


Abstract
Motor proteins that translocate on nucleic acids are key players in gene expression and maintenance. While the function of these proteins is diverse, they are driven by highly conserved core motor domains. In transcription-coupled DNA repair, motor activity serves to remove RNA polymerase stalled on damaged DNA, making the lesion accessible for repair. Structural and biochemical data on the bacterial transcription-repair coupling factor Mfd suggest that this enzyme undergoes large conformational changes from a dormant state to an active state upon substrate binding. Mfd can be functionally dissected into an N-terminal part instrumental in recruiting DNA repair proteins (domains 1–3, MfdN), and a C-terminal part harboring motor activity (domains 4–7, MfdC). We show that isolated MfdC has elevated ATPase and motor activities compared to the full length protein. While MfdN has large effects on MfdC activity and thermostability in cis, these effects are not observed in trans. The structure of MfdN is independent of interactions with MfdC, implying that MfdN acts as a clamp that restrains motions of the motor domains in the dormant state. We conclude that releasing MfdN:MfdC interactions serves as a central molecular switch that upregulates Mfd functions during transcription-coupled DNA repair.
INTRODUCTION
Molecular motors acting on nucleic acids play a crucial role in DNA replication, recombination, transcription and repair. Their ability to translocate on nucleic acids driven by ATP hydrolysis is used to remodel chromatin, disrupt protein–DNA interactions; separate DNA strands and alter secondary structure of RNA. In transcription-coupled DNA repair, motor activity serves to remove RNA polymerase (RNAP) from bulky DNA lesions that have arrested transcription. Through this pathway, transcribed DNA is repaired preferentially, whereas otherwise stalled RNAP would occlude the DNA damage and inhibit repair (1). In bacteria, a single protein, Mfd, couples stalled transcription with DNA repair, by removing the polymerase through its motor activity and recruiting the nucleotide excision repair (NER) proteins UvrA, UvrB and UvrC (2). How does Mfd disrupt the highly stable elongation complex into its components DNA, RNA and RNAP? Elegant studies by Roberts and coworkers (3) have shown that Mfd binds upstream of the stalled elongation complex, and forward translocation of the polymerase occurs at the beginning of this process. This suggested that Mfd uses a DNA motor activity to push RNAP forward and off the DNA. Further work confirmed that Mfd was a DNA translocase (4), and suggested that Mfd-dependent displacement of RNAP proceeds via rewinding of the upstream part of the transcription bubble (5).
Mfd contains seven domains (6) and can functionally be divided into an N-terminal part (domains 1–3, abbreviated MfdN hereafter) and a C-terminal part (domains 4–7, abbreviated MfdC hereafter) (Figure 1). MfdN is necessary for binding to UvrA, but not for the RNAP-displacement activity (7). Domain 2 shows similarity with domain 2 of the NER protein UvrB in sequence (7) and in structure (8). In NER, domain 2 of UvrB interacts with UvrA and the complex formed by the two proteins is responsible for damage recognition (9–11). The similarity of domain 2 in UvrB and Mfd suggests that the UvrA-binding determinants of MfdN reside in domain 2, while the roles of the other two domains in MfdN are unclear. The four C-terminal domains of Mfd (MfdC) display activities associated with RNAP-displacement and regulation. Domain 4 harbors the RNAP-binding interface (6,12), whilst domains 5 and 6 contain the ATP-dependent motor activity. Mfd is classified as a superfamily (SF) 2 helicase according to high sequence similarity of its motor domains with other members of this family (13). This family of enzymes shows broad substrate specificity, with substrates ranging from single-stranded and double-stranded DNA and RNA to non-nucleic-acid substrates. While the mechanism of ATP hydrolysis and its coupling to domain motions is well-characterized (14–16), regulation of the core motor activity by accessory domains or accessory proteins is a question of intense study (17–21).
Figure 1.

Domains of Mfd and structure of the enzyme in its apo-form. (A) Structure of Mfd [PDB-ID 2EYQ, (6)]. Domains are color-coded and the backbone is shown as a ribbon indicating secondary structure. In the orientation shown, the ATP-binding site and the putative DNA-binding site are facing the viewer. (B) Domain organization and definition of MfdN, MfdC and linker regions. Figures 1A, 5A and 7A prepared with Molmol (46).
The closest structural relative of Mfd, RecG, plays a role in replication fork reversal through ATP-dependent translocation on duplex DNA. As is typical for DNA motor proteins, ATPase activity and DNA binding in RecG are strongly coupled, allowing the free energy of ATP hydrolysis to be utilized for thermodynamically unfavorable processes linked to translocation on DNA (22,23). Despite sharing all six conserved helicase motifs, the ATPase activity of Mfd is orders of magnitude lower than that of RecG, and is hardly stimulated by DNA (4,24). This is unexpected, given that the high similarity between the two enzymes extends beyond their core motor to include additional motifs that play a role in coupling ATP hydrolysis to DNA translocation (25), and suggests Mfd activity might be inhibited as a regulatory mechanism. Indeed, Mfd is auto-inhibited by a mechanism that involves the C-terminal domain of the protein, domain 7 (4). An Mfd mutant lacking domain 7 (MfdΔD7) displays an ATPase activity that is 5-fold higher than wild-type Mfd, and is more active than the wild-type protein in RNAP-displacement assays. Unlike wild-type Mfd the truncated mutant is active in an RNAP-independent DNA translocation assay. Support for the proposal that deletion of domain 7 recapitulates a physiologically relevant activation of Mfd comes from the observation that DNA translocation by wild-type Mfd can be detected in the presence of stalled transcription elongation complexes (4). These results suggest a dormant state of Mfd with low motor activity can be switched to activated states with higher motor activity, by binding to its physiological substrate or by removal of domain 7. Interestingly, domain 7 occludes the putative UvrA-binding interface located on domain 2 of Mfd in the apo-structure (Figure 1), suggesting that domain 7 also regulates UvrA recruitment and that Mfd has to undergo major conformational changes from the apo-form to be able to bind UvrA during transcription-coupled repair (6).
The determinants of auto-inhibition in the full length enzyme and the trigger that relieves it during transcription-coupled repair are unknown. One striking structural difference between Mfd and RecG is the degree to which their motor domains are involved in inter-domain contacts, with extensive contacts observed in Mfd between the core motor and N-terminal domains 1 and 3 (6). In order for Mfd to act as a motor, its core motor domains have to be able to move. Likewise, for the interaction of Mfd and UvrA, the relative orientation of domains 2 and 7 has to change to make the UvrA-binding site accessible. However, large scale domain reorientations appear restrained by inter-domain contacts observed in the absence of substrates (6). We propose that multiple inter-domain contacts at the MfdN:MfdC interface underlie the mechanism for auto-inhibition and activation.
