


Abstract
σ54-dependent transcription requires activation by bacterial enhancer binding proteins (bEBPs). bEBPs are members of the AAA+ (ATPases associated with various cellular activities) protein family and typically form hexameric structures that are crucial for their ATPase activity. The precise mechanism by which the energy derived from ATP hydrolysis is coupled to biological output has several unknowns. Here we use Escherichia coli PspF, a model bEBP involved in the transcription of stress response genes (psp operon), to study determinants of its contact features with the closed promoter complex. We demonstrate that substitution of a highly conserved phenylalanine (F85) residue within the L1 loop GAFTGA motif affects (i) the ATP hydrolysis rate of PspF, demonstrating the link between L1 and the nucleotide binding pocket; (ii) the internal organization of the hexameric ring; and (iii) σ54 interactions. Importantly, we provide evidence for a close relationship between F85 and the −12 DNA fork junction structure, which may contribute to key interactions during the energy coupling step and the subsequent remodelling of the Eσ54 closed complex. The functionality of F85 is distinct from that of other GAFTGA residues, especially T86 where in contrast to F85 a clean uncoupling phenotype is observed.
INTRODUCTION
In bacteria, transcription is accomplished via the multi-subunit core RNA polymerase (E), composed of 2α, β, β′ and ω subunits. Sequence and structural conservation of the core RNAP throughout all kingdoms of life allows the Escherichia coli enzyme to be used as a transcription machinery model. In bacteria, promoter specificity of this molecular machine is achieved by the binding of an additional factor, the sigma factor (σ).
Differences in primary sequence and regulatory properties has enabled bacterial σ factors to be categorized into two classes (1): the prototypical σ70-class required for transcription of ‘housekeeping’ genes and the σ54-class required for transcription of genes involved in diverse processes (2). Contrasting the σ70-RNAP holoenzyme (Eσ70), which can spontaneously form transcriptionally proficient open promoter complexes, Eσ54 [recognizing −24 (GG) and −12 (GC) consensus sequences] forms closed promoter complexes that remain transcriptionally silent. Formation of the Eσ54 closed complex (RPc) results in localized DNA melting of the base pairs immediately downstream of the −12 (GC) promoter site (−12 and −11 positions) (3), producing a repressive fork junction DNA structure. The protein-protein and nucleo-protein interactions formed around this −12 fork junction DNA structure (termed the regulatory centre) prevent open complex (RPo) formation in the absence of activation. Activation of Eσ54 transcription is achieved by bacterial enhancer binding proteins (bEBPs) at the expense of ATP hydrolysis (4). The requirement for ATP hydrolysis driven activation of Eσ54 is functionally analogous to eukaryotic RNAP II, which requires energy derived from ATP hydrolysis provided by TFIIH (5,6).
bEBPs belong to the AAA+ protein family (ATPase associated with various cellular activities), which have a common requirement to form higher-order oligomers (predominantly hexamers) to function and use the energy derived from ATP hydrolysis to remodel their substrates. bEBPs, as AAA+ proteins, share conserved motifs, including Walkers A and B (for ATP binding and hydrolysis) and the second region of homology (SRH, containing arginine fingers for inter-subunit catalysis) (7–9). bEBPs are classified within Clade 6 of the AAA+ protein family (10) and are distinguished from other subclasses of AAA+ proteins by two sequence insertions: the L1 loop containing the σ54-interacting GAFTGA motif and the L2 loop (also termed the pre sensor I insertion loop, pre-SIi loop) thought to co-ordinate movement of the L1 loop (11). Well-characterized bEBPs include: DctD, DmpR, NifA, NtrC, NtrC1, PspF, XylR and ZraR (7,12,13). In this study we focus on the E. coli phage shock protein F (PspF) as a bEBP model. PspF is composed of two domains: the catalytic AAA+ domain (residues 1–275, PspF1–275) necessary and sufficient to activate σ54 transcription in vivo and in vitro, and the DNA binding domain (residues 296–325) (Figure 1). Mutagenesis and fragmentation studies have established that the GAFTGA motif, more specifically the threonine at position 86 in PspF, directly contributes to a σ54 contact surface which is important for the initial interaction between PspF and σ54 and in coupling the ATP hydrolysis derived energy to remodelling of the closed complex (14–16). Dago et al. (17) showed that a mutation in Region I of σ54 (a glycine to leucine substitution termed G4L; σ54G4L) can complement a PspF T86S substitution, that would otherwise prevent PspF from stably interacting with σ54.
Figure 1.

PspF organization. (A) Full-length PspF consists of the AAA+ domain (residues 1–275, solid box) and a DNA binding domain (residues 296–325, dashed box). Walker A (residues 37–45), L1 loop (residues 79–94), Walker B (residues 107–111), L2 loop (residues 127–142) and the SRH (second region of homology, residues 142–169 with putative arginine fingers R162 and R168) are highlighted in PspF1–275. (B) Protein sequence alignment using the 248 annotated bEBP sequences demonstrated the following microorganisms possess a naturally occurring tyrosine substitution (highlighted by the grey bar) in the ‘GAFTGA’ motif. Ec, Escherichia coli; Ps sp., Pseudomonas sp. KL28; B sp., Burkholderia sp. RP007; Bk, Burkholderia kururiensis; Ps, Pseudomonas syringae pv. Tomato; Na, Novosphingobium aromaticivorans; Xc, Xanthomonas campestris pv. Campestris; Pi sp., Pirellula sp.; Bj, Bradyrhizobium japonicum; Bc, Bacillus cereus ATCC 14579; Bh, Bacillus halodurans; Erc, Erwinia chrysanthemi; Pf, Pseudomonas fluorescens; Tt, Thermoanaerobacter tengcongensis; Lm, Listeria monocytogenes. (C) Conservation of the ‘GAFTGA’ motif residues represented by Weblogo.
