

Abstract
Chronic kidney disease is often complicated by uremic cardiomyopathy that consists of left ventricular hypertrophy and interstitial fibrosis. It is thought that hypertension and volume overload are major causes of this disease, but here we sought to identify additional mechanisms using a mouse model of chronic renal insufficiency. Mice with a remnant kidney developed an elevated blood urea nitrogen by 1 week, as expected, and showed progressive cardiac hypertrophy and fibrosis at 4 and 8 weeks even though their blood pressures were not elevated nor did they show signs of volume overload. Cardiac extracellular signal-regulated kinase (ERK) was activated in the uremic animals at 8 weeks. There was also an increased phosphorylation of S6 kinase, which is often mediated by activation of the mammalian target of rapamycin (mTOR). To test the involvement of this pathway, we treated these uremic mice with rapamycin and found that it reduced cardiac hypertrophy. Reduction of blood pressure, however, by hydralazine had no effect. These studies suggest that uremic cardiomyopathy is mediated by activation of a pathway that involves the mTOR pathway.
Keywords: blood pressure, rapamycin, uremia, ventricular hypertrophy
Pathological cardiac hypertrophy is associated with a poor prognosis, the development of cardiac arrhythmias, diastolic dysfunction, and progression to overt heart failure.1 A similar condition develops in patients with severe chronic kidney disease (CKD) that is called uremic cardiomyopathy.1–4 In this condition, cardiomyocytes enlarge and excess fibrous tissue is deposited in the heart resulting in the thickening of the ventricular walls. Uremic cardiomyopathy is associated with diastolic dysfunction and also, in some cases, with contractile abnormalities. Nearly 75% of adults have left ventricular hypertrophy (LVH) at the time of initiation of dialysis for end-stage renal disease.5 The development of LVH is an independent risk factor that is associated with reduced survival in patients receiving dialysis.6
The pathogenesis of uremic cardiomyopathy remains uncertain. As there is a very high incidence of hypertension in patients with severe CKD, one hypothesis is that cardiac hypertrophy develops as a result of pressure overload. However, correction of hypertension in rats with renal injury does not prevent the development of cardiac hypertrophy.7 Furthermore, Ayus et al.8 showed that ventricular wall dimensions were reduced in patients’ receiving intensive daily dialysis for 1 year when compared with those receiving dialysis three times per week, despite similar systolic blood pressures in both groups.
Volume overload may also contribute to LVH by increasing LV end-diastolic pressure.1,9 A reduction in intradialytic weight correlates with a reduction in LV mass index, but LVH may persist after normalization of wall stress.9 Anemia is frequently present in patients with CKD and is another factor implicated in the pathogenesis of uremic cardiomyopathy.1,10 An additional possibility is that the accumulation of hypertrophic ligands associated with renal insufficiency or end-stage renal disease may initiate a signal transduction cascade independent of mechanical stress. A variety of other substances accumulate in end-stage renal disease that may modulate cardiac growth and function, including endothelin-1, parathyroid hormone, tumor necrosis factor-α, leptin, interleukin-1α, and interleukin-6.11
The molecular pathways responsible for cardiac hypertrophy are dependent on their initiating stimuli. In the setting of load-induced hypertrophy, mechanical stress can be translated through a detection mechanism that is hypothesized to be based on integrins that initiate intracellular signaling in response to stretch of the extracellular matrix.12 Stretch may also promote the local release of ligands such as angiotensin II and endothelin-1 that bind to cognate receptors on the surface of cardiomyocytes to stimulate intracellular signaling pathways.13,14 Subsequent activation of the mammalian target of rapamycin (mTOR) complex activates the ribosomal S6 kinase that promotes mRNA translation.15 It is hypothesized that uremic cardiomyopathy may occur through a separate mechanism that is initiated by activation of Na/K ATPase channels. Cardiotonic steroids accumulate in patients with CKD and they interact with the α subunit of transmembrane Na/K ATPases to promote the activation of the intracellular mitogen-activated protein kinase pathway.16,17
Rapamycin, a direct inhibitor of mTOR reverses hypertrophy due to chronic pressure overload but has not been investigated in CKD.18 mTOR acts through several downstream effectors, including S6 Kinase, 4E-BP, and EF1α, that all modulate ribosomal function. The PI3 kinase-Akt1 pathway and the extracellular signal-regulated kinase (ERK) pathway both act upstream of mTOR to promote increased protein synthesis.19 In this work, we established a murine model of pressure-controlled uremic cardiomyopathy and identified mTOR as a key protein in the development of this condition.
RESULTS
Surgically induced renal injury in mice
To determine whether CKD in mice results in the development of cardiac hypertrophy, we studied the effect of surgically induced renal injury (SIRI) on 12-week-old 129/SvJ mice. In this method, the right kidney was injured by cauterization, and a left nephrectomy was performed after 2 weeks. The surgery was well tolerated by wild type 129/SvJ mice and no mortality was observed.
The metabolic status of mice was determined both 4 and 8 weeks after the completion of SIRI or sham surgery (Table 1). The serum creatinine was significantly increased in SIRI mice compared with sham animals at both 4- and 8-week time points. Four weeks after surgery, the serum creatinine was 0.71±0.006 in sham animals but was increased to 0.133±0.015 in SIRI mice (P=0.003; Table 1). Eight weeks after surgery, the serum creatinine was 0.104±0.007 in sham mice but was increased 30.7% to 0.136±0.018 in SIRI mice (P=0.016). Similarly, the BUN was significantly increased in SIRI mice at both the 4- and 8-week time points (Table 1). Four weeks after surgery, the serum BUN was 16.0±4.1 mg/100 ml in sham mice but was increased to 54.0±4 mg/dl in SIRI animals (P=0.00003). Eight weeks after surgery, the serum BUN was 22±1 mg/100 ml in sham mice but was increased to 83±3 mg/dl in SIRI animals (P=0.0001). A three- to four-fold increase in urine output was noted in SIRI animals compared with sham-operated animals at both 4 and 8 weeks after surgery (Table 1). Serum aldosterone levels were significantly increased in SIRI mice compared with sham mice at both the 4- and 8-week time points (P=0.042), and this was associated with significantly elevated urine sodium loss (P=0.041). Serum angiotensin II levels were transiently elevated in SIRI mice compared with sham mice 4 weeks after surgery (P=0.013) but were significantly reduced in SIRI mice compared with sham mice at the 8-week time point (P=0.0001; Table 1).20,21 Serum potassium was significantly decreased in SIRI mice to 3.9±0.1mmol/l, compared with 4.7±0.2mmol/l in sham-operated mice (P=0.01; Table 1).