To test this model, we removed all MfdN:MfdC interactions, either by separately expressing MfdN and MfdC or cleaving the connecting linker, and studied the consequences on enzymatic activity, thermostability, and structure. Our data suggest that transcription-coupled repair is regulated by a central switch between a dormant closed form and an active open form of Mfd that lacks some or all contacts between MfdN and MfdC. We hypothesize that contacts are disrupted in a concerted conformational switch that simultaneously upregulates the Mfd motor activity driving RNAP removal and enables recruitment of DNA repair proteins, precisely at times when these activities are required in transcription-coupled repair.
MATERIALS AND METHODS
Expression and purification of mfd, MfdN, and MfdC
The Mfd gene was amplified from Escherichia coli DH5α genomic DNA and cloned into pET23d, with a C-terminal His tag (amino-acid sequence KLAAALEHHHHHH) for efficient purification of the expressed gene product. Both MfdN and MfdC constructs (coding for residues 1–470 and 451–1148, respectively) were subcloned from this expression vector and contain the same C-terminal tag noted above, with MfdC having an extra methionine at its N-terminus. Transformation into E. coli expression strain BL21(DE3) was successful for Mfd and MfdN. However, the more stringent strain BL21(DE3)pLysS was needed for transformation of the MfdC construct. Mfd and MfdN were expressed in LB cultures (containing 100 µg/ml ampicillin) at 37°C induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 of 0.8 and harvested 3 h after induction. Cells were lysed in NT buffer (500 mM NaCl, 10 mM Tris–HCl, pH 8.2) by sonication or running through a microfluidizer, and the cleared lysate was applied to a 1 ml HisTrap HP column (GE Healthcare) charged with nickel(II). Protein was eluted at 250 mM imidazole and exchanged into NT buffer using a 5 ml HiTrap desalting column (GE Healthcare). Samples for crystallization were concentrated to 10 mg/ml by ultrafiltration (Centricon, 10 kDa MWCO, Millipore). Otherwise, protein aliquots were flash frozen in liquid nitrogen and stored at –80°C. Protein concentration was determined from absorbance at 280 nm, using a BioMate3 UV spectrophotometer (Thermo Fisher Scientific), with extinction coefficients estimated from the primary sequence by the PROTPARAM tool (26).
MfdC was expressed in LB cultures (containing 100 µg/ml ampicillin and 50 μg/ml chloramphenicol) induced with 0.5 mM IPTG at an OD600 of 0.6–0.8. The cultures were grown at 37°C before induction and cooled to 30°C at induction. The induced culture was allowed to grow for another 5–6 h at 30°C before harvest. Cells were lysed in NT buffer containing 1 mM phenylmethylsulphonyl fluoride (PMSF) by running through a microfluidizer twice, and the cleared lysate was applied to a 1 ml HisTrap HP column. Protein was washed with 20 ml NT buffer at 20 mM imidazole followed by 3 ml at 50 mM, and eluted at 200 mM. Importantly, NT buffer was pre-loaded into the collection tube for the eluate as the solubility of the protein is roughly 1 mg/ml and is lower when imidazole is present at high concentration. The combined eluate was exchanged to Q-binding buffer (250 mM NaCl, 10 mM Tris pH 8.2) using a 5 ml HiTrap desalting column and loaded onto a 1 ml HiTrap Q HP column (GE Healthcare) equilibrated with Q-binding buffer. The flow-through contains MfdC of >95% purity. Dithiothreitol (DTT) was added to the flow-through to 4 mM. Protein storage and concentration determination are the same as described above for Mfd and MfdN.
ATPase assay
NADH-coupled ATPase assays were modified from protocols described previously (27). Reactions were carried out in a total volume of 100 µl at 37°C for 10 min in a cuvette. Reactions were initiated by the addition of Mfd protein to a reaction mixture containing 40 mM HEPES (pH 7.5), 50 mM NaCl, 10 mM MgCl2, 5 mM phosphoenolpyruvate, 4% (v/v) glycerol, 4 mM DTT, 0.024 u/µl pyruvate kinase, 0.036 u/µl lactate dehydrogenase, and 0.25–8 mM ATP. NADH was used at 0.2 mM or 0.8 mM for cuvettes with a pathlength of 10 mm or 2 mm, respectively. The depletion of NADH was monitored by absorbance measurements at 340 nm (with absorbance at 420 nm taken for background correction) using a Beckman DU 800 UV-vis spectrometer, and the corrected absorbance values were recorded every 15 s. Triplicate measurements were performed for every ATP concentration, and the linear decrease of absorbance from 2–10 min was taken to calculate the rate of NADH oxidation (which is equal to that of ATP hydrolysis) using a molar extinction coefficient for NADH of 6.22 mM−1·cm−1. For Mfd and MfdC, the observed rates at different ATP concentrations were fitted to Michaelis–Menten kinetics to obtain KM and kcat. For other Mfd variants, rates were measured at a single ATP concentration of 4 mM.
In DNA stimulation experiments, reactions were carried out as above except that ATP was provided at a concentration of 5 mM and 45-bp blunt-ended duplex DNA was present at different molar ratios to Mfd proteins (0 : 1–4 : 1 for full length Mfd and 0 : 1–8 : 1 for MfdC). For each protein, the observed rates were normalized to the rate obtained in the absence of DNA (0 : 1 molar ratio of DNA:Mfd) and the resulting relative rates were fitted to a one-step equimolar binding model to obtain the dissociation constant and the DNA stimulation factor (ATPase rate of DNA:enzyme complex versus free enzyme).
RNAP displacement assays
The displacement of stalled transcription complexes by Mfd and MfdC was analyzed as described (4). Briefly, transcription complexes were stalled by nucleotide starvation on a 32P-labelled linear DNA fragment carrying the T7A1 promoter, generated from plasmid pSRT7A1 (4). RNAP [purified with a His-tag at the C-terminus of the ß subunit (4)] was present at 10 nM, DNA at 0.4 nM and ATP at 2 mM, CTP and GTP at 10 µM and ApU at 100 µM. After 15 min incubation at 37°C Mfd or MfdC were added to the reaction at a final concentration of 250 nM. The protein concentrations used are expected to be saturating, based on the estimated Kd values for the RNAP–T7A1 promoter interaction (28) and the Mfd–DNA interaction (7, and this work). Samples were removed at intervals and were analyzed by electrophoretic mobility shift assays using 4.5% acrylamide/1× TBE gels run at 4°C. The fraction of DNA present in elongation complexes was quantified using a Molecular Dynamics Typhoon phoshorimager and ImageQuant software.