Recent cryo-electron microscopy studies suggest that more than one L1 loop may contribute to the σ54-interacting surface since (i) the L1 loops of adjacent PspF1–275 protomers are appropriately placed to contact σ54 and (ii) can be fitted within the electron density connecting with σ54. When promoter DNA was modelled into the structure of the PspF1–275·Eσ54 complex, the L1 loop was located close to DNA (18,19). Taken together these results strongly suggest that more than one GAFTGA motif may be required to engage the RPc (and form a stable DNA·Eσ54·PspF1–275 complex in which PspF1–275 is in a hexameric state) and that particular GAFTGA motif residues contact σ54 and potentially the promoter DNA.
Sequence alignments of the GAFTGA motif from 248 annotated bEBP proteins revealed a high level of conservation of the phenylalanine residue (position 85 in PspF). A small number of naturally occurring tyrosine substitutions at this position were identified (16 of the 248 bEBPs sequences, Figure 1). The function of the GAFTGA motif phenylalanine (F85 in PspF) remains largely unknown. Its proximity to both T86 (a σ54 contact determinant) and the −12 promoter DNA (9), suggests three potential roles for residue F85: (i) a direct or indirect L1 loop–σ54 interaction and/or (ii) stabilization of the −12 fork junction DNA structure or (iii) its nucleation by base stacking. However, to date there is no direct evidence confirming an interaction between F85 and DNA.
Here we investigate the role of the PspF GAFTGA motif residue F85 in Eσ54 transcription activation. We systematically substituted F85 with 10 other amino acid residues in the context of PspF1–275. Substituting F85 drastically affects PspF1–275 functionality. Binding assays demonstrate that F85 supports σ54-activator complex formation, working in conjunction with T86 to stabilize the L1 loop–σ54 interaction. Notably, we observed that substituting F85 resulted in severely compromised ATPase activity, indicating that a communication pathway exists between the GAFTGA containing L1 loop and the distant ATP hydrolysis site. A functional correlation between the GAFTGA motif residue F85 and the self-association interface was observed. More importantly, we provide evidence that F85 interacts with the repressive −12 fork junction DNA structure (20), further suggesting a direct role of the GAFTGA motif in promoter DNA interactions.
MATERIALS AND METHODS
Nucleotides
ATP, dATP and ADP were purchased from Sigma at the highest purity level. Radiolabelled nucleotides were purchased from Perkin Elmer. Linear promoter sequences encompassing the Sinorhizobium meliloti nifH −60 to +28 promoter region were purchased from Eurofins MWG Operon.
Site-directed mutagenesis
PspF1–275 containing single amino acid substitutions (F85A, F85C, F85E, F85H, F85I, F85L, F85Q, F85R, F85W and F85Y) was constructed using site directed mutagenesis using pPB1 (containing the E. coli pspF1–275 WT sequence with an N-terminal in-frame (His)×6 fusion) as a template (21).
Protein purification
PspF1–275 WT, F85A, F85C, F85E, F85H, F85I, F85L, F85Q, F85R, F85W and F85Y variants were overproduced and purified as previously described (22). The proteins were stored in TGED buffer (20 mM Tris–HCl, pH 8.0, 50 mM NaCl, 1 mM DTT, 0.1 mM EDTA and 5% glycerol) at −80°C. Protein concentrations were determined by the Folin-Lowry method (23). Klebsiella pneumoniae σ54 was purified as described (24). Escherichia coli core RNAP was purchased from Cambio.
ATPase activity
Reactions were carried out in 10 μl final volumes. Four micromolar PspF1–275 was pre-incubated with the ATPase buffer (20 mM Tris–HCl, pH 8.0, 50 mM NaCl, 15 mM MgCl2, 0.1 mM EDTA and 0.01 μM DTT) at room temperature for 10 min. ATP hydrolysis was started upon addition of the ATP mixture (1 mM ATP and 0.06 μCi/μl [α-32P] ATP (3000 Ci/mmol)) and incubated at 37°C for different lengths of time. Reactions were quenched by five volumes of 2 M formic acid. [α-32P] ADP was separated from [α-32P] ATP using thin-layer chromatography (Polygram Cel 300 PEI). Radioactivity was detected by PhosphorImager (Fuji Bas-5000) and quantified using AIDA image analysis software. The ATPase activity of each F85 variant was expressed as a percentage of PspF1–275WT turnover (kcat). Each reaction was performed (at least) in triplicate.
Gel filtration through Superdex 200 column
High (63 μM) and low (20 μM) concentrations of PspF1–275 WT and variants were pre-incubated at room temperature with gel filtration buffer (20 mM Tri–HCl, pH 8.0, 50 mM NaCl, 15 mM MgCl2, ± 0.4 mM ADP) for 5 min. A superdex 200 column (10/300, 24 ml, GE Healthcare) assembled on the AKTA system was equilibrated with buffer ± 0.4 mM ADP. Chromatography was carried out at a flow rate of 0.5 ml/min at room temperature. Calibration was with the following globular proteins: thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), bovine serum albumin (66 kDa) and carbonic anhydrase (29 kDa).
Native gel mobility shift assay
Reactions were performed in 10 μl final volumes. Ten micromolar PspF1–275 was pre-incubated with STA buffer (25 mM Tris–acetate, pH 8.0, 8 mM Mg–acetate, 10 mM KCl, 1 mM DTT, 3.5% w/v PEG 8000), nucleotides [ADP or AMP (4 mM)], 5 mM NaF, 2.35 μM σ54 (WT or G4L), and/or 0.15 μM core RNAP at 37°C for 20 min. Where appropriate, 20 nM 32P-labelled linear DNA probes were added. After addition of 0.4 mM AlCl3, the reactions were further incubated for 10 min at 37°C to allow the in situ formation of the metal-fluoride analogues. Samples were run on a 4.5% native gel (acrylamide/bisacrylamide, 37.5 : 1) in TG buffer (25 mM Tris–HCl, pH 8.3, 192 mM glycine) and protein complexes were detected by either Coomassie staining or PhosphorImager (Fuji Bas-5000) and analysed using the AIDA software.
