a way that the total number of decoys M generated by threading would not exceed a “recognition threshold” Mmax (see Fig. 5).
Generally the number of allowed gaps and insertions (and hence the number of allowed decoys) should be chosen to achieve a maximum value of ɛ. This corresponds to maximizing the probability of a correct prediction (34). Indeed, when gaps/insertions are not restricted, ɛ may be small because the number of decoys M is large. On the other hand, restrictions on gaps/insertions that are too severe may lead to elimination of native-like conformations from the threading set of alignments leading to lower ɛ because of loss of alignments with low energy (higher apparent EN). Note that achieving maximal ɛ does not reduce the number of decoys to simple minimization: their restriction carries the risk of eliminating analogs of the native structure of the threaded sequence from the ensemble. Thus the strategy of setting optimal threading simulations adjusts gap penalties/number of fragments to achieve the maximal number of decoys that still allows recognition of the native structure by existing potentials.
Acknowledgments
L.A.M. was sponsored by the Harvard Society of Fellows. A.V.F. acknowledges the support of the Howard Hughes Medical Institute International Research Scholar Award No. 75195-544702 and of the Russian Basic Research Foundation Award No. 98-04-49303.
Abbreviations
- EVD
extreme value distribution
- REM
random energy model
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.160271197.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.160271197
References
- 1.Atschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abagyan R A, Batalov S. J Mol Biol. 1997;273:355–368. doi: 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- 3.Levitt M, Gerstein M. Proc Natl Acad Sci USA. 1998;95:5913–5920. doi: 10.1073/pnas.95.11.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finkelstein A V, Reva B A. Nature (London) 1991;351:497–499. doi: 10.1038/351497a0. [DOI] [PubMed] [Google Scholar]
- 5.Panchenko A, Marchler-Bauer A, Bryant S H. Proteins. 1999;37:133–140. doi: 10.1002/(sici)1097-0134(1999)37:3+<133::aid-prot18>3.3.co;2-4. [DOI] [PubMed] [Google Scholar]
- 6.Panchenko A R, Marchler-Bauer A, Bryant S H. J Mol Biol. 2000;296:1319–1331. doi: 10.1006/jmbi.2000.3541. [DOI] [PubMed] [Google Scholar]
- 7.Lathrop R H. J Comp Biol. 1999;6:405–418. doi: 10.1089/106652799318355. [DOI] [PubMed] [Google Scholar]
- 8.Mirny L A, Shakhnovich E I. J Mol Biol. 1998;283:507–526. doi: 10.1006/jmbi.1998.2092. [DOI] [PubMed] [Google Scholar]
- 9.Bienkowska J R, Rogers R G, Smith T F. J Comp Biol. 1999;6:299–311. doi: 10.1089/106652799318283. [DOI] [PubMed] [Google Scholar]
- 10.Thiele R, Zimmer R, Lengauer T. J Mol Biol. 1999;290:757–779. doi: 10.1006/jmbi.1999.2893. [DOI] [PubMed] [Google Scholar]
- 11.Gumbel E J. Statistics of Extremes. New York: Columbia Univ. Press; 1958. [Google Scholar]
- 12.Derrida B. Phys Rev B. 1981;24:2613–2624. [Google Scholar]
- 13.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shakhnovich E, Gutin A. Biophys Chem. 1989;34:187–199. doi: 10.1016/0301-4622(89)80058-4. [DOI] [PubMed] [Google Scholar]
- 15.Shakhnovich E I, Gutin E M. J Theor Biol. 1991;149:537–546. doi: 10.1016/s0022-5193(05)80097-9. [DOI] [PubMed] [Google Scholar]
- 16.Bryngelson J D. J Chem Phys. 1994;103:6038–6045. [Google Scholar]
- 17.Pande V, Grosberg A, Tanaka T. Biophys J. 1997;73:3192–3210. doi: 10.1016/S0006-3495(97)78345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutin A, Sali A, Abkevich V, Karplus M, Shakhnovich E. J Chem Phys. 1998;108:6466–6483. [Google Scholar]
- 19.Finkelstein A V, Gutin A, Badretdinov A. Proteins. 1995;23:151–162. doi: 10.1002/prot.340230205. [DOI] [PubMed] [Google Scholar]
- 20.Finkelstein A V. Phys Rev Lett. 1999;80:4823–4825. [Google Scholar]
- 21.Sfatos C, Gutin A M, Shakhnovich E I. Phys Rev E. 1993;48:465. doi: 10.1103/physreve.48.465. [DOI] [PubMed] [Google Scholar]
- 22.Pande V, Grosberg A, Joerg C, Tanaka T. Phys Rev Lett. 1996;76:3987–3990. doi: 10.1103/PhysRevLett.76.3987. [DOI] [PubMed] [Google Scholar]
- 23.Kuznetsov D V, Grosberg A Yu. Macromolecules. 1992;25:1970–1977. [Google Scholar]
- 24.Abkevich V, Gutin A, Shakhnovich E. J Mol Biol. 1995;252:460–471. doi: 10.1006/jmbi.1995.0511. [DOI] [PubMed] [Google Scholar]
- 25.Mirny L, Shakhnovich E. J Mol Biol. 1996;264:1164–1169. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
- 26.Leadbetter H, Lindgren M R, Rootzen G. Extremes and Related Properties of Random Sequences and Processes. Berlin: Springer; 1983. [Google Scholar]
- 27.Bryant S H, Altschul S F. Curr Opin Struct Biol. 1995;5:236–244. doi: 10.1016/0959-440x(95)80082-4. [DOI] [PubMed] [Google Scholar]
- 28.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 29.Shakhnovich E I, Gutin A. Proc Natl Acad Sci USA. 1993;90:7195–7199. doi: 10.1073/pnas.90.15.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reva B A, Skolnick J, Finkelstein A V. Proteins. 1999;35:353–359. [PubMed] [Google Scholar]
- 31.Goldstein R, Luthey-Schulten Z A, Wolynes P. Proc Natl Acad Sci USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abkevich V, Gutin A, Shakhnovich E I. J Chem Phys. 1994;101:6052–6062. [Google Scholar]
- 33.Dinner A, Abkevich V, Karplus M, Shakhnovich E I. Proteins Struct Funct Genet. 1999;35:34–40. doi: 10.1002/(sici)1097-0134(19990401)35:1<34::aid-prot4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 34.Chiu T L, Goldstein R A. Folding Des. 1998;3:223–228. doi: 10.1016/S1359-0278(98)00030-3. [DOI] [PubMed] [Google Scholar]