

Abstract
Monomeric sarcosine oxidase (MSOX) binds the L-proline zwitterion (pKa = 10.6). The reactive substrate anion is generated by ionization of the ES complex (pKa = 8.0). Tyr317 was mutated to Phe to determine whether this step might involve proton transfer to an active site base. The mutation does not eliminate the ionizable group in the ES complex (pKa = 8.9) but does cause a 20-fold decrease in the maximum rate of the reductive half-reaction. Kinetically determined Kd values for the ES complex formed with L-proline agree with results obtained in spectral titrations with wild type or mutant enzyme. Unlike wild type enzyme, Kd values with the mutant enzyme are pH-dependent, suggesting that the mutation has perturbed the pKa of a group that affects the Kd. As compared with wild type enzyme, an increase in charge transfer band energy is observed for mutant enzyme complexes with substrate analogs while a 10-fold decrease in the charge transfer band extinction coefficient is found for the complex with L-proline anion. The results eliminate Tyr317 as a possible acceptor of the proton released upon substrate ionization. Since previous studies rule out the only other nearby base, we conclude that L-proline is the ionizable group in the ES complex and that amino acids are activated for oxidation upon binding to MSOX by stabilization of the reactive substrate anion. Tyr317 may play a role in substrate activation and optimizing binding, as judged by the effects of its mutation on the observed pKa, reaction rates and charge transfer bands.
Monomeric sarcosine oxidase (MSOX)1 is a flavoprotein that contains covalently bound FAD [8a-(S-cysteinyl)FAD] (1). The enzyme catalyzes the oxidation of the methyl group in sarcosine (N-methylglycine) and the equivalent C-N bond in other secondary amino acids, such as L-proline. The crystal structure of free MSOX from Bacillus sp. B-0618 and complexes of the enzyme with various inhibitors have been determined (2–4). MSOX is a two-domain, 46 KDa protein with an overall topology similar to D-amino acid oxidase.
MSOX is a member of a growing family of enzymes that contain covalently bound flavin and catalyze similar oxidation reactions with different amine substrates (5–8). Despite considerable attention, important questions regarding the mechanism of flavin-dependent amine oxidation reactions remain unresolved. Postulated mechanisms differ with respect to: 1) the requirement for an active site base; 2) the acceptor of the α-hydrogen removed from the carbon atom in the C-N bond undergoing oxidation; 3) geometric constraints on the orientation of flavin, substrate and a putative active site base (Scheme 1). In the hydride transfer mechanism, flavin N(5) acts as the acceptor of the α-hydrogen in a reaction that does not require an active site base. Single electron transfer (SET) mechanisms are initiated by electron transfer from substrate amino group to generate a flavin/substrate radical pair (9). Polar mechanisms involve reversible nucleophilic addition of substrate amino group at flavin C(4a) to yield a covalent flavin-substrate intermediate (10). An active site base is required for the versions of the SET and polar mechanisms shown in Scheme 1 but base-independent variants have been suggested where the α-hydrogen is transferred to flavin N(5) as a hydrogen atom (11) or proton (12), respectively.
Scheme 1.

Possible mechanisms for sarcosine oxidation by MSOX. IA is the imino acid product of sarcosine oxidation (CH2=NH+-CH2-CO2−); FADH− is the 1,5-dihydroflavin anion.
With the possible exception of hydride transfer (13), it is generally agreed that the unprotonated amino group is the reactive species in flavin-dependent amine oxidation reactions. Since biologically relevant amines typically exhibit pKa values considerably above physiological pH, an active site base might be required to generate the unprotonated amine. Several observations indicate that MSOX binds the zwitterionic form of amino acid ligands. Although the enzyme is specific for secondary amines, tertiary amines like dimethylglycine act as competitive inhibitors. The crystallographic, spectral and theromodynamic properties of the MSOX·dimethylglycine complex strongly suggest that the inhibitor is bound as a zwitterion (4). The anaerobic half-reaction of MSOX with L-proline, a slow substrate, is rate-limiting during turnover and proceeds via a rapidly attained equilibrium (Kd) between free MSOX and the enzyme·substrate complex, followed by an essentially irreversible flavin reduction step (klim) (14) (Scheme 2). The kinetically determined Kd for the enzyme·L-proline complex is pH-independent in the pH range from 6.5 to 9.0. This indicates that MSOX binds the zwitterionic form of L-proline, the predominant species in solution at slightly acidic or alkaline pH values.
Scheme 2.
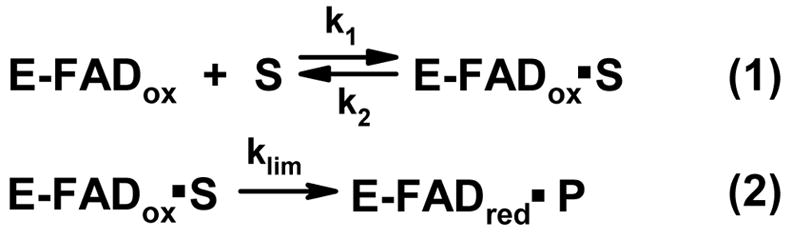
Proposed mechanism for the reductive half-reaction of MSOX with L-proline.
In contrast, observed values for klim are strongly pH-dependent, indicating that an ionizable group in the enzyme·L-proline complex must be unprotonated prior to flavin reduction. The oxidized enzyme·L-proline complex exhibits an absorption band at longer wavelengths that is attributable to flavin-ligand charge transfer interaction. This feature is expected only for a complex of oxidized flavin with the electron-rich L-proline anion. Significantly, development of the charge transfer band is pH-dependent, exhibiting a spectrally detectable pKa (pKa = 7.9) that is virtually identical to the kinetically determined pKa for an ionizable group in the enzyme·substrate complex (pKa = 8.0) (14).
