


Abstract
Ribosomal RNA (rRNA) genes are probably the most frequently used data source in phylogenetic reconstruction. Individual columns of rRNA alignments are not independent as a consequence of their highly conserved secondary structures. Unless explicitly taken into account, these correlation can distort the phylogenetic signal and/or lead to gross overestimates of tree stability. Maximum likelihood and Bayesian approaches are of course amenable to using RNA-specific substitution models that treat conserved base pairs appropriately, but require accurate secondary structure models as input. So far, however, no accurate and easy-to-use tool has been available for computing structure-aware alignments and consensus structures that can deal with the large rRNAs. The RNAsalsa approach is designed to fill this gap. Capitalizing on the improved accuracy of pairwise consensus structures and informed by a priori knowledge of group-specific structural constraints, the tool provides both alignments and consensus structures that are of sufficient accuracy for routine phylogenetic analysis based on RNA-specific substitution models. The power of the approach is demonstrated using two rRNA data sets: a mitochondrial rRNA set of 26 Mammalia, and a collection of 28S nuclear rRNAs representative of the five major echinoderm groups.
INTRODUCTION
Ribosomal RNAs (rRNAs) are the most widely used source of phylogenetic information, although protein-coding genes, often derived from Expressed Sequence Tag (EST) sequencing or from sequencing complete mitogenomes, have provided an increasingly large amount of new genomic data. The SSU and LSU rRNA genes have been sequenced for thousands of taxa throughout the metazoan kingdom, providing a much denser taxon coverage than what is available for any particular protein-coding gene. Since sequence conservation varies dramatically between different regions of rRNA genes, these data are informative on a wide range of phylogenetic time scales, ranging from recent to ancient splits (1,2).
This variation in substitution rates, however, is also a major technical obstacle for using rRNA in molecular phylogenetics. The correct assignment of homologous characters, i.e. alignment columns, is the crucial first step in molecular systematics on which all subsequent analyses depend. The high variability of substitution rate along the sequence, combined with similar variations in insertion and deletion rate, makes it impossible in practice to construct unambiguous alignments of the more variable regions by means of standard sequence alignment programs.
The rRNAs, however, are highly structured, with large parts of the molecules exhibiting very strong conservation of their base pairing patterns. Therefore, it is natural to improve alignment accuracy by incorporating secondary structure conservation. Indeed, this approach has been advocated repeatedly in the literature, e.g. (3–7). In practise, however, the application of this idea has remained a hard and tedious task, mostly because of the difficulties in obtaining a correct structural annotation. If good structure annotations were readily available, we could simply employ one of the alignment tools that explicitly incorporate secondary structure information (8–15).
The accuracy of thermodynamic folding algorithms falls sharply as the length of the RNA increases. Secondary structures can be computed with acceptable precision based on experimentally measured thermodynamic parameters only for short fragments with a length 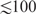 nt (16,17). This limitation is in part due to inaccuracies in the ‘nearest neighbor model’ and its parameters (18,19), in part caused by the kinetics of folding process and tertiary interactions (20). Even more importantly, the RNA and protein components of the ribosome are tightly packed and thus mutually influence their folds (21). The functional rRNA structures, therefore, cannot reasonably be expected to be identical with the structures of isolated rRNAs—which is what the thermodynamic folding algorithms compute.
nt (16,17). This limitation is in part due to inaccuracies in the ‘nearest neighbor model’ and its parameters (18,19), in part caused by the kinetics of folding process and tertiary interactions (20). Even more importantly, the RNA and protein components of the ribosome are tightly packed and thus mutually influence their folds (21). The functional rRNA structures, therefore, cannot reasonably be expected to be identical with the structures of isolated rRNAs—which is what the thermodynamic folding algorithms compute.
The latter effect can be captured at least in part by considering patterns of sequence covariation that provide information on evolutionarily conserved base pairs. The RNAalifold approach, for instance, has demonstrated that the accuracy of biological structure predictions can be increased to acceptable levels by using the consensus structures of a set of closely related sequences and by explicitly taking information on base pair covariation into account (22,23). Designed for relatively closely related sequences, RNAalifold unfortunately requires a sequence alignment as input.
RNAsalsa is designed to overcome this limitation by combining the prediction of consensus structures of closely related sequences with prior knowledge that constrains the set of acceptable structures. Consensus structures for groups of related sequences are used to generate high-quality alignments by funneling structure information into the alignment scoring function. Thus, RNAsalsa uses both structure information for adjusting and refining the sequence alignment and sequence information contained in the alignment to refine the structure predictions. We designed RNAsalsa primarily for phylogenetic applications. In this context, RNA secondary structure is of importance at two levels: first, changes in secondary structures can be useful phylogenetic markers in their own right (24). Clearly, accurate structure predictions are a necessary prerequisite to utilize structural differences in this way. Second, knowledge about conserved secondary structures allows the use of more detailed models of RNA sequence evolution. In this contribution we focus on the latter aspect.
RNA-specific substitution models (25–30) have been introduced to capture the effects of covariation among paired sites of rRNA sequences. Slightly deleterious substitutions at one side of a helix, which would disrupt the structure, are frequently compensated by a second substitution at the pairing site, so that the pairing ability (31) is restored. This leads to a strong correlation of paired positions within rRNA sequences. The corresponding alignment columns, therefore, do not display independent phylogenetic information. Since paired sites are strongly correlated but treated as independent, phylogenetic information is scored twice, leading to unjustified high support for some trees and erroneously low support for alternative trees (32,33). The application of RNA-specific substitution models for phylogenetic analyses requires a structural annotation of the input alignment. It is important that in particular the regions that are treated as paired are aligned in a structurally correct way because the effect of substitutions is evaluated with respect to specific column pairs.