Triplex-forming oligonucleotide displacement assays
DNA translocation activity was monitored using a triplex-forming oligonucleotide (TFO) displacement assay (29). This assay exploits the fact that under acidic conditions pyrimidine-rich oligonucleotides can bind to specific sites in DNA via Hoogstein base pairing in the major groove. The triplex DNA remains stable at neutral pH, but the TFO cannot rebind if displaced by a DNA translocase. The assays were performed as described in (4). Briefly, TFO-displacement reactions contained 5-nM triplex DNA (a 32P end-labelled 22 nt TFO bound to supercoiled plasmid pSRTB1), 250 nM Mfd or MfdC and 2 mM ATP in TR buffer (50 mM Tris–HCl pH 8.0, 10 mM MgCl2, 1 mM DTT). Reactions were incubated at 20°C, and samples were removed at intervals and quenched by the addition of one-fourth volume GSMB buffer [15% (w/v) glucose, 3% (w/v) SDS, 250 mM MOPS pH 5.5, 0.4 mg/ml bromophenol blue]. The triplex-containing DNA and the free TFO were separated by running the samples on 1% (w/v) agarose gels (20 mM Tris–acetate, 5 mM sodium acetate, 5 mM MgCl2, 0.1 mM EDTA, pH 5.5), and the proportion of TFO displaced from its binding site on the plasmid was quantified using a Molecular Dynamics Typhoon phosphorimager and ImageQuant software.
Cloning, expression, digestion and pull-down assay of MfdTEV450
The MfdTEV450 protein contains a tobacco etch virus (TEV) protease site (amino-acid sequence ‘ENLYFQG’) inserted after residue Leu450 in Mfd, with Gly451 already present as the last residue in the recognition site. For mutagenesis, 18 nt encoding for ENLYFQ were inserted into the Mfd expression plasmid (described above) using the Stratagene QuikChange Site-Directed Mutagenesis protocol modified for large insertions (30). Expression and purification was as for the wild-type Mfd containing a C-terminal His-tag described above. For protease digests, 0.28 mg/ml (2 µM) MfdTEV450 was incubated with 0.07 Units/µl acTEV protease (Invitrogen) for 27 h at room temperature in a buffer containing 50 mM Tris–HCl pH 8.0, 100 mM NaCl, 4 mM Mg acetate, 4 mM ATP, 0.5 mM EDTA and 1 mM DTT. Digestion products were analyzed on 10% SDS–PAGE gels. For the pull-down assay, a suspension of 50 µl of Ni-NTA beads (Qiagen) was equilibrated in 100 mM NaCl, 10 mM Tris–HCl, pH 8.2. 100 µl of digested MfdTEV450 sample was added and the suspension was incubated for 30 min at room temperature while gently agitating. The supernatant was removed and the beads were washed with 100 µl 100 mM NaCl, 10 mM Tris–HCl pH 8.2 and then with 100 µl 500 mM NaCl, 10 mM Tris–HCl pH 8.2. Bound protein was eluted with 500 mM EDTA, 500 mM NaCl, 10 mM Tris–HCl pH 8.2. Samples were analyzed on 10% SDS–PAGE gels.
Thermal denaturation monitored by circular dichroism
Circular dichroism(CD) data was measured on a Jasco Spectro polarimeter model J-715 (Jasco, Easton MD) and analyzed using the Jasco Spectra Manager software version 1.53.01. All samples were measured in quartz spectrophotometric stopper-top cells with a 1-mm pathlength (Starna, Atascadero CA). Final sample volume was 350 µl, and buffer conditions were: 500 mM NaCl, 10 mM Tris–HCl, pH 8.2. Thermal melts monitored by CD were obtained by raising the temperature from 20°C to 80°C at a rate of 1°C/min. Changes in CD signal were monitored at a wavelength of 220 nm. Sample concentrations were: 1 mg/ml for full length Mfd, 1.25 mg/ml for MfdN, 0.9 mg/ml for MfdC and 0.5 mg/ml for MfdTEV450. Samples were taken every 0.5°C or 0.2°C, with a band width of 2.0 nm, a response time of 4 s, and sensitivity set to ‘Standard’ (100 mDeg). Scans were normalized by linear fit of data points before the transition [‘f(T)’] and after the transition [‘u(T)’] and calculating normalized data according to: normalized(T) = [measured(T) – f(T)]/[u(T) – f(T)]
Crystallization, data collection and structure determination of MfdN
Crystals of MfdN were grown by sitting drop vapor diffusion at room temperature, equilibrating a mixture of 1.25 µl of protein solution (10 mg/ml MfdN in 100 mM NaCl, 20 mM HEPES pH 7.5), 1.25 µl of precipitant solution (8% w/v PEG 4000, 100 mM NaAcetate, pH 4.6) and 1 µl of 20 mM cobalt (II) chloride solution against a reservoir of 250 mM NaCl. Diffraction data at the National Synchrotron Light Source (Brookhaven, NY) were collected on beam line X26C at a wavelength of 1.072 Å. A 360 frame data set was collected at an oscillation of 0.5° per frame and a crystal-to-detector distance of 150 mM for crystals of space group P21212 and unit cell dimensions a = 84.06 Å, b = 157.65 Å and c = 35.96 Å. Data were indexed, scaled, and integrated using the HKL 2000 software (31). Structure determination and refinement was done with the CCP4 suite (32). The structure was solved by molecular replacement with MOLREP (33) using the first 470 residues of the full length protein [pdb entry 2EYQ (6)] as search model, and refined with REFMAC (34), with manual rebuilding using the ‘O’ software (35).
RESULTS
MfdC shows enhanced motor activity compared to the full length protein
To determine whether interactions between N-terminal and C-terminal domains are instrumental in downregulating Mfd in the absence of stalled RNAP, we prepared and characterized a truncated form of Mfd lacking the three N-terminal domains. Our MfdC protein contains residues 451–1188 of the E. coli protein (Figure 1) and, in contrast to wild-type Mfd, appears to be toxic when overexpressed in E. coli (transformation of MfdC into expression hosts required a system with tightly regulated expression). To assess the functional effects of removing domains 1–3 from Mfd, we compared the ATPase activity and RNAP removal activity of MfdC to that of wild-type Mfd (Figure 2). MfdC shows highly elevated ATPase activity compared to full length Mfd. Fitting the kinetic data to Michaelis–Menten kinetics, we observed a kcat that is 22-times higher for MfdC than that of the full length enzyme. The large inhibitory effect of MfdN:MfdC interactions on hydrolysis of ATP is allosteric as the ATPase active site is far removed from the MfdN:MfdC contacts. For related helicases, it has been established that ATPase activity drives large domain motions with an opening and closing of the cleft between the two motor domains (16). Our data suggest that interactions between the N-terminal and the C-terminal domains in Mfd hinder these rearrangements; the MfdC protein, which is not subject to these restraints, shows much higher activity. ATP binding, on the other hand, seems independent of MfdN:MfdC interactions as judged by the similarity in Km, suggesting that the conformational changes necessary for initial ATP binding are local in nature and not restrained by MfdN:MfdC interactions.
Figure 2.