The results suggest that the pKa of MSOX-bound L-proline may be decreased by more than 2.5 pH units as compared with free L-proline (pKa = 10.6). Alternatively, ionization of the protein-bound zwitterion may require proton transfer to an active site base with a pKa of 8.0. His269 or Tyr317 were identified as potential candidates for the active site base, as judged by the crystal structures observed for MSOX complexes with various inhibitors (2–4), including pyrrole-2-carboxylate (PCA) (Figure 1). In the MSOX·PCA complex, ND1 of His269 is located 4.17 Å from the nitrogen in PCA and 3.6 Å from the carbon atom in the inhibitor that corresponds to C(5) in L-proline or the methyl group in sarcosine. The corresponding distances for the phenolic oxygen of Tyr317 are 4.58 and 3.54 Å, respectively. Mutation of His269 to Asn reduces the rate of MSOX turnover with sarcosine or the reductive half-reaction with L-proline by nearly 2 orders of magnitude but does not eliminate the ionizable group in the enzyme·substrate complex (3). In this paper, we characterize the effect of mutating Tyr317 to Phe on MSOX catalysis.
Figure 1.
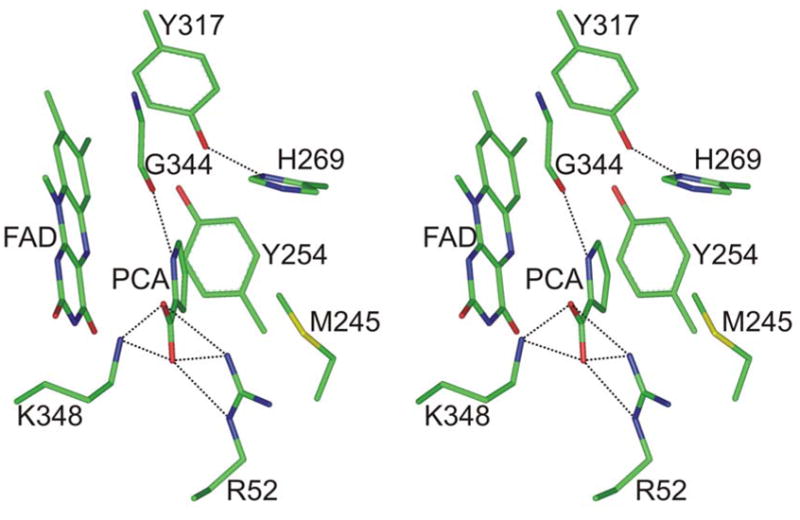
Stereoview of the active site in the MSOX complex with pyrrole-2-carboxylate (PCA). The figure was constructed using the previously reported structure (2). Atoms are colored according to the following colors: carbon, green; oxygen, red; nitrogen, blue. Hydrogen bonds to the ligand are indicated by dashed lines.
EXPERIMENTAL PROCEDURES
Materials
L-Proline was purchased from Sigma. Sarcosine, pyrrole-2-carboxylate and methylthioacetate were purchased from Aldrich. E. coli DH1 was obtained from the American Type Culture Collection (ACTT 33849). Plasmid pUC119 was from Worthington Biochemical Corporation. Primers were synthesized by MWG Biotech. dNTPs were from Qiagen. Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs.
Mutagenesis
The Tyr317Phe mutant was generated using PCR, the plasmid pMAW (1) as template and the overlap extension method described by Ho et al. (15). PCR reactions were performed using Pfu DNA polymerase (Stratagene) and a Hybaid Touchdown Thermocycler. PCR products and other DNA fragments were purified by agarose gel (1.5%) electrophoresis and recovered using the QiaQuick Gel Extraction kit (Qiagen). The left-hand fragment was generated using 5′-AGTTTTCggtaccGAATGTTATGAGCACAC-3′ as forward primer (START) and 5′-GTTTTCGTGaACATGCAGACTG-3′ as backward primer. The right-hand fragment was generated using 5′-CAGTCTGCATGTtCACGAAAAC-3′ as forward primer and 5′-CGACTCAACgaAtTCACTACTATTAGATAG-3′ as backward primer (END). The left- and right-hand fragments were combined using START and END as forward and backward primers, respectively. (Mutagenic sites in the primers are shown in lower case, codon 317 is shown in bold. The underlined bases in the primers START and END are KpnI and EcoR1 sites, respectively.) The PCR product was digested with KpnI and EcoR1, purified and subcloned between the KpnI and EcoR1 sites of plasmid pUC119. The resulting construct was used to transform E. coli DH1 cells to ampicillin resistance. For screening, plasmid DNA was isolated from randomly selected clones using the QiaPrep spin miniprep kit (Qiagen) and digested with KpnI and EcoR1. Plasmid pGZ18 exhibited the expected insert size, was isolated using the Qiagen midiprep kit (Qiagen) and sequenced across the entire insert. Sequencing was conducted by MWG Biotech.
Enzyme Purification and Assay
Wild type enzyme and the Tyr317Phe mutant were purified as previously described (1). During purification, enzyme activity was measured using the Nash assay and protein was determined using the Bio-Rad micro protein assay (1). Apparent steady-state kinetic parameters with sarcosine as substrate were obtained at 25 °C in air-saturated 100 mM potassium phosphate buffer, pH 8.0. Reaction rates were measured by monitoring hydrogen peroxide formation using a horseradish peroxidase coupled assay (16).
Spectroscopy and Data Analysis
Absorption spectra were recorded using an Agilent Technologies 8453 diode array or a Perkin-Elmer Lambda 2S spectrophotometer. Extinction coefficients were determined after enzyme denaturation with guanidine hydrochloride, as previously described (1). Spectral titrations with the Tyr317Phe mutant and methylthioacetate (MTA) or pyrrole-2-carboxylate (PCA) were conducted at 25 °C in 50 mM potassium phosphate buffer, pH 8.0. Spectral titrations with mutant or wild type MSOX and L-proline were conducted at 4 °C in various buffers, as indicated in the text. Spectra corresponding to 100% complex formation were calculated by using the observed dissociation constants and spectra recorded at the highest ligand concentration tested. The positions of charge transfer bands were estimated from difference spectra. This approach is especially useful when the charge transfer band is not well-resolved in the absolute spectrum owing to overlap with the red edge of the flavin absorption spectrum.
