

Abstract
The alveolar compartment in acute lung injury contains high levels of tissue factor (TF) procoagulant activity favoring fibrin deposition. We previously reported that the alveolar epithelium can release TF procoagulant activity in response to a proinflammatory stimulus. To test the hypothesis that the alveolar epithelium further modulates intra-alveolar fibrin deposition through secretion of an endogenous inhibitor to TF, tissue factor pathway inhibitor (TFPI), we measured TFPI levels in edema fluid (EF) from patients with acute respiratory distress syndrome. To determine whether the alveolar epithelium can release TFPI, both full-length TFPI and truncated TFPI were measured (ELISA) in pulmonary edema fluid from patients with acute respiratory distress syndrome (ARDS) and a control group of patients with hydrostatic pulmonary edema (HYDRO). TFPI protein was also measured in conditioned media (CM) and cell lysates (CL) from human alveolar epithelial cells (A549) after exposure to cytomix (TNF-α, IL-1β, IFN-γ). TFPI protein levels were higher in pulmonary edema fluid from patients with ARDS vs. HYDRO. TFPI protein was increased in CM and did not change in CL after cytomix treatment; TFPI mRNA levels (RT-PCR) did not change. Despite the high levels of TFPI, both the EF and CM retained significant TF procoagulant activity as measured by plasma recalcification time. The majority of intraalveolar TFPI was in a truncated, inactive form, whereas the majority of TFPI released from cells was full length, suggesting different mechanisms of inactivation. In summary, the alveolar epithelium releases TFPI in response to an inflammatory stimulus but does not increase TFPI gene transcription or protein production. Levels of intra-alveolar TFPI in ARDS are not sufficient to block intra-alveolar TF procoagulant activity due to truncation and inactivation of intra-alveolar TFPI.
Keywords: fibrin deposition
intra-alveolar thrombin formation and fibrin deposition are pathological hallmarks of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (41). Although the alveolar compartment in ALI/ARDS is procoagulant and antifibrinolytic (23, 25), the cellular and molecular mechanisms that modulate intra-alveolar fibrin deposition are incompletely understood. Previous studies have demonstrated that there is tissue factor (TF)-dependent procoagulant activity in bronchoalveolar lavage (BAL) fluid from patients with ARDS (4, 23, 24). The activation of TF is a critical event that results in thrombin formation. Thrombin is a key intermediate molecule with several biological functions including augmentation of vascular permeability (29) and enhancement of inflammation (12). Thrombin generation leads to fibrin polymerization and deposition with resultant formation of hyaline membranes, a pathological hallmark of ARDS. We have reported that the alveolar epithelium is one potential source of TF procoagulant activity in patients with ARDS (4), suggesting that the alveolar epithelium may actively modulate intra-alveolar fibrin deposition.
In the systemic vascular space, TF procoagulant activity is balanced by its natural inhibitor, tissue factor pathway inhibitor (TFPI). TFPI is a Kunitz-type serine protease inhibitor and is the only endogenous specific inhibitor of TF that has been described. TFPI regulates the initiation of coagulation by binding to factor Xa. The Xa-TFPI complex then binds to the tissue factor-VIIa complex, inactivating tissue factor (3). TFPI is synthesized and stored primarily in the microvascular endothelium with small amounts stored in platelets or circulating in the plasma (30). Full-length TFPI protein is very sensitive to degradation to a truncated, inactive form by the action of neutrophil elastase (21, 37), matrix metalloproteinases (5, 15), and other proteases such as chymase (20). In addition, the activity of the full-length protein is regulated at several levels. Anticoagulant activity is enhanced by glycosylphosphatidylinositol (GPI) cell surface anchoring (31), and some cells secrete a form of the TFPI protein that cannot bind GPI (31). Circulating factors such as protein S (19) and low-density lipoproteins (LDL) (36) associate with TFPI and can affect its anticoagulant potency.
The potential therapeutic role for TFPI has been studied extensively in both animal models and clinical trials in sepsis. TF-mediated fibrin deposition is important in the development of lung injury in a primate model of Escherichia coli sepsis (42), and blockade of the TF pathway through either site inactivated factor VIIa (8, 43) or antibody blockade of TF factor Xa binding attenuated lung injury and improved survival in these animals. In addition, recombinant TFPI treatment improved survival in a lethal baboon model of E. coli sepsis (7, 14) and attenuated lung injury in a rat model of E. coli sepsis (17). In human sepsis, experts have suggested that endogenous TFPI is not sufficient to overcome the intense TF procoagulant activity (22). However, systemic administration of recombinant TFPI did not improve mortality in a large, randomized trial in severe sepsis (1). In addition, among the patients with severe sepsis, the rates of ARDS were the same in the TFPI and placebo groups, and mortality was not reduced in the ARDS group.
The conflicting results in preclinical and clinical trials of recombinant TFPI highlight the need for more mechanistic studies designed to determine the specific role of TFPI in the pathogenesis and resolution of clinical ALI. The aims of our study were twofold. First, because we have previously reported that the alveolar epithelium plays a central role in modulation of intra-alveolar fibrin deposition (4, 39, 40), we hypothesized that the alveolar epithelium actively regulates TF procoagulant activity through production of TFPI. Second, based on the sensitivity of TFPI to degradation by proteases that are known to be present in the air spaces in ARDS, including neutrophil elastase (21, 37) and matrix metalloproteinases (5, 15), we hypothesized that local TFPI production is insufficient to oppose TF procoagulant activity in patients with ARDS because intra-alveolar TFPI is truncated and thus inactivated.