The MfdC protein (Mfd lacking domains 1–3) displays elevated ATPase and RNAP removal activity. (A and B) Comparison of the ATPase activity of full length Mfd and MfdC. Rates were measured at 37°C using an ATP–NADH-coupled assay, with Mfd and MfdC at a concentration of 3 and 0.1 μM, respectively. Rates, shown as the average of three independent experiments with standard error, observed at increasing ATP concentrations were fitted to a standard Michaelis–Menten curve. (C) Stimulation of ATPase activity in the presence of double-stranded DNA measured at 4 mM ATP, with Mfd at 1 μM and MfdC at 0.1 μM, respectively. Solid lines are a fit to a 1 : 1-binding isotherm. Rates are obtained as averages of three independent measurements, and are shown relative to rates observed in absence of DNA, with standard errors indicated. (D) RNAP-displacement activity monitored in vitro by EMSA. Data points represent the percentage of elongation complex remaining relative to the amount at t = 0, and are the average of three independent experiments.
Coupling between ATPase activity and DNA-binding events is crucial to DNA motor activity. For many helicases and translocases, DNA binding stimulates ATPase activity, and cofactor-binding modulates DNA affinity. In contrast, Mfd shows only moderate stimulation of ATPase activity in the presence of DNA (4,36). When we added short duplex DNA to MfdC, we found a 2-fold stimulation of ATPase activity compared to only ∼1.3-fold stimulation in the full length enzyme [Figure 2C and (4)], demonstrating that deletion of the N-terminal three domains not only affects ATP hydrolysis, but also the consequences of DNA interaction. Fitting the simplest 1 : 1 binding equation to the kinetic data, we obtain an apparent Kd of MfdC for DNA of ∼100 nM, similar to that obtained for wild-type Mfd.
To test whether the elevated ATPase activity and higher degree of DNA stimulation (observed in the absence of stalled RNAP) supports more efficient motor activity, we compared the RNAP removal activity of MfdC with that of full length Mfd (Figure 2D). While stalled RNAP is removed with a half-life of ∼80 s in the presence of full length Mfd, in the presence of MfdC the half-life is reduced to <10 s (the earliest time point available in this assay). Truncation of the three N-terminal domains of Mfd therefore increases both the ATPase activity and the RNAP-displacement activity, indicating that the MfdC protein retains the ability to couple ATP hydrolysis to motor activity.
MfdC shows a DNA translocase activity that is absent in the full length protein
DNA translocation by full length Mfd can be detected only in the presence of stalled RNAP (3,4). However, deletion of domain 7 of Mfd leads to gain of function in a translocase assay that measures the ATP-dependent TFO displacement from duplex DNA (4). We tested MfdC in this assay to see whether the enhanced ATPase phenotype reflected derepressed TFO displacement activity. Indeed, deletion of domains 1, 2 and 3 turns Mfd into a DNA motor that translocates on DNA in the absence of RNAP (Figure 3). The observation that both deletion of MfdN (this work) and deletions in MfdC (4) lead to gain of function in this assay suggests that interactions between MfdN and MfdC play an important role in regulating translocase activity. In the full length enzyme, modulating the strength of these interactions during transcription-coupled repair could act as a regulatory switch.
Figure 3.

MfdC shows gain of function in a translocase assay. A triplex-containing substrate consisting of a 32P-labeled TFO bound to a supercoiled plasmid was incubated with 250 nM full length Mfd or MfdC, and reactions were initiated by the addition of 2 mM ATP. Samples were removed at certain intervals, quenched, and analyzed by gel electrophoresis. The data show the percentage of TFO displaced from the substrate at each time point, and are the average of three independent experiments, with standard deviations shown.
Cleaving the covalent linker between MfdN and MfdC leads to enhanced ATPase activity and dissociation of the two proteolytic fragments
In the wild-type enzyme, MfdN and MfdC are covalently connected by a 23 amino-acid linker (Gly 451–Asn473). In the crystal structure of apo-Mfd, this linker shows a largely extended conformation and high temperature factors, suggesting flexibility. To test the role of the linker in maintaining MfdN:MfdC interactions, we designed a variant of Mfd with a linker cleavable by the site-specific protease from tobacco etch virus (TEV protease). Because of its high specificity (it recognizes a site spanning seven amino acids), TEV protease has been used extensively for cleaving affinity tags and analyzing conformations in vitro and in vivo (37,38). We inserted the TEV protease site after position 450 into the wild-type sequence to obtain the cleavable MfdTEV450 protein. This allows us to compare properties of a single batch of protein before and after linker cleavage. The mutation has little effect on ATPase activity (Table 1) and on thermal stability (Supplementary Figure S1). Proteolytic cleavage yielded fragments of the expected size, and ∼60% of the sample was cleaved after 27 h at room temperature (Figure 4, compare lanes 1 and 6). The ATPase activity of a MfdTEV450 sample that had undergone proteolytic digest for 2 days (>95% cleaved) was significantly higher than that of the control lacking protease treatment (Table 1). Comparison with ATPase rates for MfdC and full length Mfd measured under identical conditions demonstrate that ATPase activity is downregulated when MfdN is present in cis (intact MfdTEV450), but not when it is present in trans (cleaved MfdTEV450 with both fragments present in the assay).
Table 1.
ATPase activity of Mfd variants
| Sample | Activity (mM ATP/mM enzyme/min) |
|---|---|
| Intact MfdTEV450 | 3.3 ± 0.3 |
| Cleaved MfdTEV450 | 78 ± 14 |
| Mfd wild-type | 4.2 ± 0.3 |
| MfdC | 89 ± 4 |
ATPase activity was measured in the presence of 4 mM ATP.
Figure 4.

Upon cleavage of the covalent linker between MfdN and MfdC, the proteolytic fragments dissociate. MfdTEV450 (lane 1) treated with TEV protease for 27 hours yielded two fragments (lane 6, digest is about 60% complete). Interactions between fragments (200 µM in the assay) were probed in a pull down with Ni beads, making use of the C-terminal His-tag of MfdTEV450. Most of the N-terminal fragment is found in the supernatant (lane 2) and the low salt wash (lane 3). Lane 4: high salt wash; lane 5: elution of the C-terminal fragment and the intact MfdTEV450 protein. Numbers on the right indicate molecular weight in kDa of standard proteins in lane M.
These finding raise the question of whether MfdN:MfdC interactions are sufficiently strong to remain intact after cleavage of the linker. Using the cleaved MfdTEV450 sample, we pulled down the His-tag containing species (C-terminal fragment corresponding to MfdC, and uncleaved full length Mfd) with Nickel chelating beads. Separating and visualizing the contents of fractions on a gel shows the C-terminal fragment (and the undigested protein) bound to beads, but did not pull down the N-terminal fragment, which was detected in the supernatant (Figure 4). This result, obtained at a protein concentration of 200 nM, shows that interactions between MfdN and MfdC are too weak to be detected at these concentrations.