Table 1.
Metabolic analysis of mice after renal injury
| Sham (n=7) 4 weeks | SIRI (n=8) 4 weeks | Sham (n=7) 8 weeks | SIRI (n=8) 8 weeks | SIRI+V (n=7) 8 weeks | SIRI+Rap (n=7) 8 weeks | |
|---|---|---|---|---|---|---|
| Ser. creatinine (mg/100 ml) | 0.071±0.006 | 0.133±0.015* | 0.104±0.007 | 0.136±0.018* | 0.141±0.008* | 0.175±0.022* |
| Ser. hemoglobin (g/100 ml) | 17±0.3 | 16±0.1 | 15±1.6 | 15±1.1 | 16±0.7 | 12±2.0* |
| BUN (mg/dl) | 16±4 | 54±4* | 22±1 | 83±3* | 84±6* | 108±11* |
| 24 h urine volume (ml) | 0.530±90 | 1.730±190* | 1.110±60 | 3.420±180* | 3.640±840* | 6.422±930* |
| Urine [Na+] (mmol/l) | 151±13 | 99±6.3* | 162±19 | 108±8.5 | 58±4.2* | 48±1.5* |
| 24 h urine Na+ (mmol) | 0.0800±0.02 | 0.173±0.05* | 0.180±0.01 | 0.369±0.02* | 0.211±0.02* | 0.306±0.01* |
| Ser. potassium (mmol/l) | 4.7±0.2 | 3.9±0.1* | 3.8±0.9* | 4.2±1.0 | ||
| Ser. aldosterone (pmol/l) | 2454±196 | 3328±266* | 2745±220 | 3408±273* | 3631±290* | 3663±293* |
| Ser. angiotensin II (pg/ml) | 34.3±4 | 142±17* | 128±15 | 97.3±12* | 189±23 | 42.3±5* |
Metabolic analysis of 20-week-old mice after sham surgery (Sham), surgically induced renal injury (SIRI), SIRI with daily injections of vehicle (SIRI+V), or SIRI with daily injections of rapamcyin (SIRI+Rap). Daily injections of vehicle or rapamycin were initiated 4 weeks after SIRI and were given for 4 weeks.
P<0.05 vs sham-operated mice at the same time point.
The body weight did not change after surgery and was 25.1±0.6 g in SIRI mice compared with 24.7±0.9 g in sham animals at the 8-week time point (P=0.51; Table 2). Interestingly, SIRI mice ate 42.5% more food than sham-operated animals (Table 2). Despite their increased food intake, SIRI mice had similar body composition to sham-operated mice. Measurement of muscle and fat mass by MRI demonstrated that the lean body mass of SIRI mice was 19.2±1.2 g 8 weeks after surgery and was 20.3±0.8 g in sham-operated mice (P=0.47; Table 2). Finally, the extracellular fluid volume was nearly identical in SIRI and sham-operated mice 8 weeks after surgery (Table 2).
Table 2.
Body mass components of mice after renal injury
| Sham (n=7) 8 weeks | SIRI (n=8) 8 weeks | SIRI+V (n=7) 8 weeks | SIRI+Rap (n=7) 8 weeks | |
|---|---|---|---|---|
| Body weight (g) | 24.7±0.85 | 25.1±0.6 | 26.1±0.5 | 25.7±0.6 |
| 24 h calorie intake (kcal) | 14.1±0.75 | 20.1±1.5* | 14.1±1.0 | 20.4±1.3* |
| 24 h water intake (ml) | 1.130±186 | 3.599±757* | 3.630±510* | 6.333±646* |
| 24 h urine volume (ml) | 1.110±60 | 3.420±180* | 3.640±840* | 6.422±930* |
| Lean body mass (g) | 20.3±0.8 | 19.2±1.2 | 20.3±1.4 | 19.9±1.3 |
| Fat body mass (g) | 4.1±0.3 | 3.9±0.2 | 4.4±0.2 | 4.3±0.2 |
| Extracellular fluid volume (ml) | 4.13±1.24 | 4.10±0.89 | 4.21±0.99 | 4.02±1.16 |
| Lung weight/BW (mg/g) | 6.45±0.9 | 6.21±0.9 | 6.27±0.0.5 | 6.59±0.6 |
| Liver weight/BW (mg/g) | 26.12±2.4 | 26.82±2.0 | 27.06±4.4 | 26.55±1.9 |
| Spleen weight/BW (mg/g) | 2.52±0.3 | 2.71±0.4 | 2.93±0.3 | 2.62±0.4 |
Metabolic analysis of 20-week-old mice after sham surgery (Sham), surgically induced renal injury (SIRI), SIRI with daily injections of vehicle (SIRI+V), or SIRI with daily injections of rapamcyin (SIRI+Rap).
P<0.05 vs sham-operated mice at the same time point.
Development of cardiac hypertrophy after SIRI
To evaluate the development of cardiac hypertrophy after SIRI, sequential transthoracic echocardiography was performed on unanesthetized mice 4 and 8 weeks after SIRI or sham surgery. LV systolic function, measured as fractional shortening, was nearly identical in SIRI and sham-operated mice at both time points (Table 3). The echocardiographically determined LV mass index increased from 3.22±0.08 mg/g in sham-operated animals to 3.78±0.14 mg/g in SIRI animals 4 weeks after surgery was completed (P=0.003). Similarly, at 8 weeks after surgery, the LV mass index increased from 3.20±0.15 mg/g in sham-operated animals to 3.96±0.11 mg/g in SIRI animals (P=0.001).
Table 3.