The RNAsalsa software, which is implemented in C, is specifically optimized for phylogenetic applications. The source code as well as pre-compiled executables for various platforms, together with a detailed manual providing some guidelines for practical use, may be downloaded from http://www.rnasalsa.zfmk.de/ and http://www.bioinf.uni-leipzig.de/Software/RNAsalsa.
MATERIALS AND METHODS
Workflow and algorithms
RNAsalsa implements a workflow that makes use of several well-established algorithms for both RNA secondary structure prediction and structure enhanced alignment (Figure 1). It consists of three major stages: (i) the determination of structural constraints for each input sequence that emphasize the common features; (ii) the computation of a detailed structural model for each input sequence; and (iii) the construction of a final multiple sequence alignment together with a consensus structure.
Figure 1.

Overview of the RNAsalsa workflow. Starting from an initial sequence alignment 𝔸0 and a structure constraint σ0, a relaxed consensus constraint and then constraints for each input sequence are produced. Pairwise sequence alignments and the intersections of the two individualized constraints are used to compute a collection of constrained pairwise consensus structures τij, from which a refined individualized structure constraint τi is extracted by means of a majority voting procedure. Then τi is used as constraint in minimum energy folding to obtain the final structural annotation ψi for input sequence xi. The structure-annotated input sequences are now aligned with a structure-aware alignment method, resulting in the final alignment 𝔹 and a corresponding consensus structure ω.
The starting point for RNAsalsa is an initial alignment 𝔸0 of a collection {x1, …, xN} of homologous RNA sequences (produced e.g. simply by clustalW) and an a priori secondary structure constraint σ for a single sequence x0 which is contained in the alignment 𝔸0. The sequence x0 and its structural model are used only to initialize the structure prediction and alignment process.
In the first step, RNAsalsa checks the consistency of the initial alignment 𝔸0 and the initial constraint σ0: for each base pair in σ0, we evaluate whether the corresponding aligned positions of a sufficient number of sequences in 𝔸0 can also pair. If so, we retain the base pair, otherwise it is removed from the constraint. The resulting ‘relaxed’ constraint
| 1 |
can be seen as base pair-wise filtering of the initial constraint σ0 that removes pairs from σ0 that are largely inconsistent with the initial alignment. Projecting the relaxed constraint σ separately onto each aligned sequence (i.e. retaining only the canonical base pairs of σ that can be formed by the input sequence xi ), then produces distinct initial structure constraints for each sequence:
| 2 |
Up to this point, the result heavily depends upon the initial alignment. It may not cover the input sequences uniformly, in particular it will often be concentrated on the well-conserved (and therefore properly aligned) regions.
In the second step, RNAsalsa utilizes the improved accuracy of the predicted consensus structures. To this end, we construct a collection of pairwise sequence alignments 𝔸ij from the input sequences xi and x j. These can be constructed in different ways, either by dynamic programming alignment, or by projecting the corresponding sub-alignment of 𝔸0. For details we refer to the RNAsalsa manual, which is part of the Supplementary Data. For each of the pairwise alignments 𝔸ij we compute the consensus minimum free-energy structures
| 3 |
using the base pairs common to the projected structures σi and σj, respespectively, as constraint. This step uses the Vienna RNA Package library functions underlying RNAalifold (22) to perform the constrained folding computations.
For each sequence xi, the collection of structures {τij, i ≠ j} taken together defines a set of base pairs on xi that are both thermodynamically plausible and conserved in at least one other sequence of the input set. From this set of pairs, we select a single secondary structure
| 4 |
for sequence xi using a majority voting procedure. RNAsalsa currently implements a simple greedy procedure that selects the most frequent base pairs first and rejects pairs that would cross previously selected ones to avoid the formation of pseudo-knotted structures. Alternatively, one could also use Nussinov's Maximum Circular Matching algorithm (34) to retrieve a maximum weight sub-set of non-intersecting pairs. The base pairs of τi, which by construction typically contain most of the initial constraint-derived pairs σi, are now used as a constraint for computing the final secondary structure prediction
| 5 |
for each input sequence xi. Again, we employ the Vienna RNA Package library here.
The purpose of the entire—rather complex and computationally expensive—procedure is to use as much information as possible in guiding the last step, the computation of the secondary structure models ψi for each input sequence. This guiding information is derived from two sources: the initial structural constraint σ and the ensemble of plausible base pairs generated from all pairwise alignments.
In the next step, the sequence–structure pairs (xi, ψi) are realigned. To this end, RNAsalsa uses a hierarchical progressive alignment based on pairwise dynamic programming alignments with affine gap costs (35). The scoring function explicitly incorporates the secondary structure annotation: the (mis)match score s(xi, yj) of position i from sequence x with position j from sequence y is defined as follows:
| 6 |
where xπ(i) and yπ(j) denote the pairing partners of xi and yj in their respective secondary structures. The coefficient b0 = 1 if both xi and yj are paired nucleotides. The coefficient b1 is set to 1 if x and y share sufficient structural conservation to a certain extent that overcame the precedent filtering steps and if xπ(i) and yπ( j) are located either both upstream or both downstream of xi and yj, respectively. Otherwise the structural contribution is ignored, b1 = 0. Finally, if one xi or yj are unpaired, then b0 = b1 = 0 and c = 1. In regions without structural information we therefore use a pure nucleic acid sequence score sp, where as in structured regions, the modified scoring functions sm and sn are used. For instance, within trusted structural regions A-G is scored as a match because both may pair with U, while it is not in regions without sufficiently trusted structural information. Default scoring tables are listed in the manual and Supplementary Data. The final result is a global re-alignment 𝔹 of the input sequences which integrates all the secondary structure information obtained in the previous steps.