Reductive half-reaction studies with the Tyr317Phe mutant and L-proline were conducted at 4 °C in 100 mM potassium phosphate (pH ≤ 8.0), 100 mM sodium pyrophosphate (pH 8.2 to 9.2) or 100 mM sodium carbonate (pH ≥ 9.4) using special cuvettes with two sidearms. The cuvettes were made anaerobic by purging with argon. Reactions were initiated by rapidly tipping a small aliquot (15 μL) of a concentrated enzyme solution into a buffered solution of L-proline (585 μL) in the main compartment of the anaerobic cuvette. A single tip and return of the cuvette to the spectrophotometer was completed in less than 15 s.
Data analysis was conducted using the curve fit function in Sigma Plot (SPSS, Inc.).
RESULTS
General Properties of the Tyr317Phe Mutant
Mutation of Tyr317 to Phe does not significantly affect expression of MSOX. The mutant protein was purified to apparent homogeneity, as judged by SDS-PAGE, using the same procedure developed for wild type MSOX. The mutant enzyme contains covalently bound flavin. The absorption spectrum of the mutant exhibits a small (2 nm) hypsochromic shift of the 454 nm band but the spectral properties in the visible region are otherwise very similar to that observed for wild type MSOX (Figure 2). Although the extinction coefficient at 454 nm of the mutant enzyme is similar to that of wild type MSOX, the mutant exhibits a modest increase in the ratio A280/A454, suggesting a slightly lower incorporation of FAD (Table 1).
Figure 2.

Comparison of visible absorption spectra observed with the Tyr317Phe mutant and wild type MSOX (dashed and solid curves, respectively). Spectra were recorded in 50 mM potassium phosphate buffer, pH 8.0 at 25 °C and are normalized to the same absorbance at 454 nm.
Table 1.
Comparison of the spectral and catalytic properties of the Tyr317Phe mutant with wild type MSOX
| property | Tyr317Phe | wild type |
|---|---|---|
| absorption maxima (nm) | 372, 452 | 372, 454 |
| ε454 (M−1 cm−1) | 12,000 (12,100)a | 12,200b |
| A280/A454 | 7.2c | 5.8d |
| specific activity (U/mg)e | 8.0 | 39.3 |
| kcat (app) (min−1)f | 652 ± 3 | 2730 ± 30 |
| Km (app) (mM)f | 16.5 ± 0.2 | 4.5 ± 0.1 |
| kcat (app)/Km (app) (min−1 mM−1) | 39.5 | 607 |
The value in parentheses was obtained at 452 nm.
Value previously reported by Wagner et al. (1).
Average of values obtained with two preparations.
Average of values obtained with four preparations.
A unit of activity is defined as the formation of 1 μmol of formaldehyde/min at 37 °C, pH 8.0, with sarcosine as substrate (1).
Apparent steady-state kinetic parameters were determined in air-saturated buffer at various concentrations of sarcosine, as described in Experimental Procedures. Values for wild type enzyme were previously reported by Wagner and Jorns (16).
Turnover of the Tyr317Phe Mutant with Sarcosine
The specific activity of the mutant enzyme is 20% of that observed with wild type MSOX. Apparent steady-state kinetic parameters with sarcosine as substrate were determined at 25 °C in air-saturated buffer at pH 8.0. The mutant exhibits an apparent kcat that is 4.2-fold less and an apparent Km for sarcosine that is 3.7-fold greater than the corresponding values observed with wild type enzyme. The catalytic efficiency of the mutant is 15.4-fold lower than wild type MSOX, as estimated based on the ratio kcat/Km (Table 1).
Complexes of the Tyr317Phe Mutant with Substrate Analogs
Methylthioacetate (MTA) can be regarded as analog of sarcosine where the amino nitrogen is replaced by a sulfur atom. Pyrrole-2-carboxylate (PCA) is an aromatic analog of the substrate L-proline. Both ligands form charge transfer complexes with the Tyr317Phe mutant, as judged by the appearance of a new absorption band at longer wavelengths (Figure 3). The positions of the charge transfer bands were estimated based on maxima observed in the corresponding difference spectra (Figure 3, insets). The charge transfer bands observed for the mutant complexes with MTA and PCA are shifted hypsochromically as compared to those seen for the corresponding complexes with wild type MSOX. The hypsochromic shift with MTA is relatively modest (6 nm) but a much larger shift (63 nm) is observed with PCA. The dissociation constants of the mutant complexes with MTA and PCA are increased (14- and 21-fold, respectively) as compared to wild type MSOX (Table 2).
Figure 3.

Complexes formed with the Tyr317Phe mutant (22.3 μM, upper panel) or wild type MSOX (21.9 μM, lower panel) and methylthioacetate (MTA) or pyrrole-2-carboxylate (PCA). In each panel, the solid black line shows the absorption spectrum of the ligand-free enzyme in 50 mM potassium phosphate buffer, pH 8.0, at 25 °C. Spectra of the complexes with MTA and PCA (solid green and magenta lines, respectively) are calculated for 100% complex formation, as described in Experimental Procedures. The insets show the difference spectra used to estimate the positions of the charge transfer bands with PCA (top panel) or MTA (bottom panel). In each inset, the solid and dashed lines are difference spectra obtained with the Try317Phe mutant and wild type MSOX, respectively. The data for wild type MSOX were previously reported by Zhao and Jorns (3).
Table 2.