Echocardiographic analysis of mice after renal injury
| Sham (n=12) 4 weeks | SIRI (n=15) 4 weeks | Sham (n=7) 8 weeks | SIRI (n=8) 8 weeks | SIRI+V (n=7) 8 weeks | SIRI+Rap (n=7) 8 weeks | |
|---|---|---|---|---|---|---|
| HR (bpm) | 613±58 | 618±31 | 629±12 | 637±9 | 599±12 | 616±15 |
| PWd (mm) | 0.76±0.06 | 0.89±0.09* | 0.76±0.02 | 0.92±0.02* | 0.85±0.05* | 0.78±0.02† |
| IVSd (mm) | 0.83±0.02 | 0.94±0.03* | 0.84±0.02 | 1.03±0.03* | 0.90±0.03 | 0.89±0.04† |
| LVEDD (mm) | 3.3±0.08 | 3.2±0.06 | 3.4±0.09 | 3.2±0.06 | 3.2±0.07 | 3.0±0.04 |
| LVM (mg) | 84±2.8 | 99±4.2* | 88±3.7 | 106±2.2* | 94±4.9 | 76.1±3.9† |
| LVMI (mg/g) | 3.22±0.08 | 3.78±.14* | 3.20±0.15 | 3.96±0.11* | 3.63±0.17* | 2.94±0.18† |
| FS (%) | 63±1.0 | 62±1.2 | 64±1.0 | 65±0.9 | 65±1.4 | 65±1.9 |
HR, heart rate; PWd, left ventricular posterior wall thickness in diastole; IVSd, interventricular septal wall thickness in diastole; LVEDD, left ventricular end-diastolic diameter; LVM, calculated LV mass (mg); LVMI, left ventricular mass index (LVM/body weight) mg/g; FS, fractional shortening of the left ventricle in systole.
Transthoracic echcardiographic analysis of mice 4 or 8 weeks after sham surgery (Sham), subtotal nephrectomy (SIRI), SIRI with daily injections of vehicle (SIRI+V), or SIRI with daily injections of rapamcyin (SIRI+Rap). Daily injections of vehicle or rapamycin were initiated 4 weeks after SIRI and were given for 4 weeks.
P<0.05 for all comparisons vs sham-operated mice at the same time point except LVMI, where P<0.021;
P<0.05 for all comparisons vs SIRI mice injected with vehicle at the same time point except LVMI, where P<0.021.
To confirm whether SIRI promoted cardiac hypertrophy, we weighed heart tissue of SIRI and control mice 8 weeks after surgery (Table 4). The biventricular weight-to-tibial length ratio was increased from 6.3±0.1 mg/mm in sham-operated mice to 7.4±0.3 mg/mm in SIRI animals (P=0.012). Furthermore, the LV weight-to-tibial length ratio increased from 5.0±0.18 mg/mm sham mice compared with 5.9±0.22 mg/mm in SIRI mice (P=0.006; Table 4).
Table 4.
Heart weights of mice after renal injury
| Sham (n=7) | SIRI (n=8) | SIRI+V (n=7) | SIRI+Rap (n=7) | |
|---|---|---|---|---|
| BW (g) | 24.7±1.4 | 25.4±1.1 | 26.1±0.9 | 25.7±1.3 |
| BVW (mg) | 123±2.7 | 149±6.5* | 138±4.1 | 114±3.9† |
| LV (mg) | 96.5±3.2 | 119±4.9* | 113±3.4 | 90.2±3.9† |
| RV (mg) | 25.8±1.7 | 31.0±1.0* | 29.8±2.8 | 23.7±0.9† |
| Tibia (TL) (cm) | 1.94±0.02 | 1.99±0.02 | 2.02±0.02 | 1.98±0.02 |
| BVW/BW (mg/g) | 4.98±0.2 | 5.9±0.2* | 5.3±0.1 | 4.4±0.1† |
| BVW/TL (mg/mm) | 6.3±0.1 | 7.4±0.3* | 6.8±0.2 | 5.8±0.2† |
| LV/BW (mg/g) | 3.9±0.1 | 4.7±0.2* | 4.3±0.1 | 3.8±0.3† |
| LV/TL (mg/mm) | 5.0±0.2 | 5.9±0.2* | 5.6±0.2 | 4.6±0.2† |
| RV/BW (mg/g) | 1.2±0.0 | 1.2±0.0 | 0.9±0.1 | 1.0±0.1 |
| RV/TL (mg/mm) | 1.5±0.1 | 1.5±0.2 | 1.3±0.1 | 1.2±0.1 |
BW, body weight; BVW, biventricular weight; LV, left ventricular weight; RV, right ventricular weight; Tibia, tibial length; BVW/BW, biventricular weight-to-body weight ratio; BVW/TL, biventricular weight-to-tibial length ratio; LV/BW, left ventricular weight-to-body weight ratio; LV/TL, left ventricular weight-to-tibial length ratio; RV/BW, right ventricular weight-to-body weight ratio; RV/TL, right ventricular weight-to-tibial length ratio.
Morphometric analysis of 20-week-old mice 8 weeks after sham surgery (Sham), subtotal nephrectomy (SIRI), SIRI with daily injections of vehicle (SIRI+V), or SIRI with daily injections of rapamcyin (SIRI+Rap). Daily injections of vehicle or rapamycin were initiated 4 weeks after SIRI and were given for 4 weeks.
P<0.025 vs sham-operated mice;
P<0.021 vs SIRI mice injected with vehicle.
To evaluate whether SIRI promoted the growth of individual cardiomyocytes and the deposition of extracellular matrix in cardiac tissue, we completed a histologic analysis of ventricular tissue sections. Cardiomyocyte area increased by 119% in SIRI mice when compared with sham-operated animals 8 weeks after surgery (P=0.0001; Figure 1). Furthermore, cardiac fibrosis, evaluated in Masson’s trichrome-stained ventricular sections, increased by 120% in SIRI mice 8 weeks after surgery (P=0.0001; Figure 2).
Figure 1. Determination of cardiomyocyte area in ventricular tissue obtained 8 weeks after surgery.

Ventricular tissue was fixed, embedded, sectioned at 4-micron intervals, stained with hemotoxylin and eosin, and examined by light microscopy. Scale bar=20 μm. (a) Ventricular section from sham-operated mouse. (b) Ventricular tissue section from surgically induced renal injury (SIRI) mouse. (c) Section from SIRI mouse treated with vehicle (V) for 4 weeks. (d) Section from SIRI mouse treated with rapamycin (R) for 4 weeks. (e) Quantitative evaluation of cardiomyocyte area by computerized analysis of ventricular tissue sections. Three high power fields (400×) from each of the 4 animals per group were analyzed by computerized photomicrography with ImageJ v1.24 software. Cardiomyocyte area was calculated by use of the ImageJ Particle Analyzer algorithm. *P<0.05 vs sham.
Figure 2. Evaluation of cardiac fibrosis in ventricular tissue obtained 8 weeks after surgery.