The individual folds ψi and the alignment 𝔹 are used to derive a consensus structure
| 7 |
Since we now have the trusted alignment 𝔹, we can again employ a simple voting strategy: we start from the set of all base pairs that appear sufficiently often in superposition of the ψi. Again, we use a greedy strategy to avoid conflicting base pairs (note that no conflicts can arise if we consider only base pairs that occur at least N/2 times).
Several parameters can be adjusted in the process. In particular, the stringency of the initial filtering of base pairs, Equation (1), and the two majority voting procedures, Equations (4) and (7) can be adjusted by the user to the peculiarities of the data sets. In each case, a threshold for the minimum number of consistent pairs can be specified. Some guidelines for practical use can be found in the RNAsalsa manual.
RESULTS
Secondary structure prediction
Performance of RNAsalsa's structure predictions was evaluated in comparison with three other relevant methods: MXSCARNA (15) computes pairing probabilities and considers potential stem information in the subsequent alignment process; RNAfold (17) produces individual secondary structures of RNA sequences; and RNAalifold (22) generates the consensus structure for a given input alignment. We compared RNAfold predictions with RNAsalsa's individual predictions ψi (Equation 5), while the MXSCARNA and RNAalifold results are compared with RNAsalsa's final consensus structure ω. As a reference model we employed the mammalian 16S rRNA secondary structure adopted from (36) (Figure 2).
Figure 2.

Accuracy of structure prediction. Fraction of correctly predicted helices (green bars) compared with the mammalian 16S rRNA consensus models (36). (A) RNAsalsa significantly outperforms MXSCARNA and RNAalifold (default parameter settings; three-sample test for equality of proportions without continuity correction; χ2 = 19.96, df = 2, P < 0.0001). Average tree-edit distance (37) (B) between predicted individual structures and the mammalian 16S rRNA reference model. RNAsalsa predictions conform the consensus model much better (paired sampled t-test; t = 33.46, df = 1, P < 0.0001; N = 26).
The RNAsalsa secondary structure model for the mammalian 16S rRNA sequences is highly congruent to the Bos taurus reference model proposed by Burk et al. (36), see Supplementary Data for an illustrating graphical representation. In particular, 44 of the 52 helices within the conserved core of the structure are correctly predicted. Much of the remaining discrepancy can be explained by differences in the stringency of rules (Figure 2A). While the literature reference was derived with very conservative rules, we allow more variability among the organisms, and accept stems even if there is no direct evidence from compensatory substitutions. MXSCARNA and RNAalifold capture only 27 and 23 helices, respectively. In contrast to RNAsalsa, they failed to detect in particular long-range interactions.
Furthermore, to demonstrate the capabilities of constraints in thermodynamic folding algorithms, we compared the results of unconstrained foldings by the RNAfold software (17) only with those generated by RNAsalsa. In both cases, we used the same thermodynamical parameter sets and algorithms as implemented in RNAfold. The only difference is the usage of folding constraints in the case of RNAsalsa. These structure constraints are automatically generated and optimized for each RNA sequence within the data set.
RNAsalsa's predictions of the individual structures always use an individual structure constraint that was optimized by RNAsalsa's procedure itself up to that point. Therefore, they can match the reference model much better than thermodynamic folds by RNAfold, which have been calculated without supporting constraints (Figure 2B). See Supplementary Data for a set of comparisons performed on 16S rRNA.
Structure-aware RNA sequence alignments
The impact of secondary structures on the alignments as well as the overall performance was investigated by comparison with two commonly used sequence alignment methods, the classical ClustalW (38) and the more modern MAFFT (39) approach, and with the structural alignment method MXSCARNA. For this purpose we employed both simulated and real data sets.
Manually curated alignments for rRNAs are a rare commodity. At present, only partial data sets are available. Here, we used the alignments of 26 mammalian mitochondrial rRNAs published in (40,41). We only compared the subset of alignment positions that are marked as ‘trusted’ in these alignments. For the 16S rRNAs, we find that all four alignment programs reproduce 97.5–99% of the ‘trusted’ pairwise (mis)matches. The two structural alignment programs, MXSCARNA and RNAsalsa, have a slightly increased fraction of ‘trusted’ (mis)matches, Table 1. Very similar results were observed for the 12S mitochondrial rRNAs from the same source.
Table 1.
| Method | No. of (mis)matches | ‘Trusted ’ | Fraction |
|---|---|---|---|
| Kjer reference | 492 456 | 375 062 | 0.7616 |
| clustalW | 500 357 | 366 840 | 0.7330 |
| MAFFT | 503 628 | 371 031 | 0.7367 |
| MXSCARNA | 496 447 | 370 020 | 0.7453 |
| RNAsalsa | 491 931 | 365 574 | 0.7431 |
We list the total number of (mis)matches in all pairwise sub-alignments of the multiple sequence alignment, and the subset of (mis)matches that coincide with those marked as ‘trusted’ in the reference alignments.
The relative quality of alignments was assessed following the procedures outlined in (42,43). In particular, we calculated the sum of pairs score (SPS) and the total column score (TC) as implemented in the baliscore software (42), as well as the structure conservation index (SCI) (44). Both SPS and TC are similarity metrics for multiple alignments. While SPS evaluates the number of equivalently aligned residue pairs summed over all pairs of sequences, TC counts the number of columns exactly shared by the two alignments. When one of the alignments is used as a benchmark reference, both SPS and TC can be interpreted as measures of alignment quality. The SCI, on the other hand, measures to what extent the aligned sequences form a common secondary structure in such a way that the sequence alignment also represents a structural alignment. Originally introduced as a measure of structural conservation given the alignment, it was used in (45) to measure the ability of alignment algorithms to reconstruct conservative consensus folds. Of course, the SCI can only be employed to measure alignment quality when the existence of a common global consensus structure can safely be assumed.
