

Summary
Expression of genes in the gapA operon encoding five enzymes for triose phosphate interconversion in Bacillus subtilis is negatively regulated by the Central glycolytic genes Regulator (CggR). CggR belongs to the large SorC/DeoR family of prokaryotic transcriptional regulators, characterized by an N-terminal DNA-binding domain and a large C-terminal effector-binding domain. When no glucose is present in growth media, CggR binds to its target DNA sequence and blocks the transcription of genes in the gapA operon. In the presence of glucose, binding of the known effector molecule fructose-1,6-bisphosphate abolishes this interaction. We have identified dihydroxyacetone phosphate, glucose-6-phosphate and fructose-6-phosphate as additional CggR ligands that can bind to the effector-binding site. Crystal structures of C-CggR, the C-terminal effector-binding domain of CggR, both unliganded as well as in complex with the four ligands at resolutions between 1.65 and 1.80 Å reveal unique ligand-specific structural changes in the binding site that affect the dimer interface. Binding affinities of these ligands were determined by isothermal titration calorimetry. Chemical cross-linking shows that CggR oligomerization is mediated through its effector-binding domain, and that binding of the different ligands differentially affects the distribution of oligomers. Electrophoretic mobility shift assays (EMSAs) confirmed a destabilizing effect of fructose-1,6-bisphosphate on the CggR/DNA complex, and also showed similar effects for dihydroxyacetone phosphate. Our results suggest that CggR stability and function may be modulated by various effectors in a complex fashion.
Introduction
Bacillus subtilis, like many other bacteria, can utilize various carbohydrates as sources of carbon and energy. In order to utilize these carbohydrates, specific kinases are required to produce phosphorylated intermediates that enter into general metabolic pathways, resulting in the production of energy and/or anabolism (Deutscher et al., 1995; Stulke and Hillen, 2000). Because the activity of the central catabolic pathways needs to be in balance with the anabolic activity of the cell, transcription of the genes, encoding the enzymes of glycolysis, is tightly controlled in the presence of various carbohydrates.
Carbon catabolism regulation has been studied extensively in B. subtilis (Henkin, 1996; Stulke and Hillen, 2000). In the presence of glucose in growth media, the strongest regulation was observed for the gene encoding glyceraldehyde-3-phosphate dehydrogenase (GapA) (Ludwig et al., 2001; Ludwig et al., 2002). Expression of GapA and genes for four more enzymes involved in triose phosphate interconversions located in the same operon (gapA) is controlled by two transcriptional regulators that sense the availability of glucose: (i) Catabolite control protein A (CcpA) and (ii) Central glycolytic genes Repressor (CggR). While the CcpA-induction effect on the transcription of gapA operon is indirect (Ludwig et al., 2001), CggR is a direct repressor that binds to a unique target sequence in the promoter region of the gapA operon (Doan and Aymerich, 2003).
CggR belongs to the large SorC/DeoR family of prokaryotic transcriptional regulators. These regulators are characterized by an N-terminal DNA-binding domain and a large C-terminal effector-binding domain. Orthologues of the B. subtilis CggR protein can be found in all available genomes of Bacillaceae, 10 Lactobacillus species, two Listeria, eight Clostridium, four Staphylococcus and two Thermoanaerobacter species, Enterococcus faecalis and four other bacteria belonging to Clostridia. The biological function of orthologues has not been characterized experimentally, but their high sequence similarity and position in the chromosome strongly suggest an activity similar to that of B. subtilis CggR (Doan and Aymerich, 2003). In addition, homologous proteins with unknown function can be found in many other bacterial species, including pathogenic Salmonella and Yersinia species.
CggR recognizes a unique DNA target sequence (operator) comprising two direct repeats (Doan and Aymerich, 2003). Two CggR dimers bind to the operator in a highly cooperative manner (Zorrilla et al., 2007), thereby blocking the transcription of gapA genes. Transcriptional repression by CggR is abolished when glucose is present in growth media (Ludwig et al., 2001). The glycolysis metabolite fructose-1,6-bisphosphate (FBP) has been identified as an effector whose binding modulates CggR interactions with DNA (Doan and Aymerich, 2003). In vivo studies suggested that CggR can also respond synergically to different signals from carbohydrate catabolism as well as from amino acid anabolism (Ludwig et al., 2001). It is thus likely that various molecules can function as CggR effectors.