Comparison of complexes formed with inhibitors and the Tyr317Phe mutant or wild type MSOXa
| ligand | enzyme | Kd (mM) | Charge Transfer Band
|
|
|---|---|---|---|---|
| λmax (nm) | ε (mM−1cm−1) | |||
| methylthioacetate | Tyr317Phe | 40 ± 1 | 526 | 8.1 |
|
|
wild type | 2.88 ± 0.04 | 532 | 7.7 |
|
| ||||
| pyrrole-2-carboxylate | Tyr317Phe | 33 ± 2 | 545 | 2.6 |

|
wild type | 1.60 ± 0.06 | 608 | 2.0 |
Complexes were formed in 50 mM potassium phosphate buffer, pH 8.0 at 25 °C. Values for wild type enzyme were previously reported by Zhao and Jorns (3).
Reductive Half-Reaction of the Tyr317Phe Mutant with L-Proline
The kinetics of reduction of the mutant enzyme by L-proline were monitored under anaerobic conditions by following the loss of oxidized flavin absorbance at 454 nm. The observed spectral changes were immediately reversed upon aeration. The reductive half-reactions exhibit apparent first order kinetics over a broad range of L-proline concentrations (0.03 to 1.5 M) and pH values (pH = 7.6 to 10.0). Control studies showed that the mutant enzyme was stable over the entire pH range of these studies. At each pH, the observed apparent first order rate constants (kobs) exhibit a hyperbolic dependence on L-proline concentration (Figure 4). The corresponding double-reciprocal plots are linear with a finite Y-intercept (not shown). The results are consistent with the reductive half-reaction mechanism previously proposed for wild type MSOX (Scheme 2). Values for the limiting rate of reduction at saturating L-proline (klim) and the apparent dissociation constant for the E·S complex (Kd) were estimated by fitting the data obtained at each pH value to equation 1. For the mechanism shown in Scheme 2, the kinetically determined Kd will be equal to the true Kd provided that k2 ⋘ klim. The properties observed for reductive half-reaction of L-proline with mutant or wild type enzyme satisfy criteria described by Strickland et al. (17) for the case where this condition (k2 ⋘ klim) is satisfied.
Figure 4.

Effect of pH on the observed rate of reduction of the Tyr317Phe mutant (kobs) at various concentrations of L-proline. Reactions were conducted under anaerobic conditions at 4 °C in buffered solutions containing 11.9 μM enzyme, as detailed in Experimental Procedures. The solid (or dashed) lines are fits of the data to equation 1.
| (1) |
Effect of pH on the Limiting Rate of the Reductive Half-Reaction of the Tyr317Phe Mutant with L-Proline
The observed values for klim are strongly pH dependent (Figure 5). The rates decrease at low pH and reached a quasi plateau region at high pH, followed by a small decrease at pH > 9.6. The data in the pH range from 7.6 to 9.6 gave a good fit to equation 2 where Ka is the ionization constant of a group in the E·S complex that must be unprotonated for enzyme reduction to occur (pKa = 8.9 ± 0.1) and kmax is the limiting rate at pH ⋘ pKa (kmax = 0.69 ± 0.01 min−1). The observed pH-dependence of klim with the Tyr317Phe mutant is similar to that observed for wild type MSOX (Figure 5, inset), except for a 0.9 pH unit difference in the observed pKa. The limiting rate at pH ⋘ pKa with the mutant enzyme is 5% of the value observed with wild type MSOX (Table 3), indicating the importance of Tyr317 in L-proline oxidation. However, the results clearly show that Tyr317 cannot be the ionizable group in the E·S complex formed with L-proline and wild type MSOX.
Figure 5.
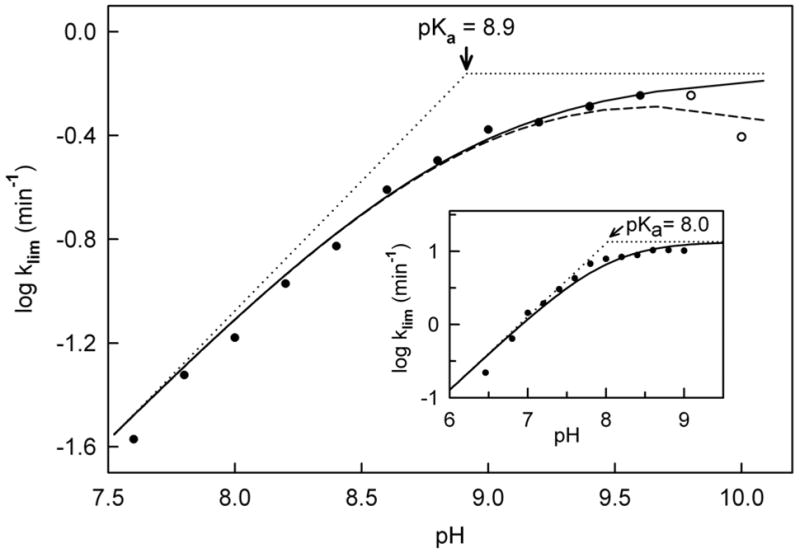
Effect of pH on the limiting rate of reduction (klim) observed with the Tyr317Phe mutant at saturating L-proline. Reactions were conducted and values for klim were determined as described in the legend to Figure 4. The solid line is a fit of the data at pH ≤ 9.6 (solid circles) to equation 2. The dashed line is a fit of all the data (solid and open circles) to equation 3 with constraints, as indicated in the text (see Table 3). The dotted lines have slopes equal to +1 or 0. The inset shows the corresponding data with wild type MSOX, as previously reported by Zhao and Jorns (14)
Table 3.