Simulated data were generated as reference alignments using the new software rnasim (46). The tool takes both a structure annotated sequence string and a pre-defined phylogenetic tree as input and simulates the evolution of RNA molecules with fitness constraints derived from thermodynamic stability along the given tree. The tool provides us with the true reference alignment of the leaf sequences at the end of each simulation run. Several sets of examples representing smaller and larger RNAs (tRNAs, mammalian 12S rRNAs, and the Saccharomyces cerevisiae 28S rRNA) were used as roots. We used various values between 1 and 100 000 for rnasim's ‘branch scaling’ option -s, whereas the default was used for all other options. Since the branch scaling parameter has a strong influence on both sequence and structure, we systematically analysed how various alignment algorithms behave as a function of divergence.
Table 2 summarizes the results of some of the RNAsim-derived data sets. For the tRNAs, all programs except clustalW perform similarly well. In case of the 28S rRNA data set, which constitutes a very good example with highly divergent sequences, RNAsalsa performs slightly better than its competitors, albeit on a low level. These examples illustrate a general pattern: most programs, including RNAsalsa, perform with comparable scores on a wide range of test cases.
Table 2.
Benchmark results for different alignment programs on rnasim-simulated data sets initialized with tRNAs and 28S rRNAs, respectively
| Methoda | tRNAs |
LSU rRNA |
||||
|---|---|---|---|---|---|---|
| SPS | TC | SCI | SPS | TC | SCIb | |
| RNAsalsa | 0.92 | 0.69 | 0.92 | 0.57 | 0.11 | 0.18 |
| MAFFT | 0.89 | 0.65 | 0.80 | 0.55 | 0.05 | 0.11 |
| ClustalW | 0.84 | 0.51 | 0.38 | 0.55 | 0.06 | 0.13 |
| MXSCARNA | 0.94 | 0.77 | 0.88 | NA | NA | NAc |
aAll algorithms were started with default set-ups.
bAll applied score values approach 1 as the alignments become identical with the reference.
cWe could not get results using MXSCARNA with LSU rRNA.
As a function of the branch length scaling parameter, we observe that all alignment algorithms perform comparably well or poorly in terms of the SPS, TC and SCI values. As expected, when the alignment problem becomes harder, i.e. for large scaling values, the performance metrics decrease uniformly (Supplementary Data). The resulting alignment then becomes more and more divergent between different methods.
We also used the BRAliBase-II set of structural alignments (http://projects.binf.ku.dk/pgardner/bralibase/bralibase2.html) (42), which covers group II introns, 5S rRNA, tRNA, U5 and SRP RNA [following (42), we did not use the SRP RNA alignments]. Again, we observe no major differences in the performance metrics (Supplementary Data). As in the case of the simulated data, the alignments become more divergent between methods as the problems become harder.
It appears that they tend to deviate from the reference in different features. RNAsalsa avoids poorly supported (mis)matches and instead tends to introduce more gaps in regions where the alignment becomes ambiguous. While this feature does not positively influence the scores in usual performance metrics, it does have a desirable effect for phylogenetic reconstruction, because gap-rich regions tend to be down-weighted or even excluded in typical phylogenetic applications. In that sense, RNAsalsa apparently reduces conflicting information arising from spurious (mis)matches. The RNAsalsa alignments emphasize the well-supported regions, and—as we have seen in the previous section—lead to quite accurate assignments of the most conserved secondary structure elements.
Exemplary applications in phylogeny reconstruction
In order to demonstrate the usefulness of RNAsalsa in phylogenetic applications, we consider two distinct data sets in detail. For each alignment, we performed phylogenetic analyses using a likelihood-based approach and compared the results with the published analyses of the data set. After individually aligning the LSU and SSU sequences, the alignments were concatenated. Then the program Aliscore (47), a method to identify ambiguously aligned regions in multiple sequence alignments, was used to extract the informative parts of the alignment. Aliscore identifies ambiguously aligned regions in multiple sequence alignments based on comparisons of aligned sequences in a sliding window and a Monte Carlo (MC) approach. The MC re-sampling compares the score of the originally aligned sequences in a given window position with scores of randomly drawn sequences of similar character composition. Maximum likelihood (ML) analysis was conducted using the RAxML software under a GTR+Γ model set up. For further details we refer to the Supplementary Data.
Primates
Tree reconstruction results of the mammalian data are shown in Figures 3 and 4. We focus here on the phylogeny of primates and, in particular, on the exact phylogenetic position of one of the most basal primates, the tarsier. To this end, we re-evaluate a data set specifically compiled for this purpose (48).
Figure 3.

Bayesian tree inferred from the combined mammalian 12S rRNA and 16S rRNA. (A) Analysis with GTR + Γ model in simple DNA mode. (B) Analysis with GTR + Γ model in RNA mode for paired positions and DNA mode for loop regions. Numbers indicate Bayesian posterior probabilities. The scale bar denotes the estimated number of substitutions per site.
Figure 4.

Phylogenies inferred from combined analyses of the mammalian 12S rRNA and 16S rRNA. Sequences are aligned with (A) RNAsalsa, (B) MAFFT, (C) ClustalW and (D) MXSCARNA. Tree reconstruction is based on ML analyses with GTR + Γ model. Numbers indicate Bootstrap support values (1000 replicates). The scale bar denotes the estimated number of substitutions per site.