The conformation of MfdN is independent of interactions with MfdC
To understand the conformational changes underlying Mfd function, it is important to know whether MfdN is flexible or acts as a rigid unit, and whether its structure depends on interactions with MfdC. The MfdN:MfdC contacts observed in the crystal structure of full length Mfd are substantial (Figure 1), suggesting that they might play a role in the stability of MfdN and MfdC. We already established that independently expressed MfdC is functional, implying that it folds properly, and now explore whether MfdN adopts the native fold independently of MfdC. We cloned MfdN as a construct encoding for domains 1 through 3, plus the larger part of the linker that connects domain 3 with domain 4 (up to residue 470, cf. Figure 1). MfdN expresses at high levels in E. coli (>40 mg/l medium) and is soluble to at least 15 mg/ml. These observations are consistent with the idea that MfdN and MfdC could act as individual but tethered entities during transcription-coupled repair. To study the potential for conformational change and the role of inter-domain contacts in the three N-terminal domains of Mfd, we have determined the crystal structure of MfdN. In contrast to a structure of residues 1–333 of Mfd reported previously (8), which lacks parts of domain 1 and all of domain 3, the MfdN structure reported here is a complete view of how the three N-terminal domains fold independently of interactions with MfdC. Comparing the MfdN structure with the full length Mfd structure (6) will show whether MfdN:MfdC interactions have any effect on the structure of domains 1 through 3.
Crystals of MfdN diffracted to 1.92 Å, and the structure was determined by molecular replacement (Table 2). Residues 451 through 470 (and the C-terminal His-tag) are missing from the structure due to disorder. This suggests that the linker does not have a preferred orientation with respect to MfdN, and is positioned in the apo-Mfd structure through its interactions with MfdC and the constraints imposed by fixing its two ends. The refined MfdN model superposes well with the corresponding part of the apo-Mfd structure (Figure 5A), with an RMSD of 449 corresponding Cα atoms of 1.1 Å (the RMSD of the same set of Cα atoms of the two copies present in the apo-structure is 0.9 Å). Aside from small local differences, which might be due to differences in resolution or packing, the folds are identical. Even though surface contacts of MfdN are significantly different than in the apo-Mfd crystal structure (Supplementary Figure S1), no large conformational change is observed, i.e. the relative domain orientations are highly similar. The interface between domains 1 and 3, dominated by hydrophobic interactions and organized around the N-terminal end of an α-helix (residues 26–30), is well-ordered in the MfdN structure (Figure 5B). In the related UvrB fold, the corresponding residues are part of the ATP-binding P-loop, the cleft between domains 1 and 3 is largely hydrophilic, and multiple conformations are observed in multiple crystal forms (39). In the context of the compact apo-conformation of Mfd (6), the invariable structure of MfdN suggests that as long as inter-domain contacts between the N-terminal and C-terminal domains are maintained, the N-terminal domains 1 through 3 form a rigid clamp restraining the activity of the C-terminal motor domains.
Table 2.
Data collection and refinement statistics
| Data collectiona | |
| Resolution of data (Å) | 50–1.92 (1.99–1.92) |
| Completeness (%) | 99.9 (99.0) |
| Redundancy | 6.9 (5.9) |
| Rmerge | 0.055 (0.649) |
| I/σ(I) | 27.9 (2.6) |
| Refinement | |
| Resolution (Å) | 50–1.95 |
| Reflections used in refinement | 34 090 |
| Rcryst | 0.208 |
| Rfree | 0.257 |
| R.m.s.d. bond lengths (Å) | 0.013 |
| R.m.s.d. bond angles (degrees) | 1.38 |
| No. of protein atoms | 3569 |
| No. of solvent atoms | 318 |
| No. of ligand atoms | 1 |
| Ramachandran statisticsb (%) | 90.1/9.7/0.3/0.0 |
aData in parentheses refer to the highest resolution shell.
bRamachandran statistics indicate the fraction of residues in the most favored, additionally allowed, generously allowed and disallowed regions, respectively, of the Ramachandran diagram as defined by PROCHECK (47).
Figure 5.

The crystal structure of MfdN suggests that the N-terminal domains of Mfd form a rigid unit. (A) Crystal structure of MfdN superimposed with corresponding residues in the structure of full length Mfd. Colored thick lines: Cα trace of MfdN; thin gray lines: Cα trace of the corresponding residues in chain A and chain B of full length Mfd, PDB ID 2EYQ (6). Domains 1, 2, 3 of MfdN are shown in yellow, orange and green, respectively. Although residues 451–470 are present in the MfdN construct, they are not visible in the electron density due to disorder. (B) Detail of the interactions between domain 1 (yellow) and domain 3 (green) in MfdN. The 2FO–FC electron density (contoured at 1 σ) around selected residues forming the interface is shown, along with the atomic model in all atom representation. Figure 5B was prepared with Pymol (W. L. DeLano, DeLano Scientific, 2002).
Intramolecular interactions with MfdN enhance the thermal stability of MfdC in cis, but not in trans
Domains 1 through 3 in MfdN contact both motor domains 5 and 6 and the regulatory domain 7. Comparison of melting curves of isolated MfdN and MfdC with that of wild-type Mfd can serve as a measure of the strength of MfdN:MfdC interactions in the full length enzyme. Thermal denaturation of samples, monitored by CD spectroscopy, was non-reversible in all cases. Instead of deriving thermodynamic parameters from the data, we therefore compare the observed transition midpoints as a measure of stability. Stability increases in the order of MfdC, full length Mfd, and MfdN, with ∼15°C difference in transition midpoints of the two parts of Mfd (Figure 6A). The large difference between the transition midpoints of full length Mfd and MfdC (∼8°C) suggests that MfdN:MfdC interactions significantly stabilize the motor domains in Mfd.
Figure 6.

MfdN stabilizes MfdC in cis, but not in trans. (A) Comparison of thermal stabilities of Mfd, MfdN and MfdC monitored by CD spectroscopy. (B) Thermal stability of an equimolar mixture of MfdN and MfdC. The calculated melting curve was computed as the weighted average of the MfdN and MfdC data from the panel (A) and, apart from some differences early in the melting process, coincides well with the data for the equimolar mixture, which is very different from the melting curve of full length Mfd shown in panel (A).
To test whether MfdN could enhance the thermal stability of MfdC in trans, we measured the melting curves of equimolar mixtures of separately expressed MfdN and MfdC (Figure 6B). The mixture showed two inflection points close to the single inflection points observed for the individual measurements of the components. The experimental curve corresponded closely to one calculated from the melting curves of MfdN and MfdC measured separately (averaged with weighting factors of 0.35 and 0.65 to account for the different signal of the two components, Figure 6B), and was clearly different from that of the full length Mfd protein (Figure 6A). Combined with pull-down (Figure 4) and ATPase data (Table 1), this shows that MfdN and MfdC do not interact significantly in the absence of a covalent tether.