Ventricular tissue was fixed, embedded, sectioned at 4-micron intervals, stained with Masson’s trichrome, and examined by light microscopy. (a) Ventricular section from sham-operated mouse. (b) Ventricular tissue section from surgically induced renal injury (SIRI) mouse. (c) Section from SIRI mouse treated with vehicle (V) for 4 weeks. (d) Section from SIRI mouse treated with rapamycin (R) for 4 weeks. (e) Quantitative evaluation of fibrotic area/total area by computerized analysis of ventricular tissue sections. Three selected high power fields (400×) from each of the 4 animals per group were analyzed by computerized photomicrography with ImageJ v1.24 software. Fibrotic area was calculated by use of the ImageJ Particle Analyzer algorithm. *P<0.05 vs sham.
To better evaluate cardiac systolic and diastolic function, we performed cardiac catheterization on anesthetized SIRI and control mice 8 weeks after surgery was completed. These studies demonstrated that LV systolic function, measured by determining +dP/dTmax, was unchanged in SIRI mice (8844±667mmHg/s) when compared with sham-operated mice (8758±856mmHg/s) 8 weeks after surgery (Table 5). Furthermore, diastolic function, measured as τ (Glantz), was similar in both SIRI and sham-operated mice 8 weeks after surgery. Systolic blood pressure, measured by determining the peak LV systolic pressure, was 139±11mmHg in sham-operated mice and was similar at 141±12mmHg in SIRI mice (P=NS). Furthermore, SIRI mice were found to be normotensive by tail cuff blood pressure measurements and also by determination of carotid artery pressures with a Millar catheter (Table 5).
Table 5.
Invasive hemodynamic analysis of mice after renal injury and rapamycin treatment
| Sham (n=7) | SIRI (n=8) | SIRI+V (n=7) | SIRI+R (n=7) | |
|---|---|---|---|---|
| HR (bpm) | 457±12 | 453±10 | 439±12 | 439±11 |
| LVPmax (mm Hg) | 139±11 | 141±12 | 132±12 | 125±9 |
| LVEDP (mm Hg) | 5.3±1.5 | 5.5±0.8 | 4.3±0.71 | 3.6±0.9 |
| Tau (Glantz) | 11.8±0.4 | 12.5±1.5 | 12.0±0.8 | 14.2±1.4 |
| +dP/d tmax (mm Hg/s) | 8758±856 | 8752±693 | 8971±100 | 7624±956 |
| Systolic TCP (mm Hg) | 129±4 | 123±5 | 124±4 | 127±2 |
| Diastolic TCP (mm Hg) | 82±3 | 75±4 | 71±3 | 73±4 |
| Systolic CAP (mm Hg) | 133±11 | 134±10 | 138±15 | 144±14 |
HR, heart rate obtained during cardiac catheterization; LVPmax, peak systolic LV pressure; LVEDP, left ventricular end-diastolic pressure; Tau, time constant of isovolumic change; +dP/dtmax, the maximal change in LV pressure per unit time; TCP, tail cuff pressure; CAP, carotid artery pressure.
Invasive hemodynamic analysis of 20-week-old mice 8 weeks after sham surgery (Sham), subtotal nephrectomy (SIRI), subtotal nephrectomy+vehicle (SIRI+V), or subtotal nephrectomy+rapamycin (SIR+R) by cardiac catheterization.
To determine whether a reduction in blood pressure would affect the development of cardiac hypertrophy after SIRI, mice were administered the arterial vasodilator hydralazine. Mice were administered hydralazine in their drinking water for 8 weeks after the completion of surgery to deliver a daily dose of 0.001 mg/g. Systolic blood pressure, determined by cardiac catheterization (LVPmax), was significantly lower in hydralazine-treated SIRI animals (101±11mmHg) when compared with untreated SIRI animals (141±12mmHg; P=0.037) or untreated sham-operated animals (139±11mmHg; P=0.032; Table 6). However, the development of cardiac hypertrophy in SIRI mice was unaffected by hydralazine administration. Indeed, the LV weight-to-tibial length ratio was 5.8±0.15 mg/mm in hydralazine-treated SIRI mice, compared with 5.0±0.2 mg/mm in untreated sham-operated mice (P=0.004; Table 6).
Table 6.
Analysis of mice treated with hydralazine after renal injury
| Sham (n=7) | SIRI+H (n=7) | SIRI+H/Sham | |
|---|---|---|---|
| BW (g) | 24.7±1.4 | 24.9±0.7 | 1.01 |
| BVW (mg) | 123±2.7 | 139±2.8 | 1.13* |
| LV (mg) | 96.5±3.2 | 115±2.8 | 1.20* |
| Tibia (TL) (cm) | 1.94±0.02 | 1.99±0.1 | 1.02 |
| BVW/BW (mg/g) | 4.98±0.2 | 5.6±0.2 | 1.12* |
| BVW/TL (mg/mm) | 6.3±0.1 | 7.0±0.15 | 1.10* |
| LV/BW (mg/g) | 3.9±0.1 | 4.6±0.12 | 1.19* |
| LV/TL (mg/mm) | 5.0±0.2 | 5.8±0.15 | 1.17* |
| HR (bpm) | 457±12 | 414±26 | 0.90 |
| LVPmax (mm Hg) | 139±11 | 101±11 | 0.73* |
| LVEDP (mm Hg) | 5.3±1.5 | 6.14±0.76 | 1.17 |
| Tau (Glantz) | 11.8±0.4 | 12.7±1.0 | 1.1 |
BW, body weight; BVW, biventricular weight; LV, left ventricular weight; Tibia, tibial length; BVW/BW, biventricular weight-to-body weight ratio; BVW/TL, biventricular weight-to-tibial length ratio; LV/BW, left ventricular weight-to-body weight ratio; LV/TL, left ventricular weight-to-tibial length ratio; HR, heart rate; LVPmax, peak systolic LV pressure; LVEDP, left ventricular end-diastolic pressure; Tau, time constant of isovolumic change.
Morphometric and invasive hemodynamic analysis of 20-week-old mice 8 weeks after surgically induced renal injury (SIRI) treated with hydralazine for 8 weeks (SIRI+H). Each value is then compared with sham mice in simple ratio. Seven animals subject to SIRI were treated with hydralazine for 8 weeks after surgery. Hydralazine was added to the animals’ drinking water to provide 0.001 mg/g of drug per day.
P<0.021 vs sham-operated mice.