Molecular studies on mitochondrial and nuclear DNA data of primates have so far lead to incongruent results. Nuclear DNA data favour haplorrhines (‘dry-nosed’ primates), i.e. the grouping of anthropoids (human-like apes) and tarsier (49). This hypothesis has gained strong support by the discovery of haplorrhine-specific SINES (50,51). Mitochondrial data, in contrast, mostly support the Prosimian hypothesis that postulates a sister group relationship of Tarsius and strepsirrhines (‘wet-nosed’ primates) (52,53,48).
The ML analysis based on the RNAsalsa alignment shows well-supported monophyletic primates (Figure 4). In contrast, primates do not appear monophyletic in analyses that use other alignments. The MAFFT alignment does not provide any phylogenetic signal to display relationships between anthropoids, the strepsirrhine representative Nycticebus, Tarsius and all remaining mammalian groups. The ClustalW analysis groups Tupaia, a scandentian representative, within primates as sister taxon to Tarsius, both forming the sister clade to Nycticebus and anthropoids. In the MXSCARNA analysis, primates appear paraphyletic with nested Rodentia.
Within primates, Tarsius appears as sister taxon to anthropoids in the RNAsalsa alignments, although with weak bootstrap support. This is also the case for the MAFFT alignment, albeit on the background of largely unresolved mammals. The MXSCARNA alignment leads to well-supported haplorrhines.
Although the placement of the tarsier is only weakly corroborated in the RNAsalsa analysis, these results show that the inclusion of good secondary structure models into the alignment procedure can make a significant difference for phylogeny reconstruction. RNAsalsa performs better than both purely sequence-based alignment approaches and sequence–structure alignments that are based directly on thermodynamic structure predictions.
The non-monophyletic appearance of primates with nested Scandentia and Rodentia in the MAFFT, ClustalW and MXSCARNA analyses must be interpreted as erroneous. A few studies based on mitochondrial genes propose paraphyletic primates with nested Dermoptera (53,54), but this observation has been explained as an effect of base composition bias in the mitochondrial markers (55). Scandentia or even Rodentia never appeared within primates to our knowledge.
An analysis of the whole mitochondrial genome of mammals revealed that heterogeneous substitution rates among different mammalian groups lead to misleading phylogenetic signals in mitochondrial genes (48). Their support for the prosimian hypothesis is thus likely an artefact. RNAsalsa apparently corrects this effect and leads to phylogenies from mitochondrial RNAs that are congruent with the results for nuclear genes.
We compared RNA-specific substitution models with simpler DNA models to determine to what extent they influence topology and/or node support of phylogenetic trees. Unfortunately, RNA substitution models are not implemented in any of the available ML software. They can, however, be used in Bayesian inference software. Here we used the parallel version of MrBayes (version 3.1.2) (56) with the GTR version of the Schoeniger and von Haeseler RNA model as described in (25) to account for character covariance. This model can be used to take interdependence of stem regions in ribosomal sequences into account. It assumes that a base pairing is converted into another one by means of a two-step process via an intermediate state. As this includes no double substitutions, other nucleotide changes are accounted for according to standard DNA models. Character covariation is considered, as the survival rate of a mutation in stem positions depends on the partner nucleotide, i.e. the substitution of the G in a UG pair by an A has a higher survival rate than the substitution by a C. As we understand it, this model currently represents a realistic and applicable approach to reflect nucleotide interdependencies in rRNA sequences. We have, however, opted for MrBayes over the alternative, PHASE (32,33,57), mostly because MrBayes provides a parallel version, which significantly reduces computation time in practice.
Bayesian inference results of the mammalian data set are shown in Figure 3. Application of simple DNA models led to a paraphyletic appearance of primates. Tupaia is a sister taxon to the strepsirrhine Nycticebus, both forming the first branching clade within the paraphyletic primates. In contrast, the application of mixed RNA/DNA models shows monophyletic primates with at least moderate nodal support. In both analyses, Tarsius appears highly supported as the sister taxon to anthropoids, forming monophyletic haplorrhines. Again, the monophyly of primates in the mixed model analysis can be interpreted as a hint that this approach performs better than the application of simple DNA models. These results corroborate the previously proposed superiority of the mixed model approach over simple DNA models (33).
Echinoderms
Our second example tackles the question of inter-class relationships in Echinodermata (Figure 5). This phylum is composed of five extant classes, the Crinoidea (sea lilies), Ophiuroidea (brittle stars), Asteroidea (starfishes), Holothuroidea (sea cucumbers) and Echinoidea (sea urchins). Monophyly in these five classes is well founded. The relationships between the five classes remain subject of ongoing discussion, however.
Figure 5.

Phylogenies inferred from analyses of the echinoderm 28S rRNA. Sequences are aligned with (A) RNAsalsa, (B) MAFFT, (C) ClustalW and (D) MXSCARNA. Tree reconstruction is based on ML analyses with GTR + Γ model. Numbers indicate Bootstrap support values (1000 replicates). The scale bar denotes the estimated number of substitutions per site.
Several contradicting hypotheses of inter-class phylogeny in Echinodermata have been raised in the past, based on morphological and molecular data. Nevertheless, there is some consensus regarding major aspects of echinoderm phylogeny (58–60). Crinoids are mostly seen as the most basal split within Echinodermata, forming the sister group to the four remaining classes (Eleutherozoa). Furthermore, there is a strong support for a sister group relationship of echinoids and holothurians (Echinozoa). Debates on the phylogenetic position of the stellate forms (starfishes and brittle stars) recently ended up in two competing hypotheses: are the ophiurids alone sister group to Echinozoa (61,62) or do asteroids and ophiuroids form a clade (Asterozoa), which is then the sister taxon to Echinozoa (60)?
Likelihood analyses based on different alignment methods are congruent only in parts of the resulting phylogenies. The sea lily species Florometra is the first split within monophyletic Echinodermata, and the two sea urchins Arbacia and Strongylocentrotus correctly appear monophyletic with highest bootstrap support.