In order to gain structural information on effector recognition and binding in the structurally sparsely characterized SorC/DeoR family of proteins, we determined the crystal structure of the C-terminal effector-binding domain of CggR. Besides the previously recognized effector FBP, we identified dihydroxyacetone phosphate (DHAP), glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P) as additional ligands and characterized their binding to CggR. Crystal structures of CggR in complex with the four ligands reveal structural changes accompanying their binding. Chemical cross-linking was used to characterize the effect of ligands on the oligomeric state of CggR and electrophoretic mobility shift assays (EMSAs) was employed to study the effects of ligands on DNA binding to CggR in vitro. Our results show that CggR is able to bind several different small, phosphorylated carbohydrates, intermediates of the glycolytic pathway, and that ligand binding is accompanied by changes in the structure of CggR as well as its oligomeric state.
Results
Overall C-CggR structure
The crystal structure of the C-terminal effector-binding domain of CggR (residues 89–340, referred to as C-CggR) was determined by single-wavelength anomalous dispersion (SAD) using the selenomethionyl-substituted variant of C-CggR, and was refined using data to 1.65 Å resolution. The orthorhombic crystal form contains two molecules in the asymmetric unit with a solvent content of 41%. The final crystallographic model contains two molecules of C-CggR with residues 87–338 each. The two N-terminal residues, Asn87 and Ala88, represent a cloning artefact that remained after proteolytic removal of the N-terminal His6 tag. The two C-terminal residues in both molecules in the asymmetric unit as well as residues 180–182 of molecule A could not be located in the electron density map and were thus not included in the final model. Data collection and model refinement statistics are shown in Tables 3 and 4 in Experimental procedures.
Table 3.
Data collection and phasing statistics.
| Data collection statistics |
|||||
|---|---|---|---|---|---|
| Free (SeMet) | FBP co-crystal | GAP/DHAP co-crystal | G6P co-crystal | F6P co-crystal | |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell parameters (Å) | 52.3, 83.3, 116.7 | 51.4, 83.2, 116.5 | 55.5, 83.8, 113.7 | 55.5, 83.8, 113.6 | 55.5, 83.6, 113.7 |
| Molecules in asymmetric unit | 2 | 2 | 2 | 2 | 2 |
| Wavelength (Å) | 0.979 | 0.979 | 1.542 | 0.979 | 0.977 |
| Resolution (Å) | 34–1.65 | 23.64–1.70 | 35.9–1.80 | 49.8–1.80 | 49.8–1.85 |
| (1.66–1.65) | (1.76–1.70) | (1.86–1.80) | (1.86–1.80) | (1.92–1.85) | |
| Unique reflections | 113 636 | 50 944 | 49 172 | 49 657 | 45,227 |
| Redundancy | 5.3 (4.6) | 4.7 (4.2) | 4.9 (2.9) | 6.7 (3.2) | 6.3 (5.5) |
| Completeness (%) | 97.1 (99.2) | 91.4 (94.2) | 99.2 (95.6) | 99.0 (90.9) | 99.2 (97.2) |
| Rmergea | 8.7 (62.7) | 7.3 (66.6) | 6.0 (51.0) | 7.5 (48.9) | 10.0 (42.9) |
| Average I/σ(I) | 52.2 (3.6) | 21.86 (2.0) | 38.7 (2.1) | 24.1 (1.9) | 21.82 (3.0) |
| Wilson B (Å2) | 23.0 | 29.0 | 33.1 | 25.5 | 20.2 |
| Phasing statistics | ||||||||
|---|---|---|---|---|---|---|---|---|
| Dataset used for phasing | Free (SeMet) | |||||||
| Se sites (expected/found) | 16/16 | |||||||
| Resolution shell limit (Å) | 10.72 | 6.01 | 4.17 | 3.19 | 2.59 | 2.18 | 1.88 | 1.65 |
| Phasing powerb | 0.83 | 0.91 | 1.26 | 1.1 | 1.1 | 0.69 | 0.36 | 0.28 |
| Rcullisc | 0.82 | 0.79 | 0.64 | 0.68 | 0.66 | 0.88 | 1.00 | 0.98 |
The data in parentheses refer to the highest-resolution shell.
Rmerge = (|Ihkl – ⟨I⟩|)/Ihkl, where the average intensity ⟨I⟩ is taken over all symmetry equivalent measurements and Ihkl is the measured intensity for any given reflection.