DISCUSSION
Regulation of Mfd motor activity
We observed that deleting domains 1–3 of Mfd increases its ATPase activity more than 20-fold, and that the deletion mutant has elevated RNAP removal activity. Deletion of domain 7 has a similar effect on Mfd (4). It is remarkable that deletions in two distinct parts of the Mfd protein lead to similar changes in activity. What might be the mechanism of this derepression caused by deletion of MfdN or domain 7? The ATPase active site located on motor domains 5 and 6 is at a distance from the deleted domains, i.e. the activation mechanism is allosteric. Smith et al. (4) suggested that deletion or movement of domain 7 might affect the ATPase activity of Mfd by conformational changes transmitted within MfdC. An alternative mechanism suggested by our current work in the context of motor domain movements observed in related helicases is that MfdN:MfdC interactions may inhibit Mfd by restraining the movement of the protein’s two motor domains.
Motor domain movements are an integral part of ATP hydrolysis by SF1 and SF2 helicases: whilst the N-terminal motor domain (domain 5 in Mfd) is sufficient for ATP binding via its P-loop, conserved residues in the C-terminal motor domain (domain 6 in Mfd) are essential for ATP hydrolysis. The structural basis for this requirement is evident in the crystal structure of the SF2 helicase Vasa, where the interface between the motor domains closes upon binding of a non-hydrolysable ATP analog, enabling multiple interactions between the C-terminal motor domain and ATP (14). Specifically, a conserved arginine of helicase motif VI (corresponding to R905 in Mfd) approaches the scissile bond of ATP, resulting in a Cα–Cα distance of 4.1 Å to a conserved glycine (corresponding to G631 in Mfd) in helicase motif I. In the apo-Mfd structure, the corresponding distance is 18 Å, suggesting that the motor domains must reorient substantially to reduce this distance by ∼12 Å. In Mfd, domain reorientations have to maintain backbone connectivity (Figure 1) and are restrained by non-covalent inter-domain contacts. There are three inter-domain contacts between MfdN and MfdC (1 : 6, 2 : 7, 3 : 5, highlighted in Figure 7A), amounting to an area of more than 1500 Å2 [calculated from PDB ID 2EYQ, (6)]. This is in contrast to many other structures of helicases and translocases, in which the C-terminal motor domain appears less restrained by inter-domain contacts [e.g. in UvrB (40) or in PcrA (41)]. Based on the ATP-bound Vasa structure (14), we modeled an Mfd conformation in which helicase motifs I and VI are in close proximity while treating MfdN as a rigid unit, guided by our data on the structure and thermal stability of MfdN (Figures 5 and 6). This led to substantial disruption of MfdN:MfdC contacts (Supplementary Figure S3). We conclude that while MfdN:MfdC interactions are in place, MfdN will act as a clamp that restrains any movement necessary for ATP hydrolysis (Figure 7B).
Figure 7.
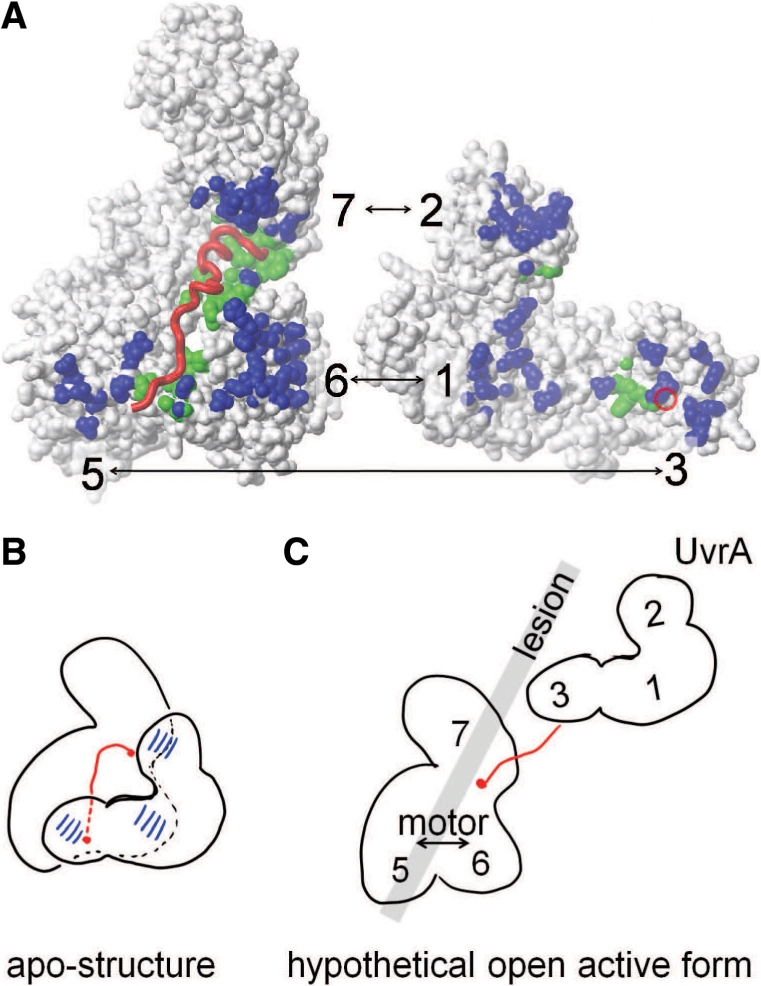
Mechanistic proposal for the role for MfdN:MfdC contacts in regulating Mfd. (A) Interface between MfdN, linker and MfdC in the apo-structure (2EYQ). To afford a view into the interface, coordinates of MfdN were rotated by 180° about a vertical axis and shifted to the right. MfdN and MfdC are shown as white surfaces, and the linker as a red worm. The red circle on the surface of MfdN indicates the N-terminal connection of the linker. Atoms in MfdN closer than 5 Å to atoms in MfdC and vice versa are shown in blue. Atoms in MfdN and MfdC closer than 5 Å to atoms in the linker are shown in green. (B) Schematic of the inter-domain contacts and the path of the linker region through the MfdN:MfdC interface in the apo-conformation. (C) Hypothetical model of an open conformation lacking all MfdN:MfdC interactions. Mfd domains are numbered, and the gray bar indicates the putative path of DNA and the location of the lesion when Mfd binds to a stalled RNAP (the latter is omitted for clarity). In this scenario, the UvrA-binding site in domain 2 is no longer occluded and MfdN is mobile, tethered only by the linker to MfdC. The interactions between the N-terminal portion of the linker with the motor domains 5 and 6 are absent, and the motor domains are no longer restrained by contacts with MfdN. Thus, a switch to the proposed open conformation would activate UvrA recruitment and motor function in a concerted manner.