Extrapolated rate of the reductive half-reaction with mutant or wild type MSOX and L-proline at pH > pKa
| kmax (min−1) | pKa | pKb | |
|---|---|---|---|
| Tyr317Phea | 0.69 ± 0.01 | 8.9 ± 0.1 | (~10.5) |
| wild typeb | 13.4 ±1.6 | 8.0 ± 0.1 (7.9 ± 0.1) | - |
Values for kmax and pKa for the mutant are based on a fit of the data at pH ≤ 9.6 to equation 2, as described in the text. An approximate value for pKb was obtained by fitting the entire data set to equation 3 with C = kmax and pKa = 8.9.
| (2) |
The presence of a second ionizable group in the mutant E·S complex that must be protonated for enzyme reduction to occur is suggested by the small decrease in values obtained for klim near the upper limit of the pH range examined. An approximate value for this second pKa (~10.5) was obtained by fitting the entire data set to equation 3 (Table 3). Previous studies with wild type MSOX provided no evidence for a second ionizable group but were conducted at pH values (6.5 to 9.0) below that required to detect this group with the mutant enzyme.
| (3) |
Effect of pH on the Stability and Spectral Properties of the E·S Complex formed with L-Proline
The kinetically determined Kd values for the complex of the mutant enzyme with L-proline are pH-dependent and show limiting values at both low and high pH with tighter binding at low pH. The data gave a good fit to equation 4 where Ka is the ionization constant of a group in the free enzyme or substrate (pKa = 9.0 ± 0.4) (Figure 6). This pKa is attributed to a group in the enzyme since free L-proline does not exhibit a pKa value in this region. Binding of L-proline to the mutant enzyme is about 6-fold tighter when this group is protonated (Kd (low pH) = 90 ± 30) as compared with the ionized form (Kd (high pH) = 510 ± 10) (Table 4). In contrast, the kinetically determined Kd for the complex of wild type MSOX with L-proline is pH-independent (Kd = 256 ± 4) (Figure 6, inset).
Figure 6.

Effect of pH on the dissociation constant (Kd) of the ES complex formed with the Tyr317Phe mutant with L-proline. Values for Kd were estimated kinetically as described in the legend to Figure 4. The solid line is a fit of the data (solid circles) to equation 4. The inset shows data previously obtained with wild type MSOX (14). The dotted line is the average of Kd values obtained in the pH range from 6.5 to 9.0
Table 4.
Stability of the E·S complex formed with mutant or wild type MSOX and L-proline
| dissociation constant (mM)
|
|||||
|---|---|---|---|---|---|
| kinetically determined | spectral titration | ||||
| Kd | PH | pKa | Kd (mM) | pH | |
| Tyr317Phe | 90 ± 30a | ⋘ pKa | 9.0 ± 0.4a | ||
| 109 ± 8 | 7.6 | 101 ± 4 | 7.6 | ||
| 440 ± 25 | 9.6 | 420 ± 40 | 9.6 | ||
| 510 ± 10a | ⋙ pKa | ||||
| wild type | 256 ± 4b | 6.5 to 9.0 | ndc | 200 ± 6 | 7.2 |
| 230 ± 9 | 8.6 | ||||
Values were determined based on the fit of data, obtained in the pH range from 7.5 to 10.0, to equation 4, as described in the text.
Average value previously reported by Zhao and Jorns (14) based on data obtained in the pH range from 6.5 to 9.0, including Kd values observed at pH 7.2 (241 ± 12 mM ) and pH 8.6 (248 ± 16 mM).
Not detected.
| (4) |
As discussed above, the kinetically determined Kd for the binding of L-proline to mutant or wild type enzyme should correspond to a true dissociation constant. To verify this prediction, we developed a method to independently measure the Kd for the enzyme·L-proline complex based on the perturbation of the flavin absorption spectrum. Since the reductive half-reaction is fully rate-determining (14), free oxidized enzyme and the oxidized enzyme·substrate complex should be essentially the only species present in solution during the steady state. In this approach, absorption spectra are recorded within 15 s after mixing different amounts of L-proline with separate aerobic samples of mutant or wild type MSOX. Under these conditions, an isosbestic conversion of the free enzyme to the enzyme·substrate complex is observed with no evidence for flavin reduction, as expected for spectra recorded under steady-state conditions (i.e., prior to consumption of available oxygen).
Two pH values were selected for titration of the mutant enzyme with L-proline, one below (7.6) and another above (9.6) the kinetically determined pKa (8.9) for ionization of the enzyme·substrate complex. The titration at pH 7.6 results in a hypsochromic shift and increase in the intensity of the lowest energy absorption band, accompanied by a relatively small change in the higher energy band (Figure 7). The isosbestic spectral course of the titration is evident by the set of difference spectra generated at various concentrations of L-proline (Figure 7, inset A). The dissociation constant was determined by fitting absorbance changes at 441 nm to a theoretical binding curve (Figure 7, inset B). The value obtained by spectral titration (Kd = 101 ± 4 mM) is in excellent agreement with the kinetically determined value at pH 7.6 (Kd = 109 ± 8 mM) (Table 4).
Figure 7.

Titration of the Tyr317Phe mutant with L-proline at pH 7.6. Spectra were recorded immediately after mixing enzyme (19.0 μM) with L-proline in 100 mM potassium phosphate buffer, pH 7.6, at 4 °C. Separate samples were prepared for each concentration of L-proline. The main panel compares the absorption spectrum of ligand free enzyme (curve 1, black) with the spectrum of the enzyme-L-proline complex (curve 2, magenta), calculated for 100% complex formation as described in Experimental Procedures. Inset A: Difference spectra obtained at 12, 48 110 and 256 mM L-proline are shown by the cyan, green, orange and blue curves, respectively. The magenta curve is the calculated difference spectrum for 100% complex. Inset B: The observed increase in absorbance at 441 nm (solid circles) is plotted versus the L-proline concentration. The solid line is a fit of the data to a theoretical binding curve.