Phasing power = Σ|FH|calc/Σ||FPH|obs − |FPH|calc|, where |FH|calc is the calculated structure factor amplitude for the heavy atom model, and |FPH|obs and |FPH|calc are the observed and calculated, respectively, structure factor amplitudes for the derivative.
Rcullis = Σ||FPH + FP| – |FH|calc|/Σ|FPH − FP|, where |FH|calc is the calculated structure factor amplitude for the heavy atom model, and FPH and FP are the observed structure factors for derivative and protein respectively.
Table 4.
Refinement statistics.
| Refinement statistics |
|||||
|---|---|---|---|---|---|
| Free | FBP co-crystal | GAP/DHAP co-crystal | G6P co-crystal | F6P co-crystal | |
| PDB code | 2OKG | 3BXF | 3BXE | 3BXG | 3BXH |
| Resolution range (Å) | 34.0–1.65 | 23.64–1.70 | 35.9–1.80 | 49.8–1.80 | 49.8–1.85 |
| (1.69–1.65) | (1.76–1.70) | (1.86–1.80) | (1.86–1.80) | (1.92–1.85) | |
| No. of reflections in working set | 57 112 (4185) | 48 096 (3526) | 47 018 (3185) | 46 520 (2986) | 42 927 (2831) |
| No. of reflections in test set | 3089 (232) | 2570 (190) | 2517 (169) | 2491 (186) | 2284 (169) |
| R-value (%)a | 19.5 (23.1) | 20.0 (27.7) | 17.7 (24.8) | 18.2 (25.2) | 16.1 (20.3) |
| Rfree value (%)b | 23.9 (29.0) | 25.6 (33.1) | 21.5 (32.3) | 23.3 (31.3) | 20.9 (28.4) |
| RMSD bond length (Å) | 0.016 | 0.014 | 0.011 | 0.014 | 0.012 |
| RMSD angle (°) | 1.479 | 1.398 | 1.220 | 1.430 | 1.442 |
| Number of atoms in AU | 4318 | 4509 | 4473 | 4440 | 4604 |
| Protein atoms | 3890 | 3837 | 3842 | 3905 | 3943 |
| Missing residues in chain A | 180–182, 339–340 | 339–340 | 89–92 | 89–92, 340 | 89–92 |
| Missing residues in chain B | 339–340 | 89–93, 339–340 | 339–340 | 339–340 | 339–340 |
| Ligand atoms bound to protein chain A |
— | 20 (FBP) | 10 (DHAP) | 16 (G6P) | 16 (F6P) |
| Ligand atoms bound to protein chain B |
10 (DHAP) | 10 (DHAP) | 10 (DHAP) | 16 (G6P) | 16 (F6P) |
| Water molecules | 472 | 638 | 611 | 487 | 623 |
| Other non-protein atoms | 3 (Cl) | 4 (Cl) | — | — | 6 (SCN) |
| Mean B-value (Å2) | 18.5 | 31.5 | 31.3 | 26.8 | 19.0 |
| Ramachandran plot statistics: number of residues in | |||||
| Favoured regions (%) | 93.4 | 93.2 | 91.9 | 93.2 | 93.2 |
| Allowed regions (%) | 6.6 | 5.8 | 8.1 | 6.8 | 6.8 |
In parentheses are statistics for the highest resolution shell.
R-value = ||Fo| − |Fc||/|Fo|, where Fo and Fc are the observed and calculated structure factors respectively.
Rfree is equivalent to R-value but is calculated for 5% of the reflections chosen at random and omitted from the refinement process. Cl, chloride ion; SCN, thiocyanate ion.