In vitro single-round full-length transcription
Full-length transcription was performed in 10 μl final volumes containing: STA buffer, 4 μM PspF1–275, 100 nM holoenzyme (reconstituted at a 1 : 4 ratio of E:σ54), 4 mM dATP and 20 nM super-coiled (nifH, pspA or pspG) promoter at 37°C. Activation of RPo formation was allowed to proceed for different lengths of time (as specified) and challenged with 0.1 mg/ml heparin. Reactions were incubated with the elongation mix containing 100 μg/ml heparin, 1 mM ATP, CTP, GTP, 0.5 mM UTP and 0.074 μCi/μl [α-32P]UTP (3000 Ci/mmol) for 10 min and quenched upon addition of 4 μl 3× formamide stop dye. Transcripts were analysed on a 4% denaturing gel run in 1× TBE buffer, visualized by PhosphorImager and analysed using the AIDA software. The in vitro abortive transcription initiation assay was carried out as described above, except the elongation mix was replaced with an abortive mix containing 100 μg/ml heparin, 0.5 mM UpG, 5 μM GTP and 0.2 μCi/μl [α-32P]GTP.
Photo-crosslinking assays
Photo-crosslinking assays were performed as described (25,26) in a volume of 10 μl containing: 10 mM Tris–acetate (pH 8.0), 50 mM potassium acetate, 8 mM magnesium acetate, 4 mM ADP, 5 mM NaF, 1 μM σ54, 5 μM PspF1–275 WT or variants and 50 nM 32P-labelled p-azidophenacyl bromide (APAB, Sigma) conjugated phosphorothioated promoter DNA probe prepared as described (27,28). AlCl3 (0.4 mM) was added to the appropriate reactions and incubated for a further 10 min at 37°C. Reactions were then UV irradiated at 365 nm for 1 min using a UV-Stratalinker 1800 (Stratagene). A 2 μl sample of the crosslinking reaction mix was directly loaded on a native 4.5% polyacrylamide gel (acrylamide/bisacrylamide 37.5:1) and run in 1× TG buffer [25 mM Tris (pH 8.3) and 192 mM glycine]. The remaining reaction mix was diluted by addition of 4 μl of loading dye (Sigma) and 4 μl 10 M urea, heated at 95°C for 3 min and (8 μl) loaded onto a 7.5% SDS–PAGE run at 200 V for 50 min. Gels were then dried and crosslinked protein–DNA complexes visualized by PhosphorImager and analysed using the AIDA software.
RESULTS
To assess the contribution of the conserved GAFTGA motif phenylalanine (F85) residue to PspF1–275 functionality we chose to substitute F85 with: (i) alanine (A) to remove the side chain; (ii) leucine (L)/isoleucine (I) to reduce the side chain length; (iii) tyrosine (Y)/tryptophan (W) to maintain aromaticity; (iv) histidine (H)/arginine (R)/glutamate (E) to introduce a charge; (v) glutamine (Q) to introduce a polar group and (vi) cysteine (C) to introduce additional functionality.
F85Y is competent for low level transcription activation
All the F85 PspF1–275 variants (F85A, F85C, F85E, F85H, F85I, F85L, F85Q, F85R, F85W and F85Y) were successfully overproduced and purified with no large differences observed in their solubility, stability or purity. We first investigated whether the F85 substitutions introduced had any effect on the transcription activation activity of PspF1–275 using an in vitro transcription assay with a super-coiled plasmid carrying the σ54-dependent Sinorhizobium meliloti nifH promoter. The transcription activation activity of each of the F85 variants was then determined with respect to wild-type (WT) PspF1–275 activity. In contrast to WT, under standard 5 min activation conditions, we were unable to detect any transcripts (<1% of WT) in the presence of any of the PspF1–275F85 variants tested (data not shown), demonstrating a major role for residue F85 in establishing one or more of the underlying functionalities of PspF1–275 required for activation (Figure 2A and data not shown). Previously, we observed that upon increased activation time (i.e. allowing accumulation of RPos to occur), the degree of the transcription activation defect can be estimated more precisely (25). When we increased the activation time, we observed that with PspF1–275WT, the amount of full-length transcripts increased between 5 and 30 min, but decreased between 30 and 60 min, consistent with the view that at early time points sufficient ATP remains available to form new RPos and the pre-formed RPos can survive for at least 30 min. The decrease in transcripts observed at the 60 min activation time point suggests that due to depletion of ATP, the number of RPos formed has decreased and the pre-formed RPos are decaying to the RPc. When we increased the activation time with the F85 variants, we detected low-levels of full-length transcripts in the presence of F85Y, a substitution which corresponds to the most frequent naturally occurring substitution (Figure 2A).
Figure 2.

PspF1–275 F85Y slowly activates transcription from the supercoiled (sc) nifH promoter. (A) Full-length (FL) transcription from the nifH promoter as a function of increased activation time in the presence of PspF1–275 WT and PspF1–275 F85Y (PspF1–275 F85A acts as a negative control). The relative intensity of each transcript is represented graphically below the gel. (B) Abortive transcription as a function of increased activation time in the presence of PspF1–275 WT and PspF1–275 F85Y (PspF1–275 F85A acts as a negative control). RPo formation is directly correlated with the amount of abortive product (UpGpGpG) generated. All the reactions were conducted at the same time, run on the same gel and scanned with the same contrast. The lanes were rearranged for clarity. The experiment was minimally performed (independently) in triplicate and the results obtained similar.
To determine whether the defect in transcription activation observed with the F85 variants was due to a defect in open complex (RPo) formation or elongation (after escape), we performed abortive transcription assays (to measure RPo formation) using the same super-coiled DNA plasmid (Figure 2B). Since we obtained similar results to the full-length transcription assays, we conclude that the defect observed with these variants is in RPo formation and not transcript elongation.
Overall, these results demonstrate that residue F85 is critical for the transcription activation activity of PspF1–275. The (naturally occurring) F85Y variant shows a significantly reduced but measurable activation activity.