Cardiac molecular adaptations in response to SIRI
To evaluate whether the LVH observed in SIRI mice was associated with changes in cardiac gene expression, the expression of marker genes was evaluated by real-time quantitative polymerase chain reaction (PCR).22,23 The concentration of mRNAs encoding atrial natriuretic factor and β-myosin heavy chain in cardiac tissue correlate with the degree of pathological hypertrophy.22,23 The concentration of atrial natriuretic factor mRNA was increased by 94% in LV tissue obtained from SIRI mice 2 weeks after surgery (P=0.012; Figure 3). The LV concentration of β-myosin heavy chain mRNA increased by 209% 2 weeks after surgery (P=0.001). The mRNA levels of both genes returned to baseline 8 weeks after surgery (Figure 3).
Figure 3. Analysis of cardiac gene expression in ventricular tissue obtained 2 and 8 weeks after surgically induced renal injury or sham surgery.

Ventricular tissue was homogenized, RNA was purified, and the expression of the atrial natriuretic factor (ANF) and β-myosin heavy chain (β-MHC) genes was analyzed by quantitative real-time RT-PCR. The specific mRNA levels for Sham (white) vs SIRI (gray) were measured in samples from 3 or 4 mice for each condition (indicated by the number in parentheses). *P<0.01 vs sham.
To determine whether SIRI modulates the activation of intracellular signaling cascades in cardiac tissue, we obtained ventricular lysates 8 weeks after SIRI or sham surgery. Immunoblot analysis of ventricular lysates with phospho-specific antibodies was performed to evaluate the phosphorylation status of ERK1/2, ribosomal S6 protein, and 4E-BP1 (Figure 4). The phosphorylation of ribosomal S6 protein and 4E-BP1 are markers of mTOR activation.16 These experiments demonstrated that ERK1 and ERK2 activation was significantly increased in SIRI ventricular tissue 8 weeks after surgery (P=0.011 for ERK1, P=0.007 for ERK2). Furthermore, S6 protein and a subfraction of 4E-BP1 (4E-BP1.2) phosphorylation were increased 8 weeks after surgery in SIRI mice (P=0.002 for S6 protein, P=0.016 for 4E-BP1.2).
Figure 4. Analysis of signaling protein phosphorylation in ventricular tissue obtained from surgically induced renal injury (SIRI) or sham mice.

(a) Analysis of cardiac signaling protein phosphorylation in ventricular tissue obtained 8 weeks after SIRI (n=5) or sham surgery (n=4). Ventricular tissue was homogenized, protein lysates generated, and proteins were separated by SDS-PAGE followed by immunoblotting with primary antibodies directed against phosphorylated-Akt1/2 (p-Akt), phosphorylated-ERK1/2 (p-ERK1/2), phosphorylated-ribosomal S6 protein (p-S6), phosphorylated-4E-BP1, and α-actin as a loading control. (b) Scanned blot densitometry was performed with Image J (v1.24) software. *P<0.05 vs sham.
Rapamycin treatment of SIRI mice
SIRI resulted in the development of cardiac hypertrophy and was associated with the increased activation of ERK and the increased phosphorylation of ribosomal S6 protein and 4E-BP1. The increased phosphorylation of ribosomal S6 protein and 4E-BP1 in SIRI cardiac tissue suggested that mTOR was activated. To evaluate the requirement for mTOR in the development of cardiac hypertrophy in SIRI mice, mice were treated with rapamycin (sirolimus) or vehicle by daily intraperitoneal administration beginning 4 weeks after surgery. In previous work, rapamycin therapy blocked pressure overload-induced cardiac hypertrophy in mice.18
Rapamycin was tolerated by SIRI mice and no mortality was observed. Renal function deteriorated with the administration of rapamycin, but animals did not become anorexic and body weight was maintained (Table 1). Treated animals became more anemic but there was no evidence of cardiac dysfunction or dilatation. Indeed, trans-thoracic echocardiography showed that the LV ejection fraction was 65±1.9% in rapamycin-treated SIRI mice compared with 65±0.9% in vehicle-treated SIRI mice 8 weeks after surgery (P=0.73; Table 3). However, echocardiographic evaluation of mice 8 weeks after surgery revealed that rapamycin treatment blocked the development of cardiac hypertrophy in SIRI mice (Table 3). Indeed, the calculated LV mass index was 4.04±0.26 mg/g in vehicle-treated SIRI mice but was only 2.94±0.15 mg/g in rapamycin-treated SIRI mice (P=0.012). Morphological evaluation of mice 8 weeks after surgery revealed that the biventricular weight-to-tibial length ratio was 6.8±0.21 mg/g in vehicle-treated SIRI mice but was decreased by 17.8% to 5.8±0.20 mg/g in rapamycin-treated animals (P=0.001; Table 4). Similarly, the LV weight-to-tibial length ratio was 5.6±0.18 mg/g in vehicle-treated SIRI mice but was reduced to 4.6±0.2 mg/g in rapamycin-treated animals (P=0.005).
To determine whether rapamycin treatment affected cardiomyocyte size or cardiac fibrosis, we completed a histologic analysis of transverse ventricular tissue sections. Histologic evaluation of cardiomyocyte area showed that rapamycin treatment of SIRI mice resulted in an 87% decrease in cardiomyocyte area compared with vehicle treatment (P=0.001; Figure 1). The deposition of extracellular matrix was evaluated by Masson’s trichrome staining of ventricular transverse sections. Cardiac fibrosis decreased by 53% in rapamycin-treated SIRI mice compared with vehicle treatment (P<0.001; Figures 2). Analysis of ventricular cytosolic lysates revealed that phosphorylation of ribosomal S6 protein and 4E-BP1 was reduced in SIRI mice after rapamycin treatment when compared with vehicle treatment (P<0.001, P=0.008, respectively; Figure 5). Cardiac ERK1/2 and Akt phosphorylation was unaffected by rapamycin treatment (Figure 5).
Figure 5. Analysis of signaling protein phosphorylation in ventricular tissue obtained from surgically induced renal injury (SIRI) or sham mice treated with rapamycin or vehicle.