The titration of the mutant enzyme with L-proline at pH 9.6 results in a more pronounced hypsochromic shift and increase in the intensity of the lowest energy absorption band, accompanied by a sizable bathochromic shift and increase in the intensity of the higher energy absorption band (Figure 8). Significantly, complex formation at pH 9.6 also results in the appearance of absorption at longer wavelengths and the presence of a positive peak in the difference spectra at 526 nm (ε526 = 720 M−1 cm−1) (Figure 8, inset A), a feature not observed at pH 7.6. This difference is attributed to ionization of enzyme bound·L-proline (pKa = 8.9, see Figure 5) and formation of a charge transfer complex of oxidized FAD with the electron rich L-proline anion. The value obtained for the dissociation constant of the complex by spectral titration at pH 9.6 (Kd = 420 ± 40 mM) is 4-fold larger than the value obtained at pH 7.6 and in excellent agreement with the kinetically determined value at pH 9.6 (Kd = 440 ± 25 mM) (Table 4).
Figure 8.

Titration of the Tyr317Phe mutant with L-proline at pH 9.6. Spectra were recorded immediately after mixing enzyme (19.0 μM) with L-proline in100 mM sodium carbonate buffer, pH 9.6, at 4 °C. Separate samples were prepared for each concentration of L-proline. The main panel shows the absorption spectrum of ligand free enzyme (curve 1, black) and the spectrum calculated for 100% complex formation (curve 2, magenta). Inset A: Difference spectra obtained at 48 180, 352 and 980 mM L-proline are shown by the cyan, green, orange and blue curves, respectively. The magenta curve is the calculated difference spectrum for 100% complex. Inset B: The observed increase in absorbance at 438 nm (solid circles) is plotted versus the L-proline concentration. The solid line is a fit of the data to a theoretical binding curve.
The titration of wild type MSOX with L-proline was also conducted at pH values above (8.6) and below (7.2) the kinetically determined pKa (8.0) for ionization of the enzyme·substrate complex. The titration at pH 8.6 results in a bathochromic shift and increase in the intensity of the lowest energy absorption band, accompanied by a hypsochromic shift and decrease in the intensity of the higher energy absorption band (Figure 9). However, the most dramatic feature of the spectral perturbation observed at pH 8.6 is the appearance of an intense charge transfer band in the long wavelength region with an estimated maximum at 512 nm (ε512 = 6800 M−1 cm−1), as judged by the position of the peak in the difference spectrum (Figure 9, inset A). The spectrally determined dissociation constant (Kd = 230 ± 9 mM) is in good agreement with the kinetically determined value (Table 4). A similar value is obtained for the dissociation constant when the spectral titration is conducted at pH 7.2 (Kd = 200 ± 6 mM). However, the intensity of the charge transfer band at 512 nm is greatly diminished (Figure 9, inset B), consistent with the expected decrease in the fraction of enzyme·substrate complexes containing the anionic substrate at the lower pH value.
Figure 9.
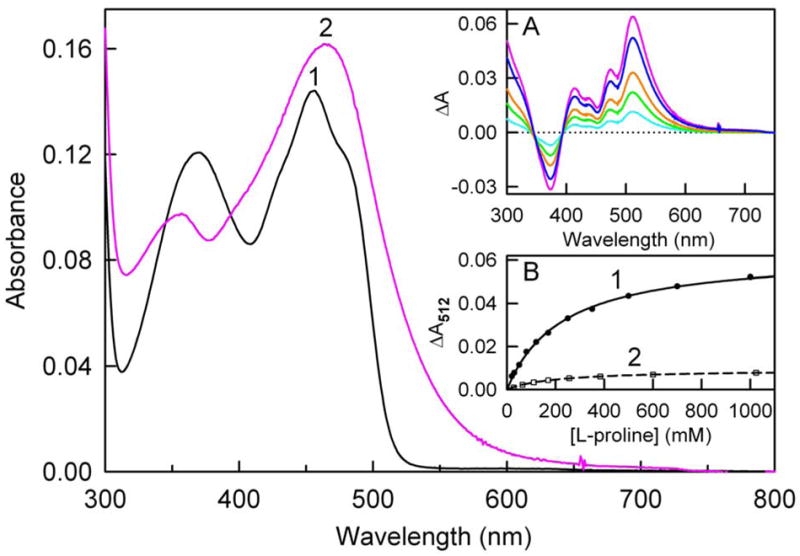
Titration of wild type MSOX with L-proline. Spectra were recorded immediately after mixing enzyme (11.8 μM) with L-proline in 100 mM sodium pyrophosphate buffer, pH 8.6 at 4 °C. Separate samples were prepared for each concentration of L-proline. The main panel shows absorption spectra of ligand free enzyme (curve 1, black) and the spectrum calculated for 100% complex formation (curve 2, magenta). Inset A: Difference spectra obtained at 50 120, 250 and 1000 mM L-proline are shown by the cyan, green, orange and blue curves, respectively. The magenta curve is the calculated difference spectrum for 100% complex. Inset B: The increase in absorbance at 512 nm (solid circles) observed at pH 8.6 is plotted versus the L-proline concentration. The solid line (curve 1) is a fit of the pH 8.6 data to a theoretical binding curve. The open squares and dashed line (curve 2) show the corresponding results obtained for a titration that was conducted at pH 7.2 in 100 mM potassium phosphate buffer.