Stable engagement of σ54 by PspF is dependent on F85
Activation of Eσ54 transcription requires PspF to interact directly with σ54. In the presence of the non-hydrolysable nucleotide analogue, ADP–AlFx (thought to represent the ATP-transition state), PspF1–275 forms a stable ‘trapped’ complex with σ54 or Eσ54 (PspF1–275·σ54·ADP–AlFx or PspF1–275·Eσ54·ADP–AlFx) proposed to represent a putative intermediate en route to the RPo (14). Using this trapping assay, we investigated whether the lack of transcription activation activity observed for the F85 variants was due to an inability of PspF1–275 to interact with σ54 or Eσ54. As shown in Figure 3 (and consistent with the transcription data in Figure 2), only the F85Y variant supported formation of a stable PspF1–275·Eσ54·ADP–AlFx complex (similar to WT), suggesting that the lack of activation activity for the other F85 variants is due to their inability to interact with σ54. In the case of F85Y, the reduced activation activity does not correlate with reduced binding to Eσ54, and so may reflect a more subtle defect in the energy coupling process between PspF and Eσ54, after the initial binding interaction has occurred.
Figure 3.

PspF1–275 F85Y is the only F85 variant interacting with σ54 or Eσ54. Native gel demonstrating that PspF1–275F85Y (in the presence of ADP–AlFx) is the only F85 variant that supports formation of a stable complex with σ54 or Eσ54. Stable complexes were visualized by Coomassie Blue staining.
F85 communicates with the protomer-protomer interface for self association
Since the majority of F85 variants failed to form a stable complex with σ54, we investigated whether the substitutions have a direct effect on the σ54-interacting motif or whether a more general defect in the organization of PspF1–275 was apparent. Structural studies have shown that six protomers of PspF1–275, arranged as a hexameric ring, interact with one σ54 subunit (19). Therefore a gross defect in hexamer formation will result in a failure to form the PspF1–275·σ54 complex. Using gel filtration, we addressed the impact of the F85 substitutions on formation of higher-order oligomers (Figure 4). Previously we established that PspF1–275WT exists in an equilibrium between dimeric and hexameric states, where the hexameric state is favoured at higher PspF1–275 concentrations or when ATP or ADP is present (22). Based on the elution profiles, the F85 variants were grouped into four distinct classes (Figure 4A): (i) WT-like (WT, F85W, F85H, F85Q, F85I and F85L), (ii) constitutive apparent octamer (F85C), (iii) concentration-dependent apparent octamer (F85Y) and (iv) defective for self-association (F85A, F85E and F85R). The apparent octameric state observed with F85Y and F85C has been reported for other PspF1–275 variants and resembles the elution profile of PspF1–275 WT in the nucleotide-bound state (22,25). In the presence of nucleotide (ADP) we observed that WT, F85W, F85L, F85Y, F85H, F85Q and F85I all formed the apparent octamer, but the F85A, F85E and F85R variants were still unable to form higher-order oligomers (Figure 4B). These results strongly suggest a structural and functional link between the F85 residue and the distant self-association interface.
Figure 4.

Gel filtration profiles of the F85 variants at room temperature. (A) The elution profiles of the F85 variants chromatographed in the absence of nucleotide. Two concentrations were used for each F85 variant, low (20 μM light grey line) and high (63 μM black line). (B) The elution profiles of the F85 variants chromatographed in the presence of 0.4 mM ADP (using the lower 20 μM protein concentration). Standard globular proteins were used for calibration: thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), bovine serum albumin (66 kDa) and carbonic anhydrase (29 kDa). Absorption unit (AU) on the scale corresponds to an A280 of 1.
The defects in hexamer formation observed with the F85A, F85E and F85R variants can account for their inability to stably interact with σ54 and demonstrate F85 has a role in supporting self-assembly. The absence of stable σ54 interactions with self-assembling F85 variants suggests another role(s) for this residue in complex formation with σ54, explored below.
F85 communicates with the ATP hydrolysis site
In bEBPs, the nucleotide binding pocket is composed of residues from adjacent protomers located at an interface region between the two subunits. Since ATP hydrolysis is necessary for activation of σ54-dependent transcription and requires formation of higher-order oligomers (22,29) and the correct functioning of the ATP hydrolysis site must be reflected in nucleotide dependent complex formation with σ54, we next investigated whether substituting F85 affected the activity and by inference the organization of the ATP hydrolysis site. We characterized (compared to WT) the ATPase activities of the F85 variants under saturating ATP conditions (Figure 5). All the F85 variants tested retained some ATPase activity and could be grouped into three distinct classes based on their ATP hydrolysis rates: (i) WT-like rate (F85W, F85H and F85I), (ii) medium rate with 20–50% of WT activity (F85Y, F85Q, F85L and F85C) and (iii) slow rate with below 10% of WT activity (F85A, F85E and F85R). As expected since oligomer formation is required for ATPase activity, the variants most defective in self-association (F85A, F85E and F85R) have the lowest ATPase activity. Some variants such as F85Q, which appear WT-like in oligomer formation, show marked defects in ATPase activity, suggesting that residue F85 favours the correct organization of the nucleotide binding pocket in the WT protein.
Figure 5.

ATPase activity of the F85 variants at 37°C in the presence of 1 mM ATP. Each assay was minimally performed in triplicate (the reproducibility of these results is indicated by the error bars) and the turnover rate expressed as a percentage of PspF1–275WT activity.
Taken together these results suggest that F85 is important for the formation, organization and activity of the PspF1–275 hexamer as an ATPase, clearly supporting the existence of a functional link between the nucleotide binding pocket and the distant surface-exposed L1 loop F85 residue.
F85 is sensitive to the –12–11 DNA region
Since the majority of F85 substitutions abolished the functionality of PspF1–275, we subsequently focused our study on the single active variant, F85Y. Based upon the location of F85 [adjacent to T86, a known σ54-interacting residue and potentially in close proximity to promoter DNA (18)], we reasoned that this residue could interact directly with the promoter DNA. Using a variety of linear DNA probes (encompassing positions −60 to +28 of the S. meliloti nifH promoter) thought to represent different DNA conformations encountered during RPo formation (24,25,30), we measured the ability of F85Y to form stable trapped DNA complexes with σ54 in order to determine whether F85 could sense the DNA structure and determine the importance of DNA conformation in PspF1–275 activation activity (Figure 6 and Figure 1S).