METHODS
Collection of pulmonary edema fluid
Samples of human pulmonary edema fluid and plasma were collected as previously described (33) from intensive care unit patients at Moffitt Long and San Francisco General Hospitals from 1995 to 2001 and at Vanderbilt University from 2003 to the present. Briefly, mechanically ventilated patients with ALI/ARDS or controls with hydrostatic pulmonary edema were identified within6hof endotracheal intubation using standard definitions (6). A standard tracheal suction catheter was inserted through the endotracheal tube and advanced slowly until resistance was met. Gentle suction was applied, and a few milliliters of undiluted pulmonary edema fluid were aspirated. Samples were placed immediately on ice and centrifuged for 10 min at 3,000 g to remove any cellular or particulate matter. Supernatants were stored at -70°C until use. The study was approved by the Committee on Human Research at the University of California San Francisco (UCSF) and the Institutional Review Board at Vanderbilt University with a waiver of informed consent. ALI/ARDS was defined by 1) bilateral pulmonary infiltrates on chest radiograph; 2) appropriate clinical setting; 3) hypoxemia with PaO2/FIO2 ratio < 300; and 4)no evidence of left heart failure according to the American European Consensus guidelines (6). Patients with hydrostatic pulmonary edema met the first three criteria but had evidence of a cardiogenic cause of pulmonary edema including acute myocardial infarction, systolic or diastolic congestive heart failure, or acute volume overload. Because of limited availability of patient samples, experiments were done with selected subsets of patients from our cohort. In addition, some patients had very limited sample volume, so we were unable to make all measurements in all patients. Some patients from UCSF have been included in prior published studies (4).
TF pathway inhibitor ELISA
An ELISA kit for TFPI (both total TFPI and the truncated form) (American Diagnostica, Stamford, CT) was used to measure protein concentrations of TFPI in human samples, cell lysates, and supernatants according to the manufacturer’s instructions. The truncated TFPI ELISA uses two different detection antibodies, one that measures both forms of TFPI (full-length and truncated) and one that is specific for full-length TFPI. Each sample was analyzed with both detection antibodies, and the difference between total TFPI and full-length TFPI was calculated to determine truncated TFPI. All measurements were done in duplicate.
Effect of A549 conditioned media on clot formation in normal plasma
Clot time was measured using a mechanical clot detection system (STart4 Coagulometer; Diagnostica Stago, Asnieres, France). Briefly, A549 cells were treated with serum-free media (control) or cytomix for either 16 or 24 h. Conditioned media was collected and centrifuged at 500 g × 10 min to remove cells and debris. Conditioned media (25 μl) was incubated with either 10 μl of PBS or TF blocking antibody (American Diagnostica) for 30 min at 37°C. Samples were then incubated with 25 μl of pooled normal human plasma (Fisher Diagnostics, Middletown, VA) for 2 min. Clot time was determined as recalcification time following the addition of 25 μl of 25 mM calcium chloride. Measurements were done in duplicate.
Immunohistochemistry study subjects
Fresh lung tissue was obtained from 12 patients admitted to the adult medical intensive care units of the University of Utah Hospital, in Salt Lake City, between 1999 and 2002, who died and underwent autopsy. The autopsy tissue was collected within 12-14 h of death using standard anatomical pathology approaches optimized to preserve tissue architecture and antigen display. Autopsy tissue was obtained from two groups of six patients each. One group died with ALI or ARDS as identified by the American European Consensus Conference definitions (6). The other group died without clinical or pathological evidence of pulmonary disease and served as controls. The autopsy protocol was approved by the Institutional Review Board at the University of Utah Health Sciences Center. The requirement for written informed consent was waived.
Tissue collection and immunohistochemistry
Two to three tissue cubes (>2 × 2 × 2 cm) were obtained from each of the three lobes of the right lung of each patient. The cubes were processed by standard methods (2). Briefly, the tissue cubes were immersion-fixed in 10% buffered neutral formalin (VWR, Media, PA) overnight at 4°C. Slices of the cubes were cut and processed for paraffin embedding for immunohistochemistry (6-μm section thickness).
For immunohistochemistry, antigen retrieval methods were used, followed by standard immunolabeling methods (2). Antigen retrieval was done using 1× Citra solution (BioGenex, San Ramon, CA). Endogenous peroxidase was quenched by 3% hydrogen peroxide in PBS for 20 min at room temperature. Between each of the steps, the tissue sections were rinsed in PBS. TFPI monoclonal antibody that recognizes both full-length and truncated forms of TFPI (American Diagnostica) was used at a dilution of 1:100 (overnight at 4°C). A standard ABC kit (Vector Laboratories, Burlingame, CA) and DAB (Sigma-Aldrich, St. Louis, MO) were used to reveal TFPI localization. All of the tissue sections were counterstained with hematoxylin, dehydrated through an ethanol series, cleared in AmeriClear, and covered by mounting medium and coverslip. Digital images were obtained using a Zeiss Axiophot (Jena, Germany) equipped with a digital camera. Brightness and contrast were adjusted to match background among the digital images; however, color adjustment and other image adjustments were not permitted. For all experiments, three negative controls were used: isotype- and species-matched irrelevant antibody (anti-insulin), omission of the primary antibody, and omission of the secondary antibody. In all cases, the negative controls demonstrated that the TFPI immunostaining was specific.
Cell culture
Alveolar epithelial cells (A549 cells; American Type Culture Collection, Manassas, VA) were grown in MEM (Cellgro, Herndon, VA) with 10% FBS (Cellgro) and 10,000 U/ml each of penicillin and streptomycin (Sigma, St. Louis, MO). For cell lysates and RNA extraction, cells were plated in a 24-well plate at 5 × 104 cells/well and maintained in culture until confluence was reached at day 5. All experiments were performed on day 5 at confluence.