DISCUSSION
MSOX binds the zwitterionic form of L-proline, the predominant species in solution around physiological pH. The reactive anionic form is generated by ionization of the enzyme·substrate complex (pKa = 8.0), as judged, in part, by the pH-dependence of the limiting rate of the reductive-half reaction observed with wild type enzyme (14). Based on the crystal structure of MSOX·inhibitor complexes, Tyr317 was identified as one of two possible active sites bases that might act as the acceptor of the proton released upon substrate ionization. Mutation of Tyr317 to Phe does not significantly affect MSOX expression, covalent flavinylation or the visible absorption spectrum of the enzyme. The pH-dependence of the limiting rate of the reductive-half reaction, observed with the mutant enzyme and L-proline, provides evidence for an ionizable group in the E·S complex that must be unprotonated for enzyme reduction to occur, similar to that seen with wild type enzyme, except for a 0.9 pH unit increase in the observed pKa. The results show that Tyr317 in wild type enzyme does not act as the acceptor of the proton released upon substrate ionization. We have previously shown that ionization of the enzyme·L-proline complex is unaffected by mutation His269, the other possible active site base (3). Therefore, we conclude that L-proline itself is the ionizable group in the enzyme·substrate complex. This outcome indicates that a substantial decrease in the pKa of the free amino acid is induced upon binding to the MSOX active site (ΔpKa = 2.6).
The results obtained with L-proline suggest that amino acid substrates are activated for MSOX oxidation by a process involving stabilization of the reactive anionic form. A similar feature has been observed for amine oxidation by trimethylamine dehydrogenase (18). How does MSOX lower the pKa of the bound amino acid? Partial stabilization is attributable to Tyr317 but not His269, as judged by the effect of mutations on the observed pKa. Specific electrostatic effects may also be important since the active site environment in MSOX is highly electropositive. There are no acidic residues in the region surrounding the flavin ring, an area that includes a number of basic residues (2). For example, Arg52 and Lys348 are located above the re face of the flavin and form hydrogen bonds with the carboxylate group in substrate analogs, like PCA (Figure 1). Arg49 and Lys265 are located above the si face. The sidechain of Arg49 is in van der Waals contact with the flavin ring (Figure 10). The 1-electron reduction potential for the flavin in wild type MSOX is 400 mV more positive than observed with free flavin (3). The electropositive environment may play a dual role in MSOX catalysis by making the flavin a more potent oxidant and by stabilizing the reactive anionic form of amino acid substrates.
Figure 10.

Stereoview of the si face of the flavin in the MSOX·PCA complex. The figure was constructed using the previously reported structure (2). Atom colors are the same as in Figure 1. Hydrogen bonds are indicated by dashed lines.
Enzyme·bound L-proline is not solvent accessible, as judged by structures of oxidized enzyme·inhibitor complexes (2, 4) and a recently observed structure of a reduced enzyme·L-proline complex2. Given that neither Tyr317 nor His269 acts as the acceptor, it is relevant to consider how the proton from the L-proline zwitterion might be ushered out of the active site. In the MSOX·PCA complex, the N(5) position of the flavin ring is located 3.73 Å from the nitrogen atom of the inhibitor. In the reduced enzyme·L-proline complex, the distance between the substrate nitrogen and flavin N(5) is 3.48 Å. We postulate that a proton relay system, involving initial proton transfer from the substrate zwitterion to flavin N(5), may provide a mechanism for proton transport. Additional components of the putative relay system include Thr48, Lys265 and four water molecules. Water 1 is hydrogen bonded to flavin N(5), the carbonyl oxygen of Thr48 and the ε-amino group of Lys265. The hydroxyl group of Thr48 is hydrogen bonded to water 2. Waters 2–4 are accessible to bulk solvent (Figure 10).
Mutation of Tyr317 to Phe causes a modest decrease (4-fold) in the apparent rate of turnover with sarcosine. A more substantial effect is observed on L-proline oxidation, as judged by the 20-fold decrease in the maximum rate of the reductive half-reaction at pH ⋘ pKa. The mutation also affects charge transfer interactions with various ligands. Complexes formed with the mutant enzyme and MTA or PCA exhibit charge transfer bands that are shifted to shorter wavelengths (higher energy) as compared with wild type MSOX. The energy difference is greater with the L-proline analog than with the sarcosine analog (Δν = 1900 and 210 cm−1 with PCA and MTA, respectively). A major difference in the intensity of the charge transfer band is observed for complexes formed with mutant or wild type enzyme and the L-proline anion (at pH > pKa). The wild type complex exhibits an intense charge transfer band with an extinction coefficient that is nearly an order of magnitude greater than the weak band observed for the mutant complex. Various studies indicate that the location and intensity of flavin charge transfer bands are dependent on the reciprocal orientation of the components of the charge transfer complex (19, 20). Interestingly, the MSOX His269Asn mutant forms charge transfer complexes with MTA, PCA or the L-proline anion that exhibit spectral properties similar to those observed for the corresponding complexes with the Try317Phe mutant. A high resolution crystal structure of the His269Asn complex with PCA reveals small differences in the ligand binding mode as compared with wild type enzyme, suggesting that His269 plays a role in optimizing substrate binding (3). A similar role for Tyr317 would be consistent with the properties observed for the Phe mutant.
The kinetically determined dissociation constant for the enzyme·L-proline complex corresponds to a true dissociation constant, as judged by the similar values obtained in spectral titrations with the Tyr317Phe mutant or wild type MSOX. The dissociation constant for the complex formed with the mutant enzyme is pH-dependent (pKa = 9.0) whereas the dissociation constant observed with wild type enzyme is pH-independent. The results suggest that mutation of Tyr317 has perturbed the pKa of an unidentified group that affects the dissociation constant of the enzyme·substrate complex.
Substrate oxidation by the polar and SET mechanisms shown in Scheme 1 requires an active site base to abstract the α-hydrogen from the carbon atom in the C-N bond undergoing oxidation. While our studies clearly show that neither Tyr317 nor His269 acts as the acceptor of the proton released during ionization of the MSOX·L-proline complex, they do not definitively rule out a possible role for either residue as the acceptor of the α-hydrogen. However, this scenario would require that His269 or Tyr317 exhibit a pKa < 5.5 since kinetic studies with wild type MSOX at pH ≥ 6.5 provide evidence for only a single ionizable group in the enzyme·L-proline complex and this pKa is attributable to substrate ionization.