Figure 6.

F85 potentially senses the DNA conformation at the −12 fork junction DNA structure. ‘Trapping’ reactions were conducted on different 32P-labelled linear DNA probes which represent the conformation of promoter DNA at different steps en route to the RPo. A–B: assays with σ54WT, C–D: assays with σ54G4L. The stable trapped complexes formed were analysed by native-PAGE and detected by PhosphorImager.
Initially, we analysed stable complex formation on the native probe (wt/wt) and established that F85Y can form stable complexes at a similar level to WT with σ54 (Figure 6A, lane 6). When we used linear probes harbouring a mismatch at −12−11 (−12−11/wt; non-template-strand mutated or wt/−12−11; template-strand mutated) that mimics the conformation of DNA within the RPc, we failed to observe stable PspF1–275·σ54·DNA complexes with F85Y (Figure 6A, lanes 7 and 8) but observed a σ54·DNA complex (similar to Figure 6A, lane 1), demonstrating that the DNA conformation of the −12−11 region is important for F85Y to bind the σ54·DNA complex. We next used a linear probe which mimics the conformation of DNA within the RPo (−10−1/wt) (31) and observed that this DNA can be accommodated within the PspF1–275·σ54 ‘trapped’ complex when either WT or F85Y is present. Since these stable trapped complexes were no longer detected in the presence of F85Y when the −10−1 template was combined with the −12−11 mutations (used earlier) (Figure 6B, compare lanes 2–5), we infer that the −12−11 sequence may be sensed and/or contacted by residue F85. However, we cannot exclude the possibility that the organization of σ54 on the −12−11 mismatch DNA probes is different (compared to the organization of σ54 on DNA probes containing wild-type −12−11 sequences) and this altered protein organization represents an unfavourable target for stable PspF1–275·σ54·DNA complex formation in the context of F85Y.
The σ54G4L variant promotes stable F85Y·σ54·DNA ‘trapped’ complex formation
The F85Y substitution prevents stable PspF1–275·σ54·DNA complex formation when the −12−11 promoter DNA region is altered. Dago et al. (17) reported that introduction of a leucine substitution (replacing the glycine residue at position 4; termed the G4L substitution) in Region I of σ54 (σ54G4L) restores the transcription activation activity of the PspF1–275 T86S variant. We reasoned that since F85 is adjacent to T86 the σ54G4L variant might also rescue the binding defects associated with F85Y. As shown in Figure 6 (compare Figure 6A and C, lanes 7 and 8 with Figure 6B and D, lanes 5 and 6) in the presence of the σ54G4L variant, F85Y forms stable PspF1–275·σ54·DNA ‘trapped’ complexes on all the DNA probes tested, demonstrating a loss of sensitivity to the DNA sequences around −12 seen when using σ54G4L. In the absence of DNA, we observed similar trapping PspF1–275·σ54 complexes using σ54WT or σ54G4L in the presence of PspF1–275 WT or F85Y (Supplementary Figure 2S), demonstrating that G4L does not simply have an overall increase in binding to F85Y.
We addressed the relationship between PspF1–275 (WT and F85Y), σ54 (WT and G4L) and promoter DNA using a UV-crosslinking experiment in the context of the altered −12−11 DNA probe (−12−11/wt). The photo-reactive DNA probes were constructed by conjugating a single, strategically placed phosphorothioate with p-azidophenacyl bromide (APAB), between positions −1/+1 (−1); the downstream edge of the transcription bubble and the transcription start-site (18,26,27). This site (−1) was specifically chosen because of its ability to crosslink PspF1–275 and σ54, allowing us to determine the proximity of both of these proteins (DNA×PspF1–275 and DNA×σ54) in the DNA trapped complex (9,32). Using the −12−11/wt DNA the σ54·DNA proximities at −1 were similar when either σ54WT or σ54G4L in the presence of PspF1–275 WT was assayed (Figure 7). When using F85Y in the absence of a stable PspF1–275·σ54·DNA complex (i.e. with σ54WT) the crosslinked DNA×PspF1–275 species is no longer evident (Figure 7, lanes 1 and 2). Whereas a clear crosslinked DNA×PspF1–275 species, similar to the one observed with PspF1–275 WT, can be observed using σ54G4L (Figure 7, lanes 3 and 4). Clearly the G4L substitution does not drastically alter the interaction PspF1–275 makes with the −1 DNA promoter region, but facilitates stable complex formation with F85Y.
Figure 7.

Crosslinking profiles of PspF1–275·σ54·DNA complexes formed on the −12−11/wt DNA. Gels showing the crosslinking profiles of the binary σ54-DNA and the trapped PspF1–275·σ54·DNA complex on the −12−11/wt DNA in the presence of σ54WT and G4L and PspF1–275WT and F85Y. The crosslinked PspF1–275 (DNA × PspF1–275) and σ54 (DNA × σ54) species are as indicated.
We showed that F85Y can form a F85Y·σ54 trapped complex in the presence of σ54WT (Figure 3) but only the presence of σ54G4L allows the F85Y·σ54 complex to accommodate the DNA. These data demonstrate that the σ54G4L variant rescues the σ54·DNA interaction defect associated with the F85Y substitution. We suggest that either a more favourable direct interaction between F85Y and the −12−11 DNA region or an indirect effect upon the interaction between T86 and σ54 could be occurring.
Cognate promoter sequences affect F85Y activation
Since the F85Y substitution severely reduces the ability to form PspF1–275·σ54·DNA complexes in a promoter sequence specific manner, we addressed whether the level of transcription activation observed with F85Y (compared to WT) could be altered in the presence of different σ54-dependent promoter sequences. Since in vivo PspF regulates the activities of the pspA and pspG promoters, we measured the in vitro Eσ54 transcription activity from the super-coiled nifH, pspA and pspG promoters (Table 1). As expected the different natural strengths of each of the promoters resulted in different amounts of full-length transcripts. However, in contrast to the rate of transcription activation by PspF1–275 WT, which varies greatly between promoters, the rate of transcription in the presence of F85Y remained fairly constant across the three different promoters. As such, the relative rate of transcription obtained in the presence of F85Y compared to WT varies significantly in a promoter-dependent manner: nifH 4.1%, pspA 8.8% and pspG 40.4%.