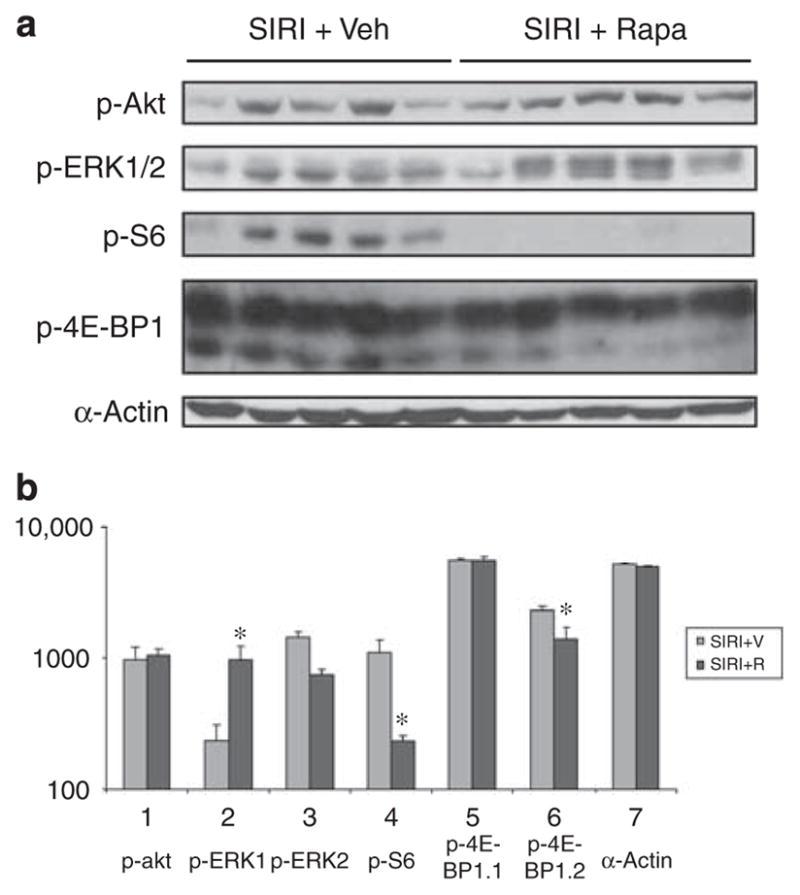
(a) Ventricular tissue obtained 8 weeks after SIRI in animals treated for 4 weeks with vehicle (Veh, n=5) or rapamycin (Rapa, n=5). Ventricular tissue was homogenized, protein lysates generated, and proteins were separated by SDS-PAGE followed by immunoblotting with primary antibodies. (b) Scanned blot densitometry was performed with Image J (v1.24) software. *P<0.05 vs SIRI+R.
DISCUSSION
Cardiac hypertrophy develops in patients with CKD and contributes to the morbidity and mortality of this condition.1 Although hypertension and volume overload may play a role in the development of uremic cardiomyopathy, more recent studies suggest that the accumulation of hypertrophic factors in serum, such as cardiotonic steroids, cytokines, growth factors, or hormones, may play a more central role.1 Previous work with animal models of uremic cardiomyopathy mainly focused on experimentation with rats. However, the genetic manipulation of rats is much more difficult and expensive than it is in mice. To date, there is one recent study that describes a mouse model of uremic cardiomyopathy with associated significant induction of hypertension.24
For this study, a hemodynamically controlled murine model of uremic cardiomyopathy was established. In the weeks following SIRI, mice developed uremia and a relatively mild reduction in glomerular filtration rate. Mice also developed a cardiomyopathy that consisted of cardiac hypertrophy, increased cardiomyocyte size, cardiac fibrosis, and altered cardiac tissue signaling pathway activation. Cardiac hypertrophy was detected by morphometry and transthoracic echocardiography. Altered cardiac signal transduction included increased activation of ERK1/2 protein kinase and increased ribosomal S6 protein and 4E-BP1 phosphorylation. The phosphorylation of S6 protein and 4E-BP1 are signs of increased mTOR activity. Despite prominent changes in cardiac structure and intracellular signal transduction, SIRI mice had systolic blood pressure that was comparable to sham controls as measured by cardiac catheterization 8 weeks after surgery.
In the murine model system described here, the left kidney was surgically removed, but a cautery technique was used to injure the right kidney so that blood flow was maintained in the remnant kidney. This technique leaves the renal artery and segmental arteries intact on the right side so that perturbation of the renin-angiotensin system is minimized.25 Injury by arterial ligation is frequently used in rat renal injury model systems and may result in more profound hypertension than the technique that was employed in the current work. For example, Griffin et al.26 demonstrated that renal mass reduction by cautery in rats did not alter blood pressure for 6 weeks after surgery. In the current work, mice subjected to SIRI did not develop systolic hypertension. Furthermore, mice treated with hydralazine developed significant cardiac hypertrophy despite a reduction in systolic blood pressure. Therefore, the cardiac hypertrophy and fibrosis that occurs in SIRI mice appears to be independent of systolic hypertension.
Although the development of cardiac hypertrophy in SIRI mice was not a consequence of pressure overload, the role of volume overload in this process must be considered.1 Chronic renal disease is associated with volume overload, and body weight is one measure of intravascular volume status. SIRI mice had similar body weights to sham-operated animals at both 4 and 8 weeks after surgery. Of note is the fact that SIRI mice had high urine output both 4 and 8 weeks after surgery.
To determine whether abnormalities in cardiac signaling in SIRI mice could help guide therapy for uremic cardiomyopathy, we treated animals with the mTOR inhibitor rapamycin. Previous work from Shioi et al.18 showed that daily intraperitoneal administration of rapamycin could block the development of cardiac hypertrophy in mice after aortic banding. Administration of rapamycin to SIRI mice from week 4 to week 8 after surgery blocked the development of cardiac hypertrophy and fibrosis. Vehicle had a minor effect on normalized heart weight. Therefore, mTOR activity is required for the development of uremic cardiomyopathy in this murine model system. The relative role of ERK activation in the pathogenesis of this condition remains uncertain. Furthermore, the present results do not address the nature of the initiating factor(s) that culminate in the development of cardiac hypertrophy, and it is unclear whether cardiotonic steroids, cytokines, growth factors, or other ligands that accumulate in CKD are of primary importance.
In conclusion, we established a murine model of normotensive uremic cardiomyopathy that developed in response to SIRI. Mice developed prominent cardiac hypertrophy with fibrosis that was associated with increased cardiac activation of the ERK and mTOR intracellular signal transduction pathways. Treatment of SIRI mice with rapamycin blocked the development of cardiac hypertrophy and fibrosis when compared with vehicle-treated animals. As organ transplant recipients with CKD often take rapamycin as an immunomodulatory drug to suppress organ rejection, it may be possible to determine whether patients on this medication exhibit reversal of established cardiac hypertrophy.