The absence of MfdN:MfdC interactions in the MfdC protein explains why MfdC displays an elevated ATP activity, while the ATPase activity of full length Mfd is much lower than that of typical SF2 motor proteins. The suggested mechanism of activation raises the questions how full length Mfd hydrolyzes ATP. We propose that the dormant closed form switches to an active open form of Mfd that lacks some or all contacts between MfdN and MfdC (Figure 7C). In our model, at least one of the motor domains would have to release its inter-domain contacts with MfdN to allow motions of the motor domains in order for ATP hydrolysis to occur. Inter-domain or inter-protein interactions with the motor domains are emerging as a common theme in the regulation of nucleic-acid helicases and translocases. For example, the C-terminal domain of the DNA repair protein UvrB inhibits its ATPase activity (42) and the DEAD-box helicase eIF4AIII is inhibited by other subunits in the exon junction complex (18,19). In both examples, the inhibitory inter-domain or inter-protein interactions occur on the face of the molecule that binds ATP and nucleic acid (the ‘business’ face of the motor), and can presumably directly interfere with these processes. In contrast, MfdN binds to MfdC on the ‘backside’, away from the conserved helicase motifs involved in ATP binding and hydrolysis (Figure 1). The arrangement of regulatory elements is reminiscent of reverse gyrase, which harbors helicase and topoisomerase domains in a single subunit, and in which deletion of the topoisomerase domain leads to 10-fold higher ATPase activity (21): the topoisomerase domain interacts with the ‘backside’ of the helicase domains and is suggested to modulate ATPase activity by coupling it to topoisomerase activity.
The truncations in MfdC and MfdΔD7 leading to higher activity are non-overlapping, but have a common feature; domain 2:domain 7 contacts are absent in both, because one of the interaction partners is missing. Therefore, both deletions could affect ATPase activity through conformational change transmitted within MfdC or through MfdN-motor domain interactions, albeit indirectly in some scenarios. If we compare MfdC and MfdΔD7, both show gain of function in the TFO removal assay, but the increase in ATPase activity is higher in MfdC (22-fold) than in MfdΔD7 (5-fold). The simplest explanation for these results is that removal of MfdN directly removes the cause of auto-inhibition while deletion of domain 7 acts indirectly through weakening of the MfdN:MfdC interface because domain 2:domain 7 interactions are absent. This would argue that the primary mechanism for auto-inhibition of full length Mfd in the absence of RNAP is the interaction of domains 1 and 3 with the motor domains. Loss of interactions between MfdN and MfdC would lift the restraints on motor domain rearrangements, and simultaneously render the UvrA-binding site located in domain 2 accessible for UvrA recruitment. Given that MfdN folds into a native conformation in the absence of interactions with MfdC, the simplest model is that all MfdN-MfdC inter-domain contacts are lost in an activated intermediate of transcription-coupled repair, with MfdN detaching as a rigid unit tethered to MfdC via an unstructured linker. This loss of all MfdN:MfdC contacts would activate motor activity and UvrA binding in a single conformational switch (Figure 7C).
Strength of MfdN:MfdC interactions
If activities of Mfd are regulated by a central conformational switch from a dormant closed form to an active open form lacking some or all inter-domain contacts between MfdN (domains 1–3) and MfdC (domains 4–7), these interactions would have to be sufficiently weak to be opened by physiological triggers. We show substantial effects of domains 1–3 on motor activity (Figure 2) and thermal stability (Figure 6) in cis, but do not observe binding (Figure 4) or effects on motor activity (Table 1) in trans. Is there a discrepancy between results obtained in cis and in trans? In general, intermolecular binding of molecules present at submicromolar concentration can be enhanced by tethering the molecules with a linker, owing to the higher effective local concentration of the binding partners in the intramolecular scenario. The effective local concentration is dependent on the length and rigidity of the tether between the binding partners (43,44). The covalent linker between MfdN and MfdC is ∼23 amino acids in length, appears rather flexible in the apo-Mfd structure with an end-to-end distance of 45Å (6) and is disordered in the MfdN structure (Figure 5). Assuming an open state in which MfdN and MfdC are tethered by this flexible linker, we can estimate the local concentration as ceffective = 1 mM by comparison to model proteins containing flexible linkers (44). An intramolecular equilibrium close to unity but favoring the closed conformation would be consistent with the stabilizing and inactivating effect of MfdN on MfdC in the full length enzyme. However, the intermolecular dissociation constant would be in the micromolar range, i.e. binding interactions after linker cleavage would be too weak to detect in our assays. Thus, the lack of effects of MfdN on MfdC in trans is not inconsistent with the strong effects observed in cis, but rather supports the idea that interactions are sufficiently weak for a switch to an open conformation as a regulatory mechanism.
Biological role of Mfd regulation
Both deletion of domains 1 through 3 and deletion of domain 7 lead to hyperactive forms of Mfd, with an elevated ATPase activity that is more stimulated by DNA, and enhanced RNAP removal activity compared to the full length enzyme. This demonstrates the possibility of activating the core motor of Mfd substantially, and these deletions appear to mimic the activation of wild-type Mfd bound to a stalled elongation complex (4). It is unknown how many rounds of ATP hydrolysis are required to remove RNAP, at which rate ATP hydrolysis occurs when Mfd acts on stalled RNAP, and whether ATP hydrolysis is the rate-determining step of RNAP removal; therefore, interpreting kinetics of RNAP displacement in terms of ATPase kinetics is not possible at this point. While it is unlikely that transcription-coupled DNA repair is completed before the next transcribing RNAP approaches the site of damage [especially for highly transcribed genes (45)], RNAP removal has to be at least faster than NER to achieve the observed preferential repair of transcribed genes. The envisioned biological role of switching between auto-inhibited and derepressed forms of Mfd is that binding of Mfd to a stalled RNAP would trigger a switch in Mfd that derepresses the DNA translocase activity and, at the same time, removes the steric inhibition on the UvrA-binding site so that UvrA recruitment is coordinated with RNAP removal. Regulation might play a role in protecting the cell from off-target translocation activity [in fact, the RNAP-independent translocase activity observed in MfdC might explain the cytotoxicity of this protein: hyperactive mutants of the SF1 helicase UvrD lead to UV sensitivity, among other phenotypes (20)]. Once both RNAP and UvrA are released Mfd will switch back to the closed, auto-inhibited conformation. This sequence of events would ensure that translocase activity does not interfere with protein–DNA interactions in general. It also would link Mfd-UvrA interactions to RNAP removal, ensuring that UvrA is not inhibited by untimely interactions with Mfd.
ACCESSION NUMBER
PDB 3HJH.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
American Cancer Society [RSG-08-055-01 to K.T.]; Biotechnology and Biological Sciences Research Council [BB/E004695 to N.J.S.]. Funding for open access charge: American Cancer Society [RSG-08-055-01 to K.T.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Jennifer Doebbler for help with diffraction data collection.