There are, however, striking differences in many other aspects. The RNAsalsa alignment shows monophyletic Echinozoa with Cucumaria as sister taxon to the two echinoids. The starfish Asterias appears as sister taxon to the brittle star Ophioderma. These monophyletic Asterozoa are the sister clade to Echinozoa. All mentioned relationships gain highest bootstrap support. The MAFFT alignment also show monophyletic Asterozoa but with lesser support. Furthermore, there is no phylogenetic signal to resolve relationships between Asterozoa, Echinoidea and Holothuroidea. The ClustalW analysis does not show monophyletic Asterozoa and Echinozoa. Instead, there is a closer relationship between echinoids and Asterias. Within Eleutherozoa, the ophiuriod Ophioderma is the first split, followed by Cucumaria and the Asterias + Echinoidea clade.
The results of the MXSCARNA analyses are comparable with those of RNAsalsa. Eleutherozoa, Echinozoa and Asterozoa are monophyletic, the latter ones with lesser support than in the RNAsalsa analyses. Compared with the previous studies on echinoderm phylogeny, the results of the structural alignment methods must be seen as more reasonable. In particular, based on monophyletic Echinozoa their superiority over the two exclusively sequence-based alignment methods is pointed out. Both of those fail to recover monophyletic Echinozoa and the ClustalW alignment erroneously show Asterias as sister taxon to the sea urchins.
Overall, we find that structure-aware alignments yield more plausible results than purely sequence-based alignments. RNA-specific substitution models yield better results with the RNAsalsa alignments (which incorporate some prior knowledge on the structure) than structural alignments that are based entirely on unconstrained thermodynamic folding.
DISCUSSION
ML analyses and Bayesian inference both revealed a remarkable influence of rRNA secondary structure consideration on both the sequence alignment and on the subsequent tree reconstruction. This phenomenon is well-known in molecular systematics and has already led to the development of RNA-specific substitution models. The application of these models, however, is confined to a few studies (3,5,32,33,63,64), mostly because of the lack of a convenient way to construct usable secondary structure models and alignments for newly sequenced rRNAs. Structural inference is not only difficult but also very tedious, and hence avoided in the overwhelming majority of published studies.
As a tool for simultaneously computing structure annotation and structure-aware sequence alignments of large RNA molecules, RNAsalsa has been designed specifically to overcome this barrier. While it can also be useful for other tasks, its primary domain of application is phylogenetic inference. Here the relatively large computational cost of the structure prediction (compared with other, less accurate tools) is of little concern, since it is dwarfed by the demands of subsequent ML or Bayesian computations. Extensive tests, and two real-world applications, demonstrate that RNAsalsa can lead to significant improvements in reconstructed phylogenies, positively affecting both tree stability and tree topology. These improvements can be traced back to two sources: alignments that emphasize the unambiguous regions improve the phylogenetic signal; second, more exact automatically generated consensus structures enhance the benefit of RNA-specific substitution models. As our examples show, both types of improvements can offset the problems incurred by unequal substitution rates and long branches.
The modular structure of RNAsalsa lends itself to incorporate further improvements. For example, it is likely beneficial to use a Sankoff-style algorithm such as foldalign (65) or locarna (13) to construct the pairwise alignments 𝔸ij and/or the final alignment 𝔹 and its consensus structure ω. The current version of RNAsalsa consistently generates alignments and consensus structures of acceptable quality (compared with the extremely tedious manual curation of such data). It is therefore suitable for routine applications in molecular phylogenetics based on structured RNAs, in particular rRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (under the auspices of SPP-1174 ‘Deep Metazoan Phylogeny’, Projects STA 850/2-1, STA 850/3-1, as well as MI 649/3 and MI 649/6.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Woese CR. Bacterial evolution. Microbiol. Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hillis DM, Dixon MT. Ribosomal DNA: molecular evolution and phylogenetic inference. Q. Rev. Biol. 1991;66:411–453. doi: 10.1086/417338. [DOI] [PubMed] [Google Scholar]
- 3.Kjer KM. Use of ribosomal-RNA secondary structure in phylogenetic studies to identify homologous positions – an example of alignment and data presentation from the frogs. Mol. Phylogenet. Evol. 1995;4:314–330. doi: 10.1006/mpev.1995.1028. [DOI] [PubMed] [Google Scholar]
- 4.Mallatt J, Sullivan J. 28S and 18S rDNA sequences support the monophyly of lampreys and hagfishes. Mol. Biol. Evol. 1998;15:1706–1718. doi: 10.1093/oxfordjournals.molbev.a025897. [DOI] [PubMed] [Google Scholar]
- 5.Buckley TR, Simon C, Flook PK, Misof B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Mol. Biol. 2000;9:565–580. doi: 10.1046/j.1365-2583.2000.00220.x. [DOI] [PubMed] [Google Scholar]
- 6.Misof B, Niehuis O, Bischoff I, Rickert A, Erpenbeck D, Staniczek A. A hexapod nuclear SSU rRNA secondary-structure model and catalog of taxon-specific structural variation. J. Exp. Zool. Part B Mol. Dev. Evol. 2006;306B:70–88. doi: 10.1002/jez.b.21040. [DOI] [PubMed] [Google Scholar]