METHODS
SIRI in mice
All research involving the use of mice were performed in strict accordance with protocols approved by the Animal Studies Committee of Washington University School of Medicine. At 12 weeks of age, male 129/SvJ mice were randomized to undergo SIRI (SIRI; n=31) or sham surgery (n=14). Mice were anesthetized, and an incision in the right flank was performed followed by surgical dissection to expose the right kidney. Capsulectomy preceded unilateral ablation of 60–70% of the cortical surface of the right kidney by Bovie cauterization.25 After a 2-week recovery period, a left flank incision was made and left total nephrectomy in anesthetized mice was performed. Sham procedures involved abdominal incisions in the same flank locations of approach.
Quantitation of body fluid constituents and body composition
Serum aldosterone levels were determined by the University of Michigan Diagnostic Center for Population and Animal Health in Ann Arbor, Michigan. Serum angiotensin II was measured at the Medical College of Wisconsin by radioimmunoassay following HPLC separation of angiotensin I, angiotensin II, and the primary angiotensin metabolites as previously described by Mattson and colleagues.27 Calorie intake was measured with a metabolic cage apparatus over a 24-h period. Urine sodium was measured at Advanced Veterinary Laboratories in St. Louis, Missouri. Serum creatinine was measured by Stephen Dunn at Thomas Jefferson University, Philadelphia, Pennsylvania, by use of HPLC with the following modifications: (1) column was Zorbax 300-SCX, 5 micron, 2.1×150mm; (2) mobile phase was 7.5mM sodium acetate adjusted to pH 4.4 with acetic acid (total acetate ~15mM) at a flow rate of 0.50ml/min (isocratic); (3) column temperature of 40°C; and (4) sample injection volume was 3.0 μl.28 Lean body mass was measured by EchoMRI 3-in-1 (Echo Medical Systems, Houston, TX) at 8 weeks after SIRI. Afterward, extracellular volume was estimated by measuring the volume of distribution of sucrose using dilutional technique.29–31
Rapamycin administration after SIRI
A separate population of 17 animals subject to SIRI were monitored for 4 weeks after surgery and subsequently randomized to rapamycin (LC Laboratories, Woburn, MA, USA) injection or vehicle injection. We treated mice with rapamycin beginning 4 weeks after surgery. Those receiving rapamycin were administered daily subcutaneous abdominal injections of rapamycin (2 mg/kg/day) in 50% DMSO 50% saline vehicle.18 Seven control animals were administered daily injections of 75 μl of a DMSO/saline vehicle by use of the same technique.
Murine echocardiography
Transthoracic echocardiography was performed in mice that were awake by use of an Acuson Sequoia 256 Echocardiography System equipped with a 15-MHz (15L8) transducer as described previously (Acuson, A Siemens Company, Malvern, PA, USA).22,23 The echocardiographer was blinded in all cases to the experimental status of the mice.
Murine hemodynamic monitoring
Cardiac catheterization was performed on anesthetized mice as previously described.32,33 In brief, mice were anesthetized with ketamine and xylazine and a 1.4-French Millar catheter was placed in the left carotid artery then advanced into the left ventricle.
Cardiac morphometry and histology
Mice were weighed, and hearts and tibiae were isolated. Cardiac chambers were dissected and washed in 10mM PBS. Biventricular and LV weights were obtained. Tissue microtomes were selected to approximate the maximum coronal diameter of each heart. H&E and trichrome staining was performed. Myocyte cross-sectional area was then calculated using 3–5 random high power fields in the lateroanterior region of 4 different mice per group. This was performed with an Axioskop microscope (Carl Zeiss, Inc., Jena, Germany) with ImageJ (version 1.24) software.
Gene expression analysis
Quantitative real-time RT-PCR analysis on RNA extracted from heart lysates with Trizol reagent (Invitrogen Corp, Carlsbad, CA, USA) was carried out with the Taqman master mix kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s specifications. The measured abundances of atrial natriuretic factor and β-myosin heavy chain mRNA were normalized to GAPDH in each sample as an internal loading control.
Protein analysis
Cytosolic protein lysates from cardiac ventricular tissue were obtained as previously described.22,23
In brief, ventricular tissue was harvested from mice and prepared for immunoblot analysis. Primary antibodies employed included murine monoclonal anti-phospho-p42/44 (Thr202/Tyr204), anti-phospho- Akt (Ser473), anti-phospho-ribosomal s6 (Ser235/236), anti-phospho- ribosomal 4E binding protein 1 (4E-BP1) (Thr37/46), and anti-alpha-actin antibody. Filters were extensively washed in TBS/T and were then incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody (Amersham, a GE Healthcare Company, Pittsburgh, PA, USA). Bands were visualized by use of the ECL system (Amersham).
Statistical analysis
All statistical analysis was performed with SigmaStat-3.1 (Systat Software, Inc., San Jose, CA, USA). Statistical relationships were determined by the Student’s t-test, where significance was achieved when a population’s mean value was separated from controls by two or more standard deviations (P=0.05). All P values calculated were two-sided. Bonferroni correction was used in analysis of echocardiographic and morophometric data requiring 2.5 or more standard deviations from control values (P=0.021).