REFERENCES
- 1.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 2.Savery NJ. The molecular mechanism of transcription-coupled DNA repair. Trends Microbiol. 2007;15:326–333. doi: 10.1016/j.tim.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Park JS, Marr MT, Roberts JW. E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 4.Smith AJ, Szczelkun MD, Savery NJ. Controlling the motor activity of a transcription-repair coupling factor: autoinhibition and the role of RNA polymerase. Nucleic Acids Res. 2007;35:1802–1811. doi: 10.1093/nar/gkm019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JS, Roberts JW. Role of DNA bubble rewinding in enzymatic transcription termination. Proc. Natl Acad. Sci. USA. 2006;103:4870–4875. doi: 10.1073/pnas.0600145103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deaconescu AM, Chambers AL, Smith AJ, Nickels BE, Hochschild A, Savery NJ, Darst SA. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 7.Selby CP, Sancar A. Structure and function of transcription-repair coupling factor. I. structural domains and binding properties. J. Biol. Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- 8.Assenmacher N, Wenig K, Lammens A, Hopfner KP. Structural basis for transcription-coupled repair: the N terminus of Mfd resembles UvrB with degenerate ATPase motifs. J. Mol. Biol. 2006;355:675–683. doi: 10.1016/j.jmb.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 9.Truglio JJ, Croteau DL, Skorvaga M, DellaVecchia MJ, Theis K, Mandavilli BS, Van Houten B, Kisker C. Interactions between UvrA and UvrB: the role of UvrB's; domain 2 in nucleotide excision repair. EMBO J. 2004;23:2498–2509. doi: 10.1038/sj.emboj.7600263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pakotiprapha D, Inuzuka Y, Bowman BR, Moolenaar GF, Goosen N, Jeruzalmi D, Verdine GL. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol. Cell. 2008;29:122–133. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pakotiprapha D, Liu Y, Verdine GL, Jeruzalmi D. A structural model for the damage-sensing complex in bacterial nucleotide excision repair. J. Biol. Chem. 2009;284:12837–12844. doi: 10.1074/jbc.M900571200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith AJ, Savery NJ. RNA polymerase mutants defective in the initiation of transcription-coupled DNA repair. Nucleic Acids Res. 2005;33:755–764. doi: 10.1093/nar/gki225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989;17:4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein drosophila vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 15.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 16.Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008;37:317–336. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
- 17.Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, Herdy B, Sonenberg N, Wagner G. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell. 2009;136:447–460. doi: 10.1016/j.cell.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 19.Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Centore RC, Leeson MC, Sandler SJ. UvrD303, a hyperhelicase mutant that antagonizes RecA-dependent SOS expression by a mechanism that depends on its C terminus. J. Bacteriol. 2009;191:1429–1438. doi: 10.1128/JB.01415-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.del Toro Duany Y, Jungblut SP, Schmidt AS, Klostermeier D. The reverse gyrase helicase-like domain is a nucleotide-dependent switch that is attenuated by the topoisomerase domain. Nucleic Acids Res. 2008;36:5882–5895. doi: 10.1093/nar/gkn587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGlynn P, Mahdi AA, Lloyd RG. Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res. 2000;28:2324–2332. doi: 10.1093/nar/28.12.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahdi AA, Briggs GS, Sharples GJ, Wen Q, Lloyd RG. A model for dsDNA translocation revealed by a structural motif common to RecG and mfd proteins. EMBO J. 2003;22:724–734. doi: 10.1093/emboj/cdg043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 25.Chambers AL, Smith AJ, Savery NJ. A DNA translocation motif in the bacterial transcription–repair coupling factor, mfd. Nucleic Acids Res. 2003;31:6409–6418. doi: 10.1093/nar/gkg868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. pp. 571–607. [Google Scholar]
- 27.Barnett RE. Effect of monovalent cations on the ouabain iniibition of the sodium and potassium ion activated adenosine triphosphatase. Biochemistry. 1970;9:4644–4648. doi: 10.1021/bi00826a004. [DOI] [PubMed] [Google Scholar]
- 28.Johnson RS, Chester RE. Stopped-flow kinetic analysis of the interaction of escherichia coli RNA polymerase with the bacteriophage T7 A1 promoter. J. Mol. Biol. 1998;283:353–370. doi: 10.1006/jmbi.1998.2101. [DOI] [PubMed] [Google Scholar]
- 29.Firman K, Szczelkun MD. Measuring motion on DNA by the type I restriction endonuclease EcoR124I using triplex displacement. EMBO J. 2000;19:2094–2102. doi: 10.1093/emboj/19.9.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange site-directed mutagenesis. BioTechniques. 1999;26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022. [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47 (Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 36.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 37.Nallamsetty S, Kapust RB, Tozser J, Cherry S, Tropea JE, Copeland TD, Waugh DS. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr. Purif. 2004;38:108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 38.Gouffi K, Gerard F, Santini CL, Wu LF. Dual topology of the Escherichia coli TatA protein. J. Biol. Chem. 2004;279:11608–11615. doi: 10.1074/jbc.M313187200. [DOI] [PubMed] [Google Scholar]
- 39.Theis K, Skorvaga M, Machius M, Nakagawa N, Van Houten B, Kisker C. The nucleotide excision repair protein UvrB, a helicase-like enzyme with a catch. Mutat. Res. 2000;460:277–300. doi: 10.1016/s0921-8777(00)00032-x. [DOI] [PubMed] [Google Scholar]
- 40.Theis K, Chen PJ, Skorvaga M, Van Houten B, Kisker C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999;18:6899–6907. doi: 10.1093/emboj/18.24.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, DellaVecchia MJ, Skorvaga M, Croteau DL, Erie DA, Van Houten B. UvrB domain 4, an autoinhibitory gate for regulation of DNA binding and ATPase activity. J. Biol. Chem. 2006;281:15227–15237. doi: 10.1074/jbc.M601476200. [DOI] [PubMed] [Google Scholar]
- 43.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J. Am. Chem. Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou HX. Quantitative account of the enhanced affinity of two linked scFvs specific for different epitopes on the same antigen. J. Mol. Biol. 2003;329:1–8. doi: 10.1016/s0022-2836(03)00372-3. [DOI] [PubMed] [Google Scholar]
- 45.Li S, Waters R. Induction and repair of cyclobutane pyrimidine dimers in the escherichia coli tRNA gene tyrT: Fis protein affects dimer induction in the control region and suppresses preferential repair in the coding region of the transcribed strand, except in a short region near the transcription start site. J. Mol. Biol. 1997;271:31–46. doi: 10.1006/jmbi.1997.1154. [DOI] [PubMed] [Google Scholar]
- 46.Koradi, Billeter, Wuthrich MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski RA, MacArthur MW, David S Moss DS, Thornton JM. Procheck – a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.