- 7.Mallatt J, Winchell CJ. Ribosomal RNA genes and deuterostome phylogeny revisited: more cyclostomes, elasmobranchs, reptiles, and a brittle star. Mol. Phylogenet. Evol. 2007;43:1005–1022. doi: 10.1016/j.ympev.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 8.Hochsmann M, Voss B, Giegerich R. Pure multiple RNA secondary structure alignments: a progressive profile approach. IEEE/ACM Trans. Comput. Biol. Bioinform. 2004;1:53–62. doi: 10.1109/TCBB.2004.11. [DOI] [PubMed] [Google Scholar]
- 9.Reeder J, Giegerich R. Consensus shapes: an alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics. 2005;21:3516–3523. doi: 10.1093/bioinformatics/bti577. [DOI] [PubMed] [Google Scholar]
- 10.Dalli D, Wilm A, Mainz I, Steger G. STRAL: progressive alignment of non-coding RNA using base pairing probability vectors in quadratic time. Bioinformatics. 2006;22:1593–1599. doi: 10.1093/bioinformatics/btl142. [DOI] [PubMed] [Google Scholar]
- 11.Torarinsson E, Havgaard HJ, Gorodkin J. Multiple structural alignment and clustering of RNA sequences. Bioinformatics. 2007;23:926–932. doi: 10.1093/bioinformatics/btm049. [DOI] [PubMed] [Google Scholar]
- 12.Lindgreen S, Gardner PP, Krogh A. MASTR: multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics. 2007;23:3304–3311. doi: 10.1093/bioinformatics/btm525. [DOI] [PubMed] [Google Scholar]
- 13.Will S, Missal K, Hofacker IL, Stadler PF, Backofen R. Inferring non-coding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katoh K, Toh H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinformatics. 2008;9:212. doi: 10.1186/1471-2105-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabei Y, Kiryu H, Kin T, Asai K. A fast structural multiple alignment method for long RNA sequences. BMC Bioinformatics. 2008;9:33. doi: 10.1186/1471-2105-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuker M, Sankoff D. RNA secondary structures and their prediction. Bull. Math. Biol. 1984;46:591–621. [Google Scholar]
- 17.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;125:167–188. [Google Scholar]
- 18.Doshi K, Cannone J, Cobaugh C, Gutell R. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:105. doi: 10.1186/1471-2105-5-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Layton DM, Bundschuh R. A statistical analysis of RNA folding algorithms through thermodynamic parameter perturbation. Nucleic Acids Res. 2005;33:519–524. doi: 10.1093/nar/gkh983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding F, Sharma S, Chalasani P, Demidov VV, Broude NE, Dokholyan NV. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA. 2008;14:1164–1173. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JHD, Noller HF. Crystal structure of the ribosome at 5.5 Å resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 22.Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 23.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins LJ, Moulton V, Penny D. Use of RNA secondary structure for studying the evolution of RNase P and RNase MRP. J. Mol. Evol. 2000;51:194–204. doi: 10.1007/s002390010081. [DOI] [PubMed] [Google Scholar]
- 25.Schoeniger M, von Haeseler A. A stochastic model for the evolution of autocorrelated DNA sequences. Mol. Phylogenet. Evol. 1994;3:240–247. doi: 10.1006/mpev.1994.1026. [DOI] [PubMed] [Google Scholar]
- 26.Rzhetsky A. Estimating substitution rates in ribosomal RNA genes. Genetics. 1995;141:771–783. doi: 10.1093/genetics/141.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tillier ERM, Collins RA. Neighbor joining and maximum-likelihood with RNA sequences—addressing the interdependence of sites. Mol. Biol. Evol. 1995;12:7–15. [Google Scholar]
- 28.Stephan W. The rate of compensatory evolution. Genetics. 1996;144:419–426. doi: 10.1093/genetics/144.1.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tillier ERM, Collins RA. High apparent rate of simultaneous compensatory basepair substitutions in ribosomal RNA. Genetics. 1998;148:1993–2002. doi: 10.1093/genetics/148.4.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parsch J, Braverman JM, Stephan W. Comparative sequence analysis and patterns of covariation in RNA secondary structures. Genetics. 2000;154:909–921. doi: 10.1093/genetics/154.2.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savill NJ, Hoyle DC, Higgs PG. RNA sequence evolution with secondary structure constraints: comparison of substitution rate models using maximum-likelihood methods. Genetics. 2001;157:399–411. doi: 10.1093/genetics/157.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jow H, Hudelot C, Rattray M, Higgs PG. Bayesian phylogenetics using an RNA substitution model applied to early mammalian evolution. Mol. Biol. Evol. 2002;19:1591–1601. doi: 10.1093/oxfordjournals.molbev.a004221. [DOI] [PubMed] [Google Scholar]
- 33.Hudelot C, Gowri-Shankar V, Jow H, Rattray M, Higgs PG. RNA-based phylogenetic methods: application to mammalian mitochondrial RNA sequences. Mol. Phylogenet. Evol. 2003;28:241–252. doi: 10.1016/s1055-7903(03)00061-7. [DOI] [PubMed] [Google Scholar]
- 34.Nussinov R, Piecznik G, Griggs JR, Kleitman DJ. Algorithms for loop matching. SIAM J. Appl. Math. 1978;35:68–82. [Google Scholar]
- 35.Gotoh O. An improved algorithm for matching biological sequences. J. Mol. Biol. 1982;162:705–708. doi: 10.1016/0022-2836(82)90398-9. [DOI] [PubMed] [Google Scholar]
- 36.Burk A, Douzery EJ, Springer MS. The secondary structure of mammalian mitochondrial 16S rRNA molecules: refinements based on a comparative phylogenetic approach. J. Mammalian Evol. 2002;9:225–252. [Google Scholar]