Footnotes
This work was supported in part by Grant GM 31704 (M. S. J.) from the National Institutes of Health.
Abbreviations: MSOX, monomeric sarcosine oxidase; FAD, flavin adenine dinucleotide; SET, single electron transfer; PCA, pyrrole-2-carboxylate; MTA, methylthioacetate.
Zhao, G., Chen, Z., Silks, L.A., Ollivault-Shiflett, M., Wu, R., Mathews, F.S. and Jorns, M.S., unpublished observations.
References
- 1.Wagner MA, Khanna P, Jorns MS. Structure of the flavocoenzyme of two homologous amine oxidases: Monomeric sarcosine oxidase and N-methyltryptophan oxidase. Biochemistry. 1999;38:5588–5595. doi: 10.1021/bi982955o. [DOI] [PubMed] [Google Scholar]
- 2.Trickey P, Wagner MA, Jorns MS, Mathews FS. Monomeric sarcosine oxidase: Structure of a covalently-flavinylated secondary amine oxidizing enzyme. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhao G, Song H, Chen Z, Mathews FS, Jorns MS. Monomeric sarcosine oxidase: Role of histidine 269 in catalysis. Biochemistry. 2002;41:9751–9764. doi: 10.1021/bi020286f. [DOI] [PubMed] [Google Scholar]
- 4.Wagner MA, Trickey P, Chen Z, Mathews FS, Jorns MS. Monomeric sarcosine oxidase: 1. Flavin reactivity and active site binding determinants. Biochemistry. 2000;39:8813–8824. doi: 10.1021/bi000349z. [DOI] [PubMed] [Google Scholar]
- 5.Khanna P, Jorns MS. Characterization of the FAD-containing N-methyltryptophan oxidase from Escherichia coli. Biochemistry. 2001;40:1441–1450. doi: 10.1021/bi0024411. [DOI] [PubMed] [Google Scholar]
- 6.Venci D, Zhao G, Jorns MS. Molecular characterization of nikD, a new flavoenzyme important in the biosynthesis of nikkomycin antibiotics. Biochemistry. 2002;41:15795–15802. doi: 10.1021/bi020515y. [DOI] [PubMed] [Google Scholar]
- 7.Eschenbrenner M, Chlumsky LJ, Khanna P, Strasser F, Jorns MS. Organization of the multiple coenzymes and subunits and role of the covalent flavin link in the complex heterotetrameric sarcosine oxidase. Biochemistry. 2001;40:5352–5367. doi: 10.1021/bi010101p. [DOI] [PubMed] [Google Scholar]
- 8.Dodt G, Kim DG, Reimann SA, Reuber BE, McCabe K, Gould SJ, Mihalik SJ. L-pipecolic acid oxidase, a human enzyme essential for the degradation of L-pipecolic acid, is most similar to the monomeric sarcosine oxidases. Biochem J. 2000;345:487–494. [PMC free article] [PubMed] [Google Scholar]
- 9.Silverman RB. Radical ideas about monoamine oxidase. Acc Chem Res. 1995;28:335–342. [Google Scholar]
- 10.Kim JM, Hoegy SE, Mariano PS. Flavin chemical models for monoamine oxidase inactivation by cyclopropylamines, alpha-silylamines, and hydrazines. J Am Chem Soc. 1995;117:100–105. [Google Scholar]
- 11.Silverman RB, Zelechonok Y. Evidence for a hydrogen atom transfer mechanism or a proton fast electron transfer mechanism for monoamine oxidase. J Org Chem. 1992;57:6373–6374. [Google Scholar]
- 12.Miller JR, Edmondson DE. Structure-activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry. 1999;38:13670–13683. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 13.Harris CM, Pollegioni L, Ghisla S. pH and kinetic isotope effects in D-amino acid oxidase catalysis - Evidence for a concerted mechanism in substrate dehydrogenation via hydride transfer. Eur J Biochem. 2001;268:5504–5520. doi: 10.1046/j.1432-1033.2001.02462.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhao GH, Jorns MS. Monomeric sarcosine oxidase: Evidence for an ionizable group in the ES complex. Biochemistry. 2002;41:9747–9750. doi: 10.1021/bi020285n. [DOI] [PubMed] [Google Scholar]
- 15.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 16.Wagner MA, Jorns MS. Monomeric sarcosine oxidase: 2. Kinetic studies with sarcosine, alternate substrates and substrate analogs. Biochemistry. 2000;39:8825–8829. doi: 10.1021/bi000350y. [DOI] [PubMed] [Google Scholar]
- 17.Strickland S, Palmer G, Massey V. Determination of dissociation constants and specific rate constants of enzyme-substrate (or protein-ligand) interactions from rapid reaction kinetic data. J Biol Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- 18.Basran J, Sutcliffe MJ, Scrutton NS. Optimizing the Michaelis complex of trimethylamine dehydrogenase - Identification of interactions that perturb the ionization of substrate and facilitate catalysis with trimethylamine base. J Biol Chem. 2001;276:42887–42892. doi: 10.1074/jbc.M108296200. [DOI] [PubMed] [Google Scholar]
- 19.Shieh H-S, Ghisla S, Hanson LK, Ludwig ML, Nordman CE. Molecular complex of lumiflavin and 2-aminobenzoic acid: Crystal structure, crystal spectra and solutions properties. Biochemistry. 1981;20:4766–4774. doi: 10.1021/bi00519a037. [DOI] [PubMed] [Google Scholar]
- 20.Dmitrenko O, Thorpe C, Bach RD. Effect of a charge-transfer interaction on the catalytic activity of Acyl-CoA dehydrogenase: A theoretical study of the role of oxidized flavin. J Phys Chem B. 2003;107:13229–13236. [Google Scholar]