Table 1.
The rate of transcription in the presence of PspF1–275 WT and F85Y was determined on three different σ54-dependent promoters
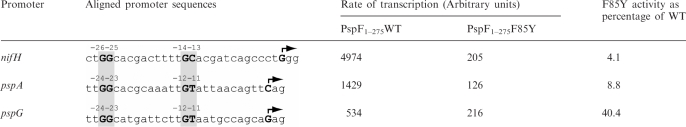 |
The rate of transcription represents the amount of transcript accumulated using full-length transcription assay divided by the time of activation before adding the elongation mix.
From these results, we propose that the role of the highly conserved F85 residue is to fix, in a promoter-specific manner, the level of Eσ54 transcription activation, potentially by sensing and/or directly interacting with the −12−11 region. At some promoters, such as pspG, a GAYTGA sequence appears adequate.
DISCUSSION
The σ54-contacting GAFTGA motif located at the tip of the flexible L1 loop is conserved amongst members of the bEBPs. Understanding the contribution of individual residues within this motif is crucial to dissecting the pathway of σ54 transcription activation. σ54-dependent transcription initiation is a multi-step process involving several conformational changes in RNAP, the bEBP and the promoter DNA. The GAFTGA motif establishes a physical interaction between PspF and σ54, to relay the consequences of the nucleotide binding interactions associated with ATP hydrolysis to conformational rearrangements in the RPc, thereby promoting RPo formation. The conserved threonine (of the GAFTGA motif) directly contributes to the σ54-interacting interface, however, the contribution of the other GAFTGA residues remains poorly understood (17,21). Using a site-directed mutagenesis approach, we assessed the contribution of the conserved phenylalanine residue (of the GAFTGA motif) to PspF functionality. Strikingly, most of the substitutions resulted in the complete lack of σ54-dependent transcription activation, which we later attributed to an inability of these variants to interact with σ54. However one variant, which represents a naturally occurring tyrosine substitution, maintained the ability to interact with σ54, but not to efficiently activate transcription from the nifH promoter, suggesting a more direct role of residue F85 in steps that occur after the initial bEBP·Eσ54·DNA interaction. Using different linear DNA probes, we showed that F85 is sensitive to the DNA sequence at the −12−11 region that is melted within the RPc (3). From our results (Figure 6) we propose that this phenylalanine may also contribute to a σ54-interacting interface but in a DNA sequence-dependent manner, potentially in part as a consequence of binding an altered σ54 target on the different DNA probes. We show that PspF is capable of modulating the transcription activity of Eσ54 in a promoter DNA-specific manner, probably by directly sensing the sequence of the −12−11 DNA. We propose that F85 could have different roles during transcription activation: (i) ensuring exposure of the σ54-interacting motif and (ii) promoting σ54 contacts either directly by forming part of the σ54-interaction interface or indirectly through the positioning of residue T86.
F85 may not constitute a direct σ54 binding determinant but senses the −12 fork junction DNA structure
In the context of a maltose binding protein (MBP) fused fragment of PspF (residues 69–93 to include the GAFTGA motif) F85A and F85L substitutions did not affect the amount of complex formation between the MBP–PspF fragment and σ54·DNA, in contrast to the outcome with T86 substitutions (21). These data suggest that F85 may not be directly involved in making a σ54 contact, in contrast to T86. In the context of the PspF1–275 protein, we demonstrated that the F85A and F85L substitutions failed to form stable PspF1–275·σ54 or PspF1–275·σ54·DNA ‘trapped’ complexes. Defects in self-association and ATPase were observed with these two variants (and many others), indicating a contribution of F85 to the underlying functionalities required to bind the RPc in a nucleotide dependent manner. We show that the trapped PspF1–275·σ54·DNA complex is observed with the F85Y variant, indicating that the structure of the L1 loop and by inference the σ54 interacting surface is not grossly affected by the tyrosine substitution. The nature of the F85 substitution seems critical in allowing PspF1–275 to form the PspF1–275·σ54·DNA trapped complex, and many F85 substitutions affect activities that are believed to underlie the correct exposure of the L1 loop.
The GAFTGA motif of the L1 loop is directly involved in stable interactions with σ54 (21) and based on recent structural studies, it appears likely that the L1 loop lies in close proximity to the −12 promoter DNA region (18). Site-specific DNA–protein crosslinking demonstrates that PspF1–275 spans the promoter DNA between positions −29 to +11 (9), although whether PspF1–275 contacts the DNA between these positions remains unknown. Results obtained using different DNA templates in the transcription assays (Table 1) strongly suggest that the L1 loop is sensitive to the −12 promoter DNA region (compare the activity of F85Y versus WT at the nifH and pspG promoters, the relative differences are striking at 4.1 and 40% activity, respectively). Notably, the level of transcription from the tested promoters in the presence of F85Y does not change drastically (2-fold), whereas with WT the level varies up to 10-fold. Activation and binding outcomes seem to depend on the −12 DNA-region functioning in concert with the F85 residue.
F85 is important in the coupling pathway relaying ATPase activity to transcription activation
F85 substitutions affect the ATPase activity of PspF1–275, which is notable given that the L1 loop and the ATP hydrolysis site are not in direct contact. Previous studies have provided evidence that a communication pathway exists between residues N64 and E108 of the nucleotide binding pocket, which serves to couple local conformational changes (as a result of nucleotide binding and hydrolysis) to remodelling of σ54 (25,33). The proposed pathway links residue N64 (of the nucleotide binding pocket) via Linker 1 and Helix 3 (which contains the L1 loop insertion) to the σ54-interacting GAFTGA motif. In the case of F85Y, the tyrosine substitution could indirectly alter the orientation of N64, thereby affecting the position of the Walker B E108 residue within the ATP hydrolysis site, resulting in reduced ATPase and transcription activation activities. In other F85 substitutions, the effect on the protein activity is even more pronounced and results in altered oligomerization properties, reduced ATPase activity and the inability to interact with σ54. Altered presentations of interface residues (including those used for ATPase activity) can explain such defects, and a mechanism for this to occur is supported by the L1 loop to ATPase site signalling pathway described above. F85 is thought (based on crystal structure data from NtrC1) to make an interaction with Helix 3 and changes in this interaction might explain the properties of some F85 variants (34).