Preparation of cell supernatants and lysates
A549 cells were grown to confluence in 24-well plates. Cells were treated with 20 ng/ml of cytomix (a combination of TNF-α, IL-1β, and IFN-γ at a concentration of 20 ng/ml each in serum-free media) for increasing lengths of time. Cells incubated with serum-free media for equivalent lengths of time served as controls. Following exposure, the cells were incubated with 250 μl of lysis buffer (50 mM Tris, 100 mM NaCl, 0.1% Triton X-100, pH 7.45) for 15 min at 37°C. The lysate was removed and placed in a clean microfuge tube and centrifuged at 500 g to remove cellular debris. The supernatants and lysates were stored at -70°C until assays were performed.
Isolation of mRNA
A549 cells were cultured in 24-well plates until confluent. Appropriate stimulus was applied in serum-free MEM to cells before mRNA isolation. Cells were then trypsinized and collected into cryotubes. Cells from three identical wells were pooled for each mRNA extraction. mRNA was extracted using a kit from Qiagen (Valencia, CA) according to the manufacturer’s instructions and stored immediately at -70°C.
Real-time PCR
Semiquantitative real-time RT-PCR was done using mRNA isolated from A549 cells using the SYBR Green method (Bio-Rad, Hercules, CA). Before the PCR reaction, cDNA was prepared using Superscript II RT (Invitrogen, Carlsbad, CA). All quantitative PCR reactions were run with a no template control for each primer set and a no cDNA control from each experimental condition. Both TFPI and β-actin primers were obtained from IDT (Coralville, IA). Semiquantitation was established using the ΔΔCt method (28). Briefly, the Ct (confidence threshold) for each well was calculated by the Bio-Rad iCycler. For each experimental condition, the Ct for β-actin was subtracted from the Ct for TFPI to generate the ΔCt. The difference between the ΔCt of the control experiment and the ΔCt of treated cells was calculated to give the ΔΔCt. Fold increase in mRNA was calculated by 2ΔΔCt as a quantitative estimate.
Statistical analysis
Statistical analysis was done using SPSS software version 13.0 for Macintosh. In the in vitro experiments, differences between groups were assessed using a one-way ANOVA followed by Tukey test for post hoc analysis. For comparison of two groups of normally distributed data, the Student’s t-test was used. Data from human samples were not normally distributed, and differences between groups were compared using the Mann-Whitney U test. For all analyses, P < 0.05 was considered statistically significant.
RESULTS
Patient characteristics
Clinical characteristics are summarized in Table 1 for 39 patients with ALI/ARDS and 23 patients with hydrostatic edema from whom pulmonary edema fluid was collected and 12 patients from whom lung tissue was obtained for immunohistochemistry. Among the patients in the edema fluid studies, mortality was significantly higher in patients with ALI/ARDS than patients with hydrostatic pulmonary edema.
Table 1. Patient characteristics.
| Patients From Whom Edema Fluid Samples Were Obtained |
Patients From Whom Lung Tissue Was Obtained For Immunohistochemistry |
|||
|---|---|---|---|---|
| Characteristic | ALI/ARDS (n = 39) | Hydrostatic (n = 23) | ALI/ARDS (n = 6) | No ALI/ARDS (n = 6) |
| Male, n (%) | 17 (44%) | 13 (57%) | 4 (73%) | 1 (16%) |
| Caucasian, n (%) | 28 (72%) | 12 (52%) | 6 (100%) | 6 (100%) |
| Age years, means (SD) | 39 (16) | 54 (20) | 41 (16) | 53 (20) |
| Current smoker, n (%) | 6 (15%) | 7 (30%) | 1 (16%) | 1 (16%) |
| Lung injury score, means (SD) | 3.0 (0.7) | 2.6 (0.7) | 3.6 (0.5) | 1.5 (1.2) |
| Tidal volume, ml/kg, means (SD) | 10.4 (3.1) | 10.2 (2.4) | ||
| SAPSII, means (SD) | 47 (21) | 40 (16) | 62 (15) | 49 (18) |
| Ventilator-free days, means (SD) | 8 (10) | 17 (10) | ||
| Hospital mortality, n (%) | 20 (51%) | 5 (22%)* | 6 (100%) | 6 (100%) |
TFPI levels in pulmonary edema fluid
Levels of total TFPI in undiluted pulmonary edema fluid were measured in 30 patients with ARDS and 20 patients with hydrostatic pulmonary edema (HYDRO). Median total TFPI levels in the pulmonary edema fluid were significantly higher in patients with ARDS (Fig. 1) {23,635 pg/ml [interquartile range (IQR) 14,978-38,793 pg/ml]} compared with patients with hydrostatic pulmonary edema [15,136 pg/ml (IQR 7,327-19,868 pg/ml), P = 0.029 by Mann Whitney U test].
Fig. 1.