C-CggR adopts an open, three-layer α/β/α sandwich architecture (Fig. 1). The central part is formed by a twisted, six-stranded, parallel β sheet flanked on both sides by a total of five α helices, adopting a typical nicotinamide adenine dinucleotide (NAD)-binding fold (Fig. 1). Three peripheral subdomains, named P1, P2 and P3, are added to this central part so that the C-CggR structure resembles a trefoil in its shape (Fig. 1). The peripheral subdomain P1 (residues 94–113 and 340–331) consists of a short β strand and two terminal helices (designated P1αA and P1αC) that pack against each other. In full-length CggR, the N-terminal helix links the effector-binding domain to the preceding DNA-binding domain. The long P1αB helix packs against the central domain but, topologically, is not part of the canonical NAD-binding fold. The peripheral subdomain P2 (residues 175–184 and 242–289) is composed of a small antiparallel β sheet, formed by P2 β strands 1–4 and flanked on one side by two short helices (P2αA and αB). A second short antiparallel β sheet is formed by P2 β strands 5 and 6, and is connected to helix αB in the central domain by an extended loop. This loop (residues 178–184), together with β strand 6 of the P2 subdomain (residues 175–177), is part of the effector-binding site, and undergoes significant structural rearrangements upon ligand binding (discussed below). The peripheral subdomain P3 is composed of an extended loop and a single helix (residues 203–221).
Fig. 1.

Overall structure of C-CggR.
A. Topology diagram of C-CggR. The central NAD-binding fold subdomain is shown in yellow, and the three peripheral subdomains P1, P2 and P3 are coloured red, blue and green respectively. β strands are numbered sequentially, and α helices are referred to by letters for each domain separately. Residue numbers are indicated, and the region undergoing structural rearrangements upon ligand binding (flap) is outlined in orange.
B. Ribbon representation of the overall structure of C-CggR. The location of the effector-binding site is indicated by an asterisk. The colour coding is identical to the one used in A.
The present study provides the first detailed structural description of a member of the large family of DeoR transcriptional regulators. Previously, structural information about this family was available only for effector-binding domains of uncharacterized putative transcriptional regulators from Streptococcus pneumoniae and E. faecalis [Protein Data Bank (PDB) codes 2GNP and 2O0M respectively] obtained by the Midwest Center for Structural Genomics. These two structures share significant similarity with C-CggR with root-mean-square deviations (RMSDs) of 2.2 Å for 225 equivalent Cα atoms of 2GNP and 1.91 Å for 220 equivalent Cα atoms of 2O0M respectively.
A structural comparison with models deposited in the Protein Data Bank using the program DALI (Holm and Sander, 1994) identified two enzymes involved in the metabolism of carbohydrates as structurally similar to C-CggR: 6-phosphogluconolactonase (PGLS) from Thermatoga maritima (PDB codes 1PBT and 1VL1) and glucosamine 6-phosphate (GlcN6P) deaminase NagB from Escherichia coli (PDB code 1DEA) (Oliva et al., 1995). This structural similarity was indeed predicted previously and used for homology modelling (Doan et al., 2008). The structures of PGLS and NagB superimpose with C-CggR with RMSDs of 3.9 Å for 176 equivalent Cα atoms and 3.6 Å for 168 equivalent Cα atoms respectively (Fig. 2). The substrates of these two enzymes (6-phosphogluconolactone and GlcN6P respectively) are structurally similar to each other and also to G6P, one of the CggR ligands we describe in this study. The location of active sites in structures of both enzymes coincides with the effector-binding site of CggR.
Fig. 2.
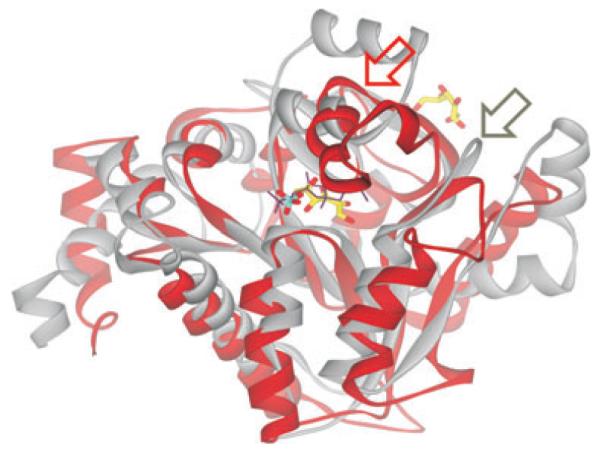
Superimposition of C-CggR and GlcN6P deaminase NagB structures Ribbon representation of C-CggR (gray) and NagB (red) structures. FBP bound to C-CggR is shown as black lines, and the reaction product fructose-6-phosphate (F6P) bound to NagB (PDB code 1FQO, Rudino-Pinera et al., 2002) is shown as a stick model with carbons in yellow, oxygen atoms in red and phosphorous in cyan. Note that the NagB structure contains a second F6P molecule that is bound to its allosteric site. Regions that undergo structural rearrangements upon substrate and ligand binding are indicated by red and gray arrows for NagB and C-CggR respectively.