F85Y holds significant regulatory value?
Because tyrosine is present in ∼7% of the annotated bEBP sequences, we considered whether co-variation between GAYTGA, σ54 and/or the promoter sequence was occurring. Promoter sequence analysis failed to identify a clear DNA motif that supported co-variation of the tyrosine residue and DNA sequence (data not shown). Alignment analysis of σ54 Region I in organisms that contained a naturally occurring bEBP GAYTGA motif, established that the σ54 residues Q39 and D42 (Klebsiella pneumoniae numbering) exist as glutamate residues (see Figure 3S), suggesting a possible σ54 adaptation linked to use of the GAYTGA motif. Mutating these σ54 residues in K. pneumoniae to glutamate (Q39E, D42E or the double substitutions Q39E/D42E), or using σ54 from Pseudomonas syringiae pv. Tomato DC3000 (an organism where GAYTGA is present in the HrpS bEBP) did not significantly increase the transcription activation activity of F85Y (data not shown), suggesting Q39E, D42E do not adapt σ54 to use of the GAYTGA motif. As the transcription activation activity observed for F85Y was substantially lower than PspF1–275 WT harbouring F85 (irrespective of the promoter DNA sequence bound), we suggest that the presence of the naturally occurring tyrosine residue in the GAYTGA motif could have a direct role in setting the necessary level of transcription activation from certain promoters. This mechanism of regulation could have significant biological relevance. Consistent with this proposal, a tyrosine containing GAYTGA motif exists in the HrpS bEBP from P. syringiae pv. Tomato DC3000 [which activates, together with HrpR, transcription from the hrpL promoter, an alternative sigma factor that transcribes genes involved in the Type III secretion system (35)]. When replaced by phenylalanine the GAFTGA form of HrpS increases σ54-dependent transcription from the hrpL promoter by approximately 50% in E. coli (M. Jovanovic, unpublished data).
Taken together, these observations strongly suggest that the phenylalanine of the GAFTGA motif could play a functionally relevant role in modulating the level of transcription activation, dependent on the nature of the −12 promoter DNA sequence, by directly influencing bEBP·σ54·DNA interactions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Wellcome Trust (grant number 084599/Z/07/Z to M.B.); the Biotechnology and Biological Sciences Research Council (grant number BB/G001278/1 to M.B.); BBSRC David Phillips Fellowship (BB/E023703/1) (to S.W.). Funding for open access charge: Wellcome Trust (grant number 084599/Z/07/Z).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr G. Jovanovic, Dr J. Schumacher and Miss E. James for valuable comments on the manuscript. We also thank the members of Prof. Buck's laboratory for helpful discussions and friendly support.
REFERENCES
- 1.Paget MS, Helmann JD. The sigma70 family of sigma factors. Genome Biol. 2003;4:203. doi: 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. The bacterial enhancer-dependent sigma(54) (sigma(N)) transcription factor. J. Bacteriol. 2000;182:4129–4136. doi: 10.1128/jb.182.15.4129-4136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris L, Cannon W, Claverie-Martin F, Austin S, Buck M. DNA distortion and nucleation of local DNA unwinding within sigma-54 (sigma N) holoenzyme closed promoter complexes. J. Biol. Chem. 1994;269:11563–11571. [PubMed] [Google Scholar]
- 4.Wigneshweraraj SR, Burrows PC, Bordes P, Schumacher J, Rappas M, Finn RD, Cannon WV, Zhang X, Buck M. The second paradigm for activation of transcription. Prog. Nucleic Acid Res. Mol. Biol. 2005;79:339–369. doi: 10.1016/S0079-6603(04)79007-8. [DOI] [PubMed] [Google Scholar]
- 5.Kim TK, Ebright RH, Reinberg D. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science. 2000;288:1418–1422. doi: 10.1126/science.288.5470.1418. [DOI] [PubMed] [Google Scholar]
- 6.Lin YC, Choi WS, Gralla JD. TFIIH XPB mutants suggest a unified bacterial-like mechanism for promoter opening but not escape. Nat. Struct. Mol. Biol. 2005;12:603–607. doi: 10.1038/nsmb949. [DOI] [PubMed] [Google Scholar]
- 7.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure—diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 8.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bose D, Joly N, Pape T, Rappas M, Schumacher J, Buck M, Zhang X. Dissecting the ATP hydrolysis pathway of bacterial enhancer-binding proteins. Biochem. Soc. Trans. 2008;36:83–88. doi: 10.1042/BST0360083. [DOI] [PubMed] [Google Scholar]
- 10.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 11.Burrows PC, Schumacher J, Amartey S, Ghosh T, Burgis TA, Zhang X, Nixon BT, Buck M. Functional roles of the pre-sensor I insertion sequence in an AAA+ bacterial enhancer binding protein. Mol. Microbiol. 2009 doi: 10.1111/j.1365-2958.2009.06744.x. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buck M, Bose D, Burrows P, Cannon W, Joly N, Pape T, Rappas M, Schumacher J, Wigneshweraraj S, Zhang X. A second paradigm for gene activation in bacteria. Biochem. Soc. Trans. 2006;34:1067–1071. doi: 10.1042/BST0341067. [DOI] [PubMed] [Google Scholar]
- 13.Studholme DJ, Dixon R. Domain architectures of sigma54-dependent transcriptional activators. J. Bacteriol. 2003;185:1757–1767. doi: 10.1128/JB.185.6.1757-1767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaney M, Grande R, Wigneshweraraj SR, Cannon W, Casaz P, Gallegos MT, Schumacher J, Jones S, Elderkin S, Dago AE, et al. Binding of transcriptional activators to sigma 54 in the presence of the transition state analog ADP-aluminum fluoride: insights into activator mechanochemical action. Genes Dev. 2001;15:2282–2294. doi: 10.1101/gad.205501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez V, Olvera L, Soberon X, Morett E. In vivo studies on the positive control function of NifA: a conserved hydrophobic amino acid patch at the central domain involved in transcriptional activation. Mol. Microbiol. 1998;28:55–67. doi: 10.1046/j.1365-2958.1998.00772.x. [DOI] [PubMed] [Google Scholar]
- 16.Rombel I, Peters-Wendisch P, Mesecar A, Thorgeirsson T, Shin YK, Kustu S. MgATP binding and hydrolysis determinants of NtrC, a bacterial enhancer-binding protein. J. Bacteriol. 1999;181:4628–4638. doi: 10.1128/jb.181.15.4628-4638.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dago AE, Wigneshweraraj SR, Buck M, Morett E. A role for the conserved GAFTGA motif of AAA+ transcription activators in sensing promoter DNA conformation. J. Biol. Chem. 2007;282:1087–1097. doi: 10.1074/jbc.M608715200. [DOI] [PubMed] [Google Scholar]
- 18.Bose D, Pape T, Burrows PC, Rappas M, Wigneshweraraj SR, Buck M, Zhang X. Organization of an activator-bound RNA polymerase holoenzyme. Mol. Cell. 2008;32:337–346. doi: 10.1016/j.molcel.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rappas M, Schumacher J, Beuron F, Niwa H, Bordes P, Wigneshweraraj S, Keetch CA, Robinson CV, Buck M, Zhang X. Structural insights into the activity of enhancer-binding proteins. Science. 2005;307:1972–1975. doi: 10.1126/science.1105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y, Lew CM, Gralla JD. Promoter opening by sigma(54) and sigma(70) RNA polymerases: sigma factor-directed alterations in the mechanism and tightness of control. Genes Dev. 2000;14:2242–2255. doi: 10.1101/gad.794800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bordes P, Wigneshweraraj SR, Schumacher J, Zhang X, Chaney M, Buck M. The ATP hydrolyzing transcription activator phage shock protein F of Escherichia coli: identifying a surface that binds sigma 54. Proc. Natl Acad. Sci. USA. 2003;100:2278–2283. doi: 10.1073/pnas.0537525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joly N, Schumacher J, Buck M. Heterogeneous nucleotide occupancy stimulates functionality of phage shock protein F, an AAA+ transcriptional activator. J. Biol. Chem. 2006;281:34997–35007. doi: 10.1074/jbc.M606628200. [DOI] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 24.Cannon WV, Gallegos MT, Buck M. Isomerization of a binary sigma-promoter DNA complex by transcription activators. Nat. Struct. Biol. 2000;7:594–601. doi: 10.1038/76830. [DOI] [PubMed] [Google Scholar]
- 25.Joly N, Rappas M, Wigneshweraraj SR, Zhang X, Buck M. Coupling nucleotide hydrolysis to transcription activation performance in a bacterial enhancer binding protein. Mol. Microbiol. 2007;66:583–595. doi: 10.1111/j.1365-2958.2007.05901.x. [DOI] [PubMed] [Google Scholar]
- 26.Burrows PC, Wigneshweraraj SR, Buck M. Protein-DNA interactions that govern AAA+ activator-dependent bacterial transcription initiation. J. Mol. Biol. 2008;375:43–58. doi: 10.1016/j.jmb.2007.10.045. [DOI] [PubMed] [Google Scholar]
- 27.Burrows PC, Severinov K, Buck M, Wigneshweraraj SR. Reorganisation of an RNA polymerase-promoter DNA complex for DNA melting. EMBO J. 2004;23:4253–4263. doi: 10.1038/sj.emboj.7600406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer AN, Barany F. Photoaffinity cross-linking of TaqI restriction endonuclease using an aryl azide linked to the phosphate backbone. Gene. 1995;153:1–8. doi: 10.1016/0378-1119(94)00752-e. [DOI] [PubMed] [Google Scholar]
- 29.Schumacher J, Zhang X, Jones S, Bordes P, Buck M. ATP-dependent transcriptional activation by bacterial PspF AAA+protein. J. Mol. Biol. 2004;338:863–875. doi: 10.1016/j.jmb.2004.02.071. [DOI] [PubMed] [Google Scholar]
- 30.Cannon W, Bordes P, Wigneshweraraj SR, Buck M. Nucleotide-dependent triggering of RNA polymerase-DNA interactions by an AAA regulator of transcription. J. Biol. Chem. 2003;278:19815–19825. doi: 10.1074/jbc.M301296200. [DOI] [PubMed] [Google Scholar]
- 31.Wedel A, Kustu S. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes Dev. 1995;9:2042–2052. doi: 10.1101/gad.9.16.2042. [DOI] [PubMed] [Google Scholar]
- 32.Burrows PC, Joly N, Cannon WV, Camara BP, Rappas M, Zhang X, Dawes K, Nixon BT, Wigneshweraraj SR, Buck M. Coupling sigma factor conformation to RNA polymerase reorganisation for DNA melting. J. Mol. Biol. 2009;387:306–319. doi: 10.1016/j.jmb.2009.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joly N, Burrows PC, Buck M. An intramolecular route for coupling ATPase activity in AAA+ proteins for transcription activation. J. Biol. Chem. 2008;283:13725–13735. doi: 10.1074/jbc.M800801200. [DOI] [PubMed] [Google Scholar]
- 34.Lee SY, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev. 2003;17:2552–2563. doi: 10.1101/gad.1125603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutcheson SW, Bretz J, Sussan T, Jin S, Pak K. Enhancer-binding proteins HrpR and HrpS interact to regulate hrp-encoded type III protein secretion in Pseudomonas syringae strains. J. Bacteriol. 2001;183:5589–5598. doi: 10.1128/JB.183.19.5589-5598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.