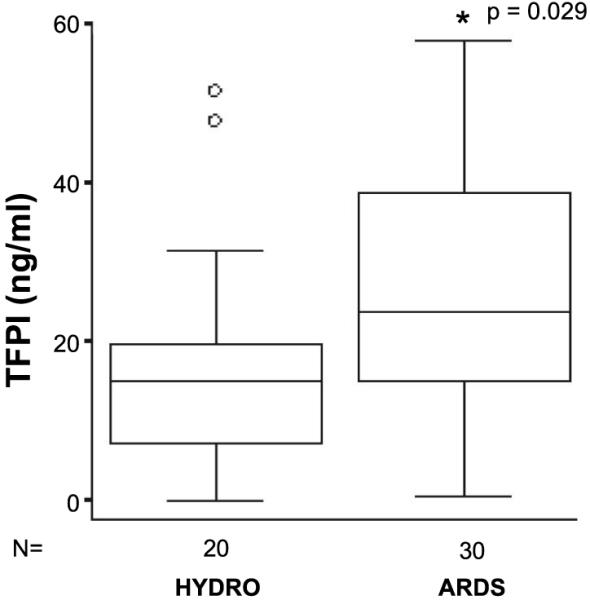
Boxplot of tissue factor pathway inhibitor (TFPI) protein levels in acute respiratory distress syndrome (ARDS) pulmonary edema fluid compared with hydrostatic edema fluid (HYDRO) as measured by ELISA. Horizontal line represents median value, box represents 25th-75th percentile, error bars represent 10th-90th percentile, open circles represent outliers. TFPI protein levels were significantly higher in ARDS vs. HYDRO (P = 0.029 by Mann-Whitney U test).
TFPI immunohistochemistry in human lung
Six patients who died with ARDS and six control patients who died without ARDS were studied. We have previously reported clinical characteristics and TF immunohistochemistry in these patients (4). The immunohistochemical results in Fig. 2 are representative of all 12 patients studied. In control patients (Fig. 2, A and B), TFPI immunostaining was seen along the alveolar membrane, in cells that histologically appear to be both type I and type II alveolar epithelial cells. The staining pattern was quite different in patients who died with ARDS (Fig. 2, C and D) with loss of alveolar septal staining and intense staining in the alveolar space, hyaline membranes, and alveolar macrophages. No TFPI immunostaining was seen in the pulmonary vascular endothelium in any of the subjects.
Fig. 2.

Immunostaining for TFPI in patients who died with ARDS compared with controls who died without evidence of pulmonary disease. In non-ARDS patients (A and B), there is linear TFPI staining along the alveolar septae, likely representing staining of both type I and type II alveolar epithelial cells. In ARDS patients, the linear staining is lost and the majority of TFPI labeling is seen in alveolar exudates (arrow in C) with some residual staining in alveolar lining cells. B and D represent a higher magnification of panels A and C.
TFPI production and release from alveolar epithelial cells
We next tested whether the alveolar epithelium was a potential source of intra-alveolar TFPI by measuring TFPI protein by ELISA and TFPI mRNA by PCR in an alveolar epithelial cell culture system (A549) both basally and in response to a proinflammatory stimulus (cytomix). A549 cells release TFPI protein into the media in a time-dependent manner (Fig. 3A) when incubated with serum-free media (P < 0.05 for 0 h vs. 16 and 24 h by ANOVA), and this release is significantly augmented by cytomix. Conversely, the amount of TFPI in cell lysates (Fig. 3B) is much lower than that in conditioned media and does not change over time in response to cytomix. Concurrently, TFPI mRNA does not change significantly over this time period (Fig. 4).
Fig. 3.

TFPI protein is released by A549 cells over time in response to cytomix (TNF-α, IL-1β, and IFN-γ) (A) with no change in TFPI protein concentration in cell lysates over the same time period (B). In both panels, light-shaded bars represent cells incubated with serum-free media, whereas the dark-shaded bars represent incubation with 20 ng/ml cytomix in serum-free media. *P < 0.001 vs. 0, 1, 3, 6 h; #P = 0.003, 0.004, 0.10, 0.03, vs. 0, 1, 3, 6 h by ANOVA with post hoc Tukey test.
Fig. 4.
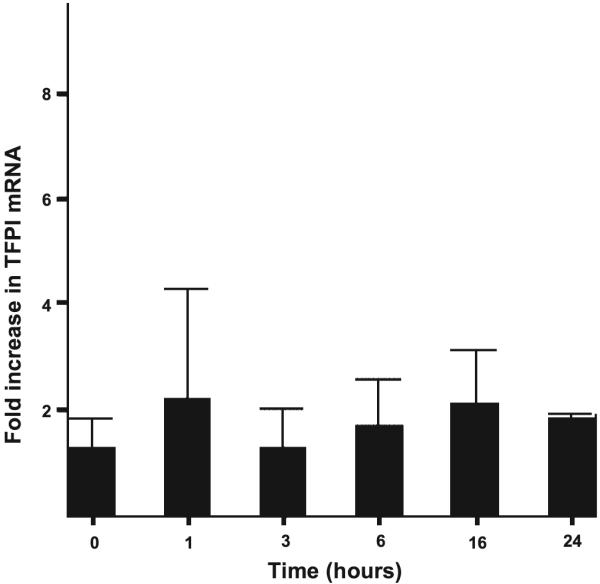
Semiquantitative fold increase in TFPI mRNA in A549 cells over time in response to 20 ng/ml cytomix. There is no change in mRNA expression over time.
Clot time of conditioned media
Clot time of conditioned media from cytomix-treated A549 cells was done using a mechanical clot detector. With cytomix treatment, A549 cells release TF procoagulant activity over time (Fig. 5). This shortening of clot time was inhibited by a blocking antibody to TF (data not shown).
Fig. 5.