Acknowledgments
This work was supported by the Burroughs Wellcome Fund (AJM) and NIH grants RO1 HL061567 (AJM), P01 HL057278 (AJM), RO1 HL 076670 (AJM), and P30 DK079333 (AS, AJM). The authors acknowledge the assistance of the DDRCC Morphology Core Facility at Washington University (NIH DDRCC P30 DK52574). The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
References
- 1.London GN. Cardiovascular disease in chronic renal failure: pathophysiologic aspects. Semin Dial. 2003;16:85–94. doi: 10.1046/j.1525-139x.2003.16023.x. [DOI] [PubMed] [Google Scholar]
- 2.Foley RN, Parfrey PS, Harnett JD, et al. The prognostic importance of left ventricular geometry in uremic cardiomyopathy. J Am Soc Nephrol. 1995;5:2024–2031. doi: 10.1681/ASN.V5122024. [DOI] [PubMed] [Google Scholar]
- 3.Levin A, Singer J, Thompson CR, et al. Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am J Kidney Dis. 1996;27:347–354. doi: 10.1016/s0272-6386(96)90357-1. [DOI] [PubMed] [Google Scholar]
- 4.Stewart GA, Gansevoort RT, Mark PB, et al. Electrocardiographic abnormalities and uremic cardiomyopathy. Kidney Int. 2005;67:217–226. doi: 10.1111/j.1523-1755.2005.00072.x. [DOI] [PubMed] [Google Scholar]
- 5.Levin A, Thompson CR, Ethier J, et al. Left ventricular mass index increase in early renal disease: impact of decline of hemoglobin. Am J Kidney Dis. 1999;34:125–134. doi: 10.1016/s0272-6386(99)70118-6. [DOI] [PubMed] [Google Scholar]
- 6.Silberberg J, Barre PE, Prichard SS, et al. Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int. 1989;36:286–290. doi: 10.1038/ki.1989.192. [DOI] [PubMed] [Google Scholar]
- 7.Rambausek M, Ritz E, Mall G, et al. Myocardial hypertrophy in rats with renal insufficiency. Kidney Int. 1985;28:775–782. doi: 10.1038/ki.1985.197. [DOI] [PubMed] [Google Scholar]
- 8.Ayus JC, Mizani M, Achinger SG, et al. Effects of short daily versus conventional hemodialysis on left ventricular hypertrophy and inflammatory markers: a prospective, controlled study. J Am Soc Nephrol. 2005;16:2778–2788. doi: 10.1681/ASN.2005040392. [DOI] [PubMed] [Google Scholar]
- 9.Yamakawa H, Imamura T, Matsuo T, et al. Diastolic wall stress and ANG II in cardiac hypertrophy and gene expression induced by volume overload. Am J Physiol Heart Circ Physiol. 2000;279:H2939–H2946. doi: 10.1152/ajpheart.2000.279.6.H2939. [DOI] [PubMed] [Google Scholar]
- 10.Harnett JD, Kent GM, Foley RN, et al. Cardiac function and hematocrit level. Am J Kidney Dis. 1995;25(4 Suppl 1):S3–S7. doi: 10.1016/0272-6386(95)90673-8. [DOI] [PubMed] [Google Scholar]
- 11.Winchester JF, Audia PF. Extracorporeal strategies for the removal of middle molecules. Semin Dial. 2006;19:110–114. doi: 10.1111/j.1525-139X.2006.00135.x. [DOI] [PubMed] [Google Scholar]
- 12.Yutao X, Geru W, Xiaojun B, et al. Mechanical stretch-induced hypertrophy of neonatal rat ventricular myocytes is mediated by beta(1)-integrin-microtubule signaling pathways. Eur J Heart Fail. 2006;8:16–22. doi: 10.1016/j.ejheart.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Yamazaki T, Komuro I, Kudoh S, et al. Angiotensin II partly mediates mechanical stress-induced cardiac hypertrophy. Circ Res. 1995;77:258–265. doi: 10.1161/01.res.77.2.258. [DOI] [PubMed] [Google Scholar]
- 14.Yamazaki T, Komuro I, Kudoh S, et al. Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem. 1996;271:3221–3228. doi: 10.1074/jbc.271.6.3221. [DOI] [PubMed] [Google Scholar]
- 15.Proud CG. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc Res. 2004;63:403–413. doi: 10.1016/j.cardiores.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy DJ, Vetteth S, Periyasamy SM, et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495. doi: 10.1161/01.HYP.0000202594.82271.92. [DOI] [PubMed] [Google Scholar]
- 17.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol. 2007;293:C509–C536. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 18.Shioi T, McMullen JR, Tarnavski O, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- 19.Ma L, Teruya-Feldstein J, Bonner P, et al. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007;67:7106–7112. doi: 10.1158/0008-5472.CAN-06-4798. [DOI] [PubMed] [Google Scholar]
- 20.Chen LY, Li P, He Q, et al. Transgenic study of the function of chymase in heart remodeling. J Hypertens. 2002;20:2047–2055. doi: 10.1097/00004872-200210000-00025. [DOI] [PubMed] [Google Scholar]
- 21.Xu J, Carretero OA, Lin CX, et al. Role of cardiac overexpression of ANG II in the regulation of cardiac function and remodeling postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H1900–H1907. doi: 10.1152/ajpheart.00379.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren J, Zhang S, Kovacs A, et al. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005;38:617–623. doi: 10.1016/j.yjmcc.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Harris IS, Zhang S, Treskov I, et al. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 2004;110:718–723. doi: 10.1161/01.CIR.0000138190.50127.6A. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy DJ, Elkareh J, Shidyak A, et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol. 2008;294:F450–F454. doi: 10.1152/ajprenal.00472.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gagnon RF, Gallimore B. Characterization of a mouse model of chronic uremia. Urol Res. 1988;16:119–126. doi: 10.1007/BF00261969. [DOI] [PubMed] [Google Scholar]
- 26.Griffin KA, Picken M, Bidani AK. Method of renal mass reduction is a critical modulator of subsequent hypertension and glomerular injury. J Am Soc Nephrol. 1994;4:2023–2031. doi: 10.1681/ASN.V4122023. [DOI] [PubMed] [Google Scholar]
- 27.Cholewa BC, Mattson DL. Role of the renin-angiotensin system during alterations of sodium intake in conscious mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R987–R993. doi: 10.1152/ajpregu.2001.281.3.R987. [DOI] [PubMed] [Google Scholar]
- 28.Dunn SR, Qi Z, Bottinger EP, et al. Utility of endogenous creatinine clearance as a measure of renal function in mice. Kidney Int. 2004;65:1959–1967. doi: 10.1111/j.1523-1755.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- 29.Pierson RN, Price D, Wang J, et al. Extracellular water measurements: organ tracer kinetics of bromide and sucrose in rats and man. Am J Physiol. 1978;235:254–264. doi: 10.1152/ajprenal.1978.235.3.F254. [DOI] [PubMed] [Google Scholar]
- 30.Caster WO, Simon AB. A comparison of sodium, chloride, thiocyanate, and sucrose spaces as estimates of extracellular fluid volume. Physiol Chem and Physics. 1980;12:205–213. [PubMed] [Google Scholar]
- 31.Iwakawa T, Ishihara H, Takamura K, et al. Measurements of extracellular fluid volume in highly perfused organs and lung water in hypo- and hypervolaemic dogs. Eur J Anaesthesiol. 1998;15:414–421. doi: 10.1097/00003643-199807000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Rogers JH, Tamirisa P, Kovacs A, et al. RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J Clin Invest. 1999;104:567–576. doi: 10.1172/JCI6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren J, Avery J, Zhao H, et al. Beta3 integrin deficiency promotes cardiac hypertrophy and inflammation. J Mol Cell Cardiol. 2007;42:367–377. doi: 10.1016/j.yjmcc.2006.11.002. [DOI] [PubMed] [Google Scholar]