Structures of C-CggR-ligand complexes
During the refinement of the C-CggR structure determined from selenomethionyl-substituted protein crystals, non-protein electron density indicated the presence of a small molecule in one of the two C-CggR molecules in the asymmetric unit. The electron density is compatible with glyceraldehyde-3-phosphate (GAP), DHAP and glycerol-3-phosphate (G3P). All three molecules have very similar 3-D structures and are intermediates of carbohydrate metabolism (Fig. 4). The O1 oxygen of the ligand is at a distance of 2.6 Å from the side-chain carboxyl group of Glu269, suggesting the presence of a hydrogen-bond interaction. Assuming that Glu269 is deprotonated, this hydrogen bond is only possible if the O1 oxygen in the ligand is part of a hydroxyl group. It is thus likely that the bound ligand is either DHAP or G3P and not GAP. C-CggR apparently appropriated the ligand from the cytoplasm of the expression host, E. coli, as no exogenous carbohydrate ligands were added to any solution during protein purification and crystallization. Glycerol was used as the main carbon source for E. coli cultivation, which after phosphorylation to G3P enters the glycolytic pathway through conversion to DHAP and GAP (Fig. 4). It is thus possible that a mixture of these triosephosphates is present in the crystal. DHAP was included in the final crystallographic model (Fig. 3), and for the sake of brevity, the ligand bound to this type of C-CggR crystal is referred to as DHAP throughout the remainder of the text.
Fig. 4.

Glycolysis in B. subtilis. The glycolytic pathway with the respective enzymes encoded by gapA operon and controlled by CggR (Tpi, triose phosphate isomerase; GapA, glyceraldehyde-3-phosphate dehydrogenase; Pgk, phosphoglycerate kinase; Pgm, phosphoglycerate mutase; Eno, phosphoglycerate enolase). Structural formulas of glycolytic intermediates investigated in this work are shown.
Fig. 3.

Ligands bound to the C-CggR effector-binding site in five crystal structures. The ligands, dihydroxyacetone phosphate (DHAP), fructose-1,6-bisphosphate (FBP), glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P) are shown as stick models with carbons in green, oxygen atoms in red and phosphorous in orange. 2Fo-Fc omit electron density maps for each ligand are contoured at 1.0σ. Water molecules in the unoccupied ligand binding site are shown as red spheres with corresponding 2Fo-Fc map contoured at 1.0σ.
The procurement of this ligand from E. coli and its retention by C-CggR through several chromatographic purification steps indicate that this ligand likely binds with high affinity. The finding of serendipitous ligand in C-CggR structure provided us with the first indication that CggR is capable of binding molecules involved in carbohydrate metabolism other than the previously identified FBP (Doan and Aymerich, 2003).
In order to investigate whether the crystal packing prevented ligand binding to one of the two C-CggR molecules in the asymmetric unit, native protein was crystallized in the presence of a high concentration (10 mM) of GAP, and the crystal structure of the complex was determined by difference-Fourier techniques and refined using diffraction data to 1.8 Å resolution. The resulting electron density revealed that the ligand-binding site in both molecules was occupied. Because GAP and DHAP readily interconvert and exist in equilibrium when free in solution, and in order to satisfy the hydrogen bond to Glu269 mentioned above, we modelled this ligand as DHAP (Fig. 3). This result showed that both C-CggR molecules present in the asymmetric unit are able to bind ligand in their respective effector-binding sites. The differential occupancies observed in crystals obtained with selenomethionyl-substituted protein therefore seem to be due to different affinities of the ligand-binding sites towards DHAP. The concentration of the ligand during expression of C-CggR in E. coli was apparently not sufficient to occupy both binding sites, or fraction of the ligand was lost during the subsequent purification and crystallization steps.
We then investigated whether the DHAP-binding site coincides with the binding site for the known C-CggR effector FBP (Doan and Aymerich, 2003). The native protein was co-crystallized in the presence of 90 mM FBP, and the crystal structure of the resulting complex was determined by difference-Fourier techniques, and refined using diffraction data to 1.7 Å resolution. The resulting electron density showed that a molecule of FBP was bound to only one of the two C-CggR molecules in the asymmetric unit, namely to molecule A whose ligand-binding site was unoccupied in the crystals obtained with selenomethionyl-substituted protein. The binding site of the second C-CggR molecule retained the DHAP molecule appropriated from E. coli (Fig. 3).