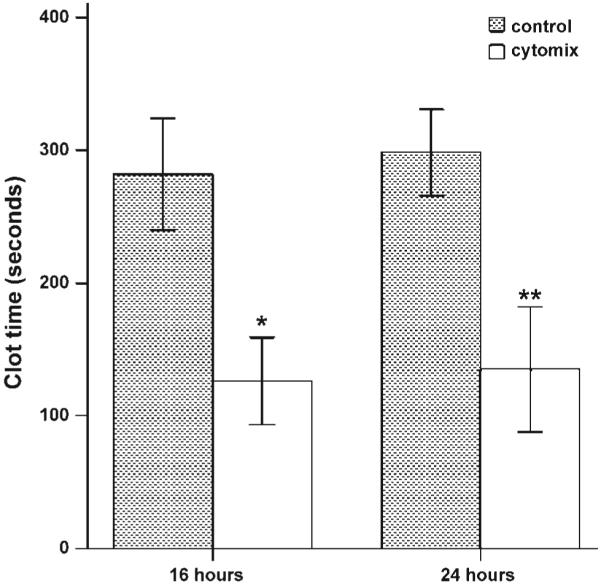
Clot time of conditioned media from A549 cells treated with cytomix (open bars) vs. controls (shaded bars). Clot time was significantly shorter in cytomix-treated cells at both time points. *P < 0.001 vs. 16-h control, **P < 0.001 vs. 24-h control by Student’s t-test.
Truncated TFPI release from A549 cells
To determine if the TFPI released from cytomix-treated A549 cells was full length or truncated, we measured total and truncated TFPI by ELISA (Fig. 6). In untreated cells (control), 100% of the TFPI released is in a full-length form. With cytomix treatment, a small percentage of TFPI, 9.8% and 9.5% at 16 and 24 h, respectively, is truncated, and the remainder is released in a fulllength form.
Fig. 6.
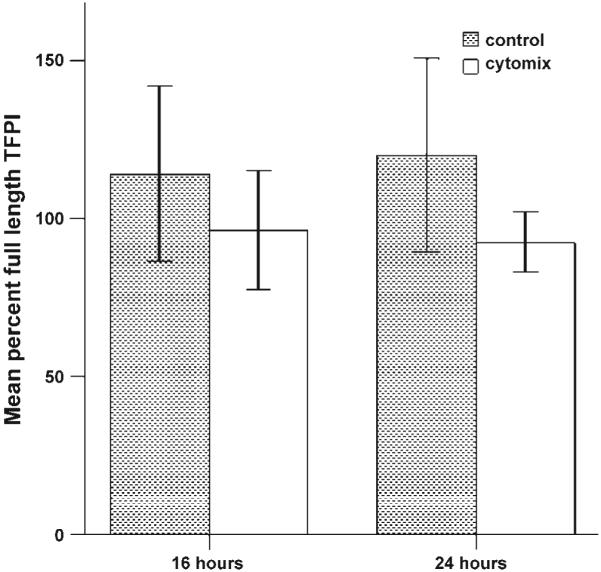
Full-length TFPI released from A549 cells. Percent full-length TFPI was calculated as full-length TFPI/total TFPI and is expressed as a mean percentage of full-length TFPI. All of the TFPI released by untreated cells was full length, whereas TFPI released from cells treated with cytomix for 16 and 24 h was 90.2% and 90.5% full-length, respectively, with the remainder being truncated.
Truncated TFPI in pulmonary edema fluid
To determine whether edema fluid TFPI was present in a full-length, active form or a truncated, inactive form, we measured truncated TFPI protein by ELISA in pulmonary edema fluid from 17 patients with ARDS (Fig. 7). We found that in all but one patient with ARDS, the majority of TFPI was truncated and likely inactive.
Fig. 7.

Full-length and truncated TFPI levels in edema fluid from individual patients with ALI/ARDS. Full bar represents total TFPI. Dark portion represents full-length TFPI and lightly shaded portion represents truncated TFPI.
DISCUSSION
Fibrin deposition is a pathological hallmark of ARDS, and the TF pathway is a key modulator of intra-alveolar fibrin deposition. TF is upregulated and activated in the alveolar compartment in ARDS (4, 23). Despite the emerging interest in the role of TF in the pathogenesis of ARDS, very little is known about the role the endogenous inhibitor of TF, TFPI, in the lung. De Moerloose et al. (16) measured procoagulant activity and TFPI levels in BAL fluid from patients with interstitial lung disease, pneumonia, Pneumocystis carinii pneumonia (PCP), and normal controls. They found a significant increase in TFPI in both pneumonia and PCP patients compared with controls; these patients still had increased BAL procoagulant activity, and there was no relationship between TFPI levels and procoagulant activity (16). Sabharwal et al. (38) showed that TFPI levels in BAL fluid of patients at risk for ARDS and those with ARDS were 7-fold and 20-fold higher than normal controls, but no other measures of procoagulant activity were made.
In this study, we first tested whether patients with ARDS had elevated levels of TFPI protein in undiluted pulmonary edema fluid compared with a control population of critically ill patients with hydrostatic edema. TFPI levels in the air spaces were significantly higher in patients with ARDS compared with controls with a median level of 23,600 pg/ml in ARDS. Despite these high levels of TFPI in the alveolar compartment, pulmonary edema fluid from patients with ARDS contains high levels of TF-dependent procoagulant activity as measured by clot formation time of normal plasma (4). Based on these observations, we hypothesized that the TFPI protein in the air spaces in patients with ARDS was in a truncated and thus inactive form. To test this hypothesis, we measured truncated TFPI protein by ELISA and found that significantly more of the TFPI protein is truncated and thus inactive in patients with ARDS compared with those with hydrostatic edema. This finding provides a mechanism for the finding that pulmonary edema fluid retains TF procoagulant activity despite high levels of TFPI protein, and this result has implications for therapeutic manipulation of intra-alveolar fibrin deposition by introducing TFPI locally into the alveolar space.
To our knowledge, no prior studies on the role of TFPI in the pathogenesis of ARDS have examined the cellular origin of TFPI. Although intra-alveolar TFPI might, in part, originate from the systemic circulation and enter the alveolar space as a result of increased alveolocapillary membrane permeability, there are also potential local sources of TFPI within the lung. The alveolar epithelium is a key regulator of the intra-alveolar environment and has many important functions in the development and resolution of ARDS including surfactant production (18), repair of the alveolar epithelial barrier (32), and modulation of fibrin deposition (26). Our group (4) has shown that the alveolar epithelium can activate coagulation through upregulation of TF and modulate coagulation via the protein C pathway (39) in response to inflammatory stimuli. In immunohistochemical studies of TFPI protein in lung sections of patients with ARDS compared with patients who died without ARDS, we found that basally, TFPI is present along alveolar septae and in the alveolar epithelium. In ARDS, TFPI is present in the air spaces, hyaline membranes, alveolar epithelium, and alveolar macrophages, and the alveolar septal staining is no longer evident. TFPI staining was specific to the cells lining the distal air spaces and was not present in the pulmonary vascular endothelium in blood vessels of any size. This is consistent with the current literature that suggests that TFPI is not produced by the pulmonary vascular endothelium. In patients with idiopathic pulmonary arterial hypertension, circulating TFPI levels are no different compared with controls (9, 13); however, in patients with parenchymal lung diseases such as idiopathic pulmonary fibrosis (10) and chronic obstructive pulmonary disease (11), TFPI levels were elevated compared with controls. These findings suggest that under basal conditions in the lung, TFPI is present only in the cells that line the alveolar space and is released into the alveolar space in the setting of acute lung injury.
We next wanted to test the hypothesis that the alveolar epithelium is a source of TFPI. We found that following stimulation with cytomix, A549 cells release TFPI protein into the media without a significant increase in TFPI mRNA or protein production. This confirmed our immunostaining findings and suggests that under basal conditions, the alveolar epithelium expresses TFPI protein that is released from the epithelium in response to an inflammatory stimulus. Although the A549 cells released high levels of TFPI, the conditioned media retained TF-dependent procoagulant activity. One possible explanation is that TFPI released from the cells is truncated, similar to what is seen in ARDS. However, in our cell culture system, less than 10% of the TFPI released was truncated, presumably because the cell culture system does not contain metalloproteases or neutrophil elastases. There are other potential explanations for the failure of the TFPI released into the conditioned media to inhibit TF-dependent procoagulant activity. TFPI anticoagulant activity is dependent on several factors including GPI cell surface binding (31) and association with other proteins such as protein S (19), or non-protein factors such as LDL (36), all of which could affect the ability of TFPI to inhibit TF procoagulant activity.
These findings are important for several reasons. First, this is the first report of TFPI protein production and release by the alveolar epithelium. Others have measured TFPI in BAL from patients with ARDS and pneumonia, but the cellular origin of the protein has not been described. Our finding is critically important because it identifies a local target in the lung (the alveolar epithelium) that can be directly targeted to modulate intra-alveolar TF activity. Second, this report demonstrates that the majority of TFPI in the air spaces in ARDS is inactive. Several investigators have identified proteases that cleave fulllength TFPI, many of which are derived from inflammatory cells and are very high in the air spaces in ARDS. This is an important observation that must be taken into consideration for future studies that attempt to modulate intra-alveolar TF activity. Even the intra-alveolar administration of TFPI might be rendered inactive by the presence of high levels of proteolytic activity.
Our study has some limitations. First, the mechanism of TFPI truncation was not explored. Others have shown that TFPI is sensitive to cleavage by neutrophil elastases (21, 37) and metalloproteases (5, 15) and that the cleaved protein is inactive. Given the abundance of these proteases in the air space in ALI/ARDS (34), it is likely that similar proteolytic mechanisms are responsible for the cleavage of TFPI in the lungs. Second, while these experiments are the first to characterize TFPI expression and release by the alveolar epithelium, the overall functional significance of alveolar epithelial TFPI expression is still unknown. Animal studies have shown lung protective effects of TF inhibition in the setting of sepsis (8, 44), but a large, randomized, placebo-controlled study of recombinant TFPI in sepsis did not show a benefit to TFPI treatment (1). These conflicting results highlight the need for a better understanding of the role of TF in the lung in ALI/ARDS and mandate continued study of the complex regulation and balance of TF and TFPI in the lung. Inhibition of the TF pathway for therapeutic benefit in ALI/ARDS could be achieved in several different ways, including recombinant TFPI, site-inactivated VIIa, blocking antibody to Xa, and other small molecule inhibitors. Which inhibitor to use, when to administer it, and whether to administer the inhibitor in the systemic circulation or directly into the air spaces of the lung are still to be determined.
In summary, we have identified a potential role for the alveolar epithelium in modulating TF-dependent fibrin deposition in the air spaces, through production and release of TFPI. However, despite very high levels of intra-alveolar TFPI, the alveolar space still has significant amounts of TF-dependent procoagulant activity because the majority of TFPI in the air spaces in ARDS is truncated and inactive. Future studies aimed at modulating intra-alveolar TF activity through induction of or administration of TFPI will need to consider the intra-alveolar milieu and the possibility that lung-specific therapeutics may be compromised by proteolytic activity.
Acknowledgments
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-081332 and HL-51856 and the Vanderbilt Physician Scientist Development Program.
REFERENCES
- 1.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, Beale R, Svoboda P, Laterre PF, Simon S, Light B, Spapen H, Stone J, Seibert A, Peckelsen C, De Deyne C, Postier R, Pettila V, Artigas A, Percell SR, Shu V, Zwingelstein C, Tobias J, Poole L, Stolzenbach JC, Creasey AA. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 2.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol. 2002;161:1783–1796. doi: 10.1016/S0002-9440(10)64455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemost. 2001;86:959–972. [PubMed] [Google Scholar]
- 4.Bastarache J, Wang L, Geiser T, Wang Z, Albertine K, Matthay M, Ware L. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62:608–616. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belaaouaj AA, Li A, Wun TC, Welgus HG, Shapiro SD. Matrix metalloproteinases cleave tissue factor pathway inhibitor. Effects on coagulation. J Biol Chem. 2000;275:27123–27128. doi: 10.1074/jbc.M004218200. [DOI] [PubMed] [Google Scholar]
- 6.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. The American-European Consensus Conference on ARDS. [DOI] [PubMed] [Google Scholar]
- 7.Carr C, Bild GS, Chang AC, Peer GT, Palmier MO, Frazier RB, Gustafson ME, Wun TC, Creasey AA, Hinshaw LB, Taylor FB, Jr, Galluppi GR. Recombinant E coli-derived tissue factor pathway inhibitor reduces coagulopathic and lethal effects in the baboon gram-negative model of septic shock. Circ Shock. 1994;44:126–137. [PubMed] [Google Scholar]
- 8.Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, Petersen LC, Piantadosi CA. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med. 2003;167:1200–1209. doi: 10.1164/rccm.200204-287OC. [DOI] [PubMed] [Google Scholar]
- 9.Cella G, Bellotto F, Tona F, Sbarai A, Mazzaro G, Motta G, Fareed J. Plasma markers of endothelial dysfunction in pulmonary hypertension. Chest. 2001;120:1226–1230. doi: 10.1378/chest.120.4.1226. [DOI] [PubMed] [Google Scholar]
- 10.Cella G, Cipriani A, Tommasini A, Rampin E, Sbarai A, Rocconi R, Mazzaro G, Luzzatto G. Tissue factor pathway inhibitor (TFPI) antigen plasma level in patients with interstitial lung disease before and after heparin administration. Semin Thromb Hemost. 1997;23:45–49. doi: 10.1055/s-2007-996069. [DOI] [PubMed] [Google Scholar]
- 11.Cella G, Sbarai A, Mazzaro G, Vanzo B, Romano S, Hoppensteadt T, Fareed J. Plasma markers of endothelial dysfunction in chronic obstructive pulmonary disease. Clin Appl Thromb Hemost. 2001;7:205–208. doi: 10.1177/107602960100700304. [DOI] [PubMed] [Google Scholar]
- 12.Cirino G, Cicala C, Bucci MR, Sorrentino L, Maraganore JM, Stone SR. Thrombin functions as an inflammatory mediator through activation of its receptor. J Exp Med. 1996;183:821–827. doi: 10.1084/jem.183.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collados MT, Velazquez B, Borbolla JR, Sandoval J, Masso F, Montano LF, Guarner V. Endothelin-1 and functional tissue factor: a possible relationship with severity in primary pulmonary hypertension. Heart Vessels. 2003;18:12–17. doi: 10.1007/s003800300002. [DOI] [PubMed] [Google Scholar]
- 14.Creasey AA, Chang AC, Feigen L, Wun TC, Taylor FB, Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–2860. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham AC, Hasty KA, Enghild JJ, Mast AE. Structural and functional characterization of tissue factor pathway inhibitor following degradation by matrix metalloproteinase-8. Biochem J. 2002;367:451–458. doi: 10.1042/BJ20020696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Moerloose P, De Benedetti E, Nicod L, Vifian C, Reber G. Procoagulant activity in bronchoalveolar fluids: no relationship with tissue factor pathway inhibitor activity. Thromb Res. 1992;65:507–518. doi: 10.1016/0049-3848(92)90202-l. [DOI] [PubMed] [Google Scholar]
- 17.Enkhbaatar P, Okajima K, Murakami K, Uchiba M, Okabe H, Okabe K, Yamaguchi Y. Recombinant tissue factor pathway inhibitor reduces lipopolysaccharide-induced pulmonary vascular injury by inhibiting leukocyte activation. Am J Respir Crit Care Med. 2000;162:1752–1759. doi: 10.1164/ajrccm.162.5.9911018. [DOI] [PubMed] [Google Scholar]
- 18.Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, Hull W, Whitsett JA, Akino T, Kuroki Y, Nagae H, Hudson LD, Martin TR. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med. 1999;160:1843–1850. doi: 10.1164/ajrccm.160.6.9901117. [DOI] [PubMed] [Google Scholar]
- 19.Hackeng TM, Sere KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci USA. 2006;103:3106–3111. doi: 10.1073/pnas.0504240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamuro T, Kido H, Asada Y, Hatakeyama K, Okumura Y, Kunori Y, Kamimura T, Iwanaga S, Kamei S. Tissue factor pathway inhibitor is highly susceptible to chymase-mediated proteolysis. FEBS J. 2007;274:3065–3077. doi: 10.1111/j.1742-4658.2007.05833.x. [DOI] [PubMed] [Google Scholar]
- 21.Higuchi DA, Wun TC, Likert KM, Broze GJ., Jr. The effect of leukocyte elastase on tissue factor pathway inhibitor. Blood. 1992;79:1712–1719. [PubMed] [Google Scholar]
- 22.Idell S. Anticoagulants for acute respiratory distress syndrome: can they work? Am J Respir Crit Care Med. 2001;164:517–520. doi: 10.1164/ajrccm.164.4.2102095. [DOI] [PubMed] [Google Scholar]
- 23.Idell S, Gonzalez K, Bradford H, MacArthur CK, Fein AM, Maunder RJ, Garcia JG, Griffith DE, Weiland J, Martin TR, McLarty J, Fair DS, Walsh PN, Colman RW. Procoagulant activity in bronchoalveolar lavage in the adult respiratory distress syndrome. Contribution of tissue factor associated with factor VII. Am Rev Respir Dis. 1987;136:1466–1474. doi: 10.1164/ajrccm/136.6.1466. [DOI] [PubMed] [Google Scholar]
- 24.Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest. 1989;84:695–705. doi: 10.1172/JCI114217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Idell S, Koenig KB, Fair DS, Martin TR, McLarty J, Maunder RJ. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 1991;261:L240–L248. doi: 10.1152/ajplung.1991.261.4.L240. [DOI] [PubMed] [Google Scholar]
- 26.Jesmin S, Gando S, Matsuda N, Sakuma I, Kobayashi S, Sakuraya F, Hattori Y. Temporal changes in pulmonary expression of key procoagulant molecules in rabbits with endotoxin-induced acute lung injury: elevated expression levels of protease-activated receptors. Thromb Haemost. 2004;92:966–979. doi: 10.1160/TH04-03-0160. [DOI] [PubMed] [Google Scholar]
- 27.Le Gall JR, Lemeshow S, Saulnier F. A new simplified acute physiology score (SAPS II) based on a European/North American multicenter study. JAMA. 1993;270:2957–2963. doi: 10.1001/jama.270.24.2957. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta CT) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 1994;267:L223–L241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- 30.Lwaleed BA, Bass PS. Tissue factor pathway inhibitor: structure, biology and involvement in disease. J Pathol. 2006;208:327–339. doi: 10.1002/path.1871. [DOI] [PubMed] [Google Scholar]
- 31.Maroney SA, Cunningham AC, Ferrel J, Hu R, Haberichter S, Mansbach CM, Brodsky RA, Dietzen DJ, Mast AE. A GPI-anchored co-receptor for tissue factor pathway inhibitor controls its intracellular trafficking and cell surface expression. J Thromb Haemost. 2006;4:1114–1124. doi: 10.1111/j.1538-7836.2006.01873.x. [DOI] [PubMed] [Google Scholar]
- 32.Matthay MA, Fukuda N, Frank J, Kallet R, Daniel B, Sakuma T. Alveolar epithelial barrier. Role in lung fluid balance in clinical lung injury. Clin Chest Med. 2000;21:477–490. doi: 10.1016/s0272-5231(05)70160-x. [DOI] [PubMed] [Google Scholar]
- 33.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- 34.Moraes TJ, Chow CW, Downey GP. Proteases and lung injury. Crit Care Med. 2003;31:S189–S194. doi: 10.1097/01.CCM.0000057842.90746.1E. [DOI] [PubMed] [Google Scholar]
- 35.Murray JF, Matthay MA, Luce JM, Flick MR. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:720–723. doi: 10.1164/ajrccm/138.3.720. [DOI] [PubMed] [Google Scholar]
- 36.Novotny WF, Girard TJ, Miletich JP, Broze GJ., Jr. Purification and characterization of the lipoprotein-associated coagulation inhibitor from human plasma. J Biol Chem. 1989;264:18832–18837. [PubMed] [Google Scholar]
- 37.Petersen LC, Bjorn SE, Nordfang O. Effect of leukocyte proteinases on tissue factor pathway inhibitor. Thromb Haemost. 1992;67:537–541. [PubMed] [Google Scholar]
- 38.Sabharwal AK, Bajaj SP, Ameri A, Tricomi SM, Hyers TM, Dahms TE, Taylor FB, Jr, Bajaj MS. Tissue factor pathway inhibitor and von Willebrand factor antigen levels in adult respiratory distress syndrome and in a primate model of sepsis. Am J Respir Crit Care Med. 1995;151:758–767. doi: 10.1164/ajrccm/151.3_Pt_1.758. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Bastarache JA, Wickersham N, Fang X, Matthay MA, Ware LB. Novel role of the human alveolar epithelium in regulating intraalveolar coagulation. Am J Respir Cell Mol Biol. 2007;36:497–503. doi: 10.1165/rcmb.2005-0425OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ware LB, Fang X, Matthay MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L514–L521. doi: 10.1152/ajplung.00442.2002. [DOI] [PubMed] [Google Scholar]
- 41.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 42.Welty-Wolf KE, Carraway MS, Idell S, Ortel TL, Ezban M, Piantadosi CA. Tissue factor in experimental acute lung injury. Semin Hematol. 2001;38:35–38. doi: 10.1053/shem.2001.29505. [DOI] [PubMed] [Google Scholar]
- 43.Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, Idell S, Piantadosi CA. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med. 2001;164:1988–1996. doi: 10.1164/ajrccm.164.10.2105027. [DOI] [PubMed] [Google Scholar]
- 44.Welty-Wolf KE, Carraway MS, Ortel TL, Ghio AJ, Idell S, Egan J, Zhu X, Jiao JA, Wong HC, Piantadosi CA. Blockade of tissue factor-factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol. 2006;290:L21–L31. doi: 10.1152/ajplung.00155.2005. [DOI] [PubMed] [Google Scholar]