- 37.Shapiro BA, Zhang K. Comparing multiple RNA secondary structures using tree comparisons. CABIOS. 1990;6:309–318. doi: 10.1093/bioinformatics/6.4.309. [DOI] [PubMed] [Google Scholar]
- 38.Thompson JD, Higgs DG, Gibson TJ. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties, and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kjer KM, Honeycutt RL. Site specific rates of mitochondrial genomes and the phylogeny of eutheria. BMC Evol. Biol. 2007;7:8. doi: 10.1186/1471-2148-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kjer KM, Swigonova Z, LaPolla JS, Broughton RE. Why weight? Mol. Phylogenet. Evol. 2007;43:999–1004. doi: 10.1016/j.ympev.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 42.Gardner PP, Wilm A, Washietl S. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res. 2005;33:2433–2439. doi: 10.1093/nar/gki541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilm A, Mainz I, Steger G. An enhanced RNA alignment benchmark for sequence alignment programs. Algorithms Mol. Biol. 2006;1:19. doi: 10.1186/1748-7188-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc. Natl Acad. Sci. USA. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gruber AR, Berhart SH, Hofacker IL, Washietl S. Strategies for measuring evolutionary conservation of RNA secondary structures. BMC Bioinformatics. 2008;9:122. doi: 10.1186/1471-2105-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo S, Wang LS, Kim J. Large-scale simulation of RNA macroevolution by an energy-dependent fitness model. Sys. Biol. 2009 http://kim.bio.upenn.edu/software/rnasim.shtml. [Google Scholar]
- 47.Misof B, Misof K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 2009;58:21–34. doi: 10.1093/sysbio/syp006. [DOI] [PubMed] [Google Scholar]
- 48.Schmitz J, Ohme M, Zischler H. The complete mitochondrial sequence of Tarsius bancanus: evidence for an extensive nucleotide compositional plasticity of primate mitochondrial DNA. Mol. Biol. Evol. 2002b;19:544–553. doi: 10.1093/oxfordjournals.molbev.a004110. [DOI] [PubMed] [Google Scholar]
- 49.Goodman M, Porter CA, Czelusniak J, Page SL, Schneider H, Shoshani J, Gunnell G, Groves CP. Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol. Phylogenet. Evol. 1998;9:585–598. doi: 10.1006/mpev.1998.0495. [DOI] [PubMed] [Google Scholar]
- 50.Zietkiewicz E, Richer C, Labuda D. Phylogenetic affinities of tarsier in the context of primate Alu repeats. Mol. Phylogenet. Evol. 1999;11:77–83. doi: 10.1006/mpev.1998.0564. [DOI] [PubMed] [Google Scholar]
- 51.Schmitz J, Ohme M, Zischler H. SINE insertions in cladistic analyses and the phylogenetic affiliations of Tarsius bancanus to other primates. Genetics. 2001;157:777–784. doi: 10.1093/genetics/157.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayasaka K, Gojobori T, Horai S. Molecular phylogeny and evolution of primate mitochondrial DNA. Mol. Biol. Evol. 1988;5:626–644. doi: 10.1093/oxfordjournals.molbev.a040524. [DOI] [PubMed] [Google Scholar]
- 53.Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O'Brien SJ. Molecular phylogenetics and the origins of placental mammals. Nature. 2001;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- 54.Arnason U, Adegoke JA, Bodin K, Born EW, Esa YB, Gullberg A, Nilsson M, Short RV, Xu X, Janke A. Mammalian mitogenomic relationships and the root of the eutherian tree. Proc. Natl Acad. Sci. USA. 2002;99:8151–8156. doi: 10.1073/pnas.102164299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmitz J, Ohme M, Suryobroto B, Zischler H. The colugo (Cynocephalus variegatus, Dermoptera): the primates' gliding sister? Mol. Biol. Evol. 2002a;19:2308–2312. doi: 10.1093/oxfordjournals.molbev.a004054. [DOI] [PubMed] [Google Scholar]
- 56.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 57.Gowri-Shankar V, Rattray M. On the correlation between composition and site-specific evolutionary rate: implications for phylogenetic inference. Mol. Biol. Evol. 2006;23:352–364. doi: 10.1093/molbev/msj040. [DOI] [PubMed] [Google Scholar]
- 58.Littlewood DT, Smith AB, Clough KA, Emson RH. The interrelationships of the echinoderm classes: morphological and molecular evidence. Biol. J. Linn. Soc. 1997;61:409–438. [Google Scholar]
- 59.Smith AB. Echinoderm larvae and phylogeny. Ann. Rev. Ecol. Syst. 1997;28:219–241. [Google Scholar]
- 60.Janies D. Phylogenetic relationship of extant echinoderm classes. Canadian J. Zool. 2001;79:1232–1250. [Google Scholar]
- 61.Smith AB. Fossil evidence for the relationship of extant echinoderm classes and their times of divergence. In: Paul CRC, Smith AB, editors. Echinoderm Phylogeny and Evolutionary Biology. Oxford: Clarendon Press; 1988. [Google Scholar]
- 62.Scouras A, Smith JM. The complete mitochondrial genomes of the sea lily Gymnocrinus richeri and the feather star Phanogenia gracilis: signature nucleotide bias and unique nad4L gene rearrangement within crinoids. Mol. Phylogenet. Evol. 2006;39:323–34. doi: 10.1016/j.ympev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 63.Kjer KM. Aligned 18S and insect phylogeny. Syst. Biol. 2004;53:506–514. doi: 10.1080/10635150490445922. [DOI] [PubMed] [Google Scholar]
- 64.Niehuis O, Naumann CM, Misof B. Identification of evolutionary conserved structural elements in the mt SSU rRNA of Zygaenoidea (Lepidoptera): a comparative sequence analysis. Org. Divers. Evol. 2006c;6:17–32. [Google Scholar]
- 65.Hull Havgaard JH, Lyngso R, Stormo GD, Gorodkin J. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40% Bioinformatics. 2005;21:1815–1824. doi: 10.1093/bioinformatics/bti279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.