The fact that DHAP and FBP both bind to the same binding site with high affinity, but are structurally significantly different, prompted us to explore the possibility that C-CggR is able to bind other carbohydrate metabolites produced in the pathway preceding the enzymatic step regulated by CggR (Fig. 4).
We succeeded in obtaining crystals suitable for structure determination of C-CggR in the presence of 10 mM G6P or 10 mM F6P, and refined the resulting complex structures using diffraction data to 1.8 Å and to 1.85 Å respectively. In these two structures, the ligands were found to occupy the effector-binding sites in both C-CggR molecules in the asymmetric unit (Fig. 3). Because the native protein used for all co-crystallization experiments originated from the same protein batch, these results showed that the DHAP molecule that copurifies with native C-CggR can be displaced by 10 mM G6P or F6P present in the crystallization experiment but not by 90 mM FBP, most likely because of the specific differences in the patterns of interaction between C-CggR and these ligands (see below). Co-crystallization with another metabolite, glucose-1-phosphate (G1P), did not yield observable electron density for the ligand (data not shown), indicating that this molecule is not able to bind to C-CggR, or, less likely, that our crystal form prevents association.
Data collection and structure refinement statistics are shown in Tables 2 and 3 in Experimental procedures.
Table 2.
Crystal interface parameters.
| Structure | Dimer | Interface | Interface area (Å2) | Stable assemblya | ΔiG P-valueb |
|---|---|---|---|---|---|
| Free | Free/DHAP | I | 771.6 | + | 0.153 |
| II | 762.4 | + | 0.039 | ||
| III | 505.2 | − | 0.672 | ||
| FBP co-crystal | FBP/DHAP | I | 748.3 | + | 0.112 |
| II | 517.5 | − | 0.062 | ||
| III | 551.6 | − | 0.457 | ||
| GAP/DHAP co-crystal | DHAP/DHAP | I | 603.6 | − | 0.053 |
| II | 841.9 | − | 0.112 | ||
| III | 504.3 | − | 0.678 | ||
| G6P co-crystal | G6P/G6P | I | 618.2 | − | 0.042 |
| II | 547.8 | − | 0.095 | ||
| III | 359.6 | − | 0.810 | ||
| F6P co-crystal | F6P/F6P | I | 602.8 | − | 0.069 |
| II | 550.4 | − | 0.090 | ||
| III | 557.4 | − | 0.864 |
This classification is based on the value of free energy of dissociation of a potential assembly (Krissinel and Henrick, 2005).
ΔiG P-value indicates the P-value of the observed solvation free energy gain upon formation of the interface. The P-value measures the probability of getting a lower ΔiG value, compared with atoms picked randomly from a protein’s surface. P > 0.5 means that the interface is less hydrophobic than it could be. Therefore, the interface is likely to be an artefact of crystal packing. P < 0.5 indicates interfaces with very high hydrophobicity, implying that the interface might be interaction-specific (Krissinel and Henrick, 2005).
Structural features of ligand binding
The ligand-binding site in C-CggR is located in a cleft between the central subdomain and the peripheral sub-domain P2 (Fig. 1B). The finding of differential DHAP binding to the two C-CggR molecules in the asymmetric unit, afforded us with an opportunity to structurally compare the free and ligand-bound states of C-CggR, and recognize structural changes induced by binding of the four different ligands.
The ligand-binding site has the shape of a groove, about 19 Å long and 5 Å wide, lined with positively charged residues, appropriately poised to attract and guide the phosphoryl-containing, negatively charged ligands to the binding site (Fig. 5D). When the CggR-binding site is unoccupied, a state referred to as ‘U’ (for unliganded), residues 174–184 form a large loop with residues 180–182 being disordered (Fig. 5A and B). When DHAP binds to the effector-binding site, it forms numerous interactions with protein residues. Most prominently, the phosphoryl group establishes ionic interactions with Arg250 and Lys310 and additional hydrogen bonds with Thr151 and Thr152 (Fig. 6A). The interactions with this phosphoryl group are common to all structurally characterized ligands. The phosphoryl group seems to serve as an anchor to position the ligands in the effector-binding site. In addition, DHAP contacts C-CggR through several non-polar as well as direct and water-mediated polar interactions involving its carbohydrate portion (Fig. 6A, Table S1 and Fig. S1).
Fig. 5.

Ligand-induced conformational changes in the CggR effector-binding site.
A. Superimposition of C-CggR shown as a ribbon model in the unliganded U conformation (coloured yellow, with residues 180–182 missing in the model) and the two liganded conformations L1 (red) and L2 (blue). Regions that undergo large structural changes are indicated with their residue numbers.
B. Details of the CggR effector-binding sites with bound DHAP and FBP. Protein and ligands are coloured as in A.
C. Details of the structural rearrangements of residues Gly149 and Gln185.
D. C-CggR solvent-accessible surfaces in the U, L1 and L2 conformations. The surfaces are coloured according to electrostatic potential (blue positive, red negative, calculated with DS ViewerPro 6.0, Accelerys Software Inc.). The ligand molecule is represented as a stick model, and its solvent accessible surface is shown in transparent yellow.
Fig. 6.

Polar interactions in the C-CggR ligand-binding site. Polar interaction network of FBP (A), DHAP (B), G6P (C) and F6P (D) in the C-CggR ligand-binding site (interactions shown as green dashed lines). Parameters describing C-CggR—ligand interactions are summarized in Table S1. The amino acid residues forming the effector-binding site and contacting ligands are highly conserved in CggR homologues (Fig. S2). We therefore surmise that ligand specificities and interactions are conserved in homologous CggR-like repressors present in other Gram-positive bacteria.
Binding of DHAP induces a series of structural changes to C-CggR. The conformation of the ligand-binding site in the presence of DHAP is referred to as ‘L2’ (for liganded). Strikingly, when DHAP binds, the segment consisting of residues 174–184 closes over the ligand, acting as a flap (Fig. 5A and B). Residue Gln185 serves as a hinge for the flap closure with its main-chain atoms taking the position of the side-chain of this residue in the unliganded structure (Fig. 5C). This transition appears to be facilitated by a peptide flip in Gly149 that allows the neighbouring Gly150 to establish a main-chain hydrogen bond with Asn184. In the DHAP-bound state, the Gly149 carbonyl oxygen is pointing towards the ligand-binding site, whereas in the unliganded state, it is pointing away from it (Fig. 5C). Residues 180–182 undergo a disorder-to-order transition, and the flap is further stabilized by an ionic interaction between Glu269 and Arg250 that is in contact with the phosphoryl group of the ligand. Additional structural changes occur in the region composed of residues 203–211, which interacts with residues in the flap (Fig. 5A and B). This region also participates in crystal packing interactions.
Binding of FBP leads to additional interactions between C-CggR residues and the second phosphoryl moiety in this ligand. Specifically, Arg175 and Arg251 establish ionic interactions, and Gln185 establishes a hydrogen bond (Fig. 6B). These interactions act as a clamp that stabilizes the flap that was observed to be fully ordered in the FBP-bound state, although it adopts essentially the same conformation as in the unliganded state. In addition to the increased order, this conformation also exhibits the Gly149 peptide flip, as in the case for DHAP binding (Fig. 5C). The conformation of the ligand-binding site in the presence of FBP is referred to as ‘L1’. As mentioned above, the FBP co-crystal structure contains FBP bound to only one C-CggR molecule. The ligand-binding site in the second molecule is occupied by a DHAP molecule. FBP cannot displace this DHAP molecule, because the required flap conformation is not compatible with the packing in our crystal form. When FBP-bound C-CggR was superimposed onto DHAP-bound C-CggR in the FBP co-crystal structure, steric clashes resulted in residues 217–218 and 221–223 of one molecule and residues 179–181 and 206–210 of the second molecule in the asymmetric unit.
Binding of F6P and G6P leads C-CggR to adopt the same L2 conformation as in the presence of DHAP. The main-chain conformation of the effector-binding site is identical in all three cases. Also, both F6P and G6P establish interactions between their O1 oxygen atoms and the side chain of Glu180. Additional interactions between the sugar moieties and C-CggR differ somewhat for these two ligands, mainly because of the different sugar rings (Fig. 6C and D). It should be noted that, while the F6P and FBP sugars adopt the β-configuration in the C-CggR effector-binding site, in the case of G6P, the electron density is compatible with both the α- and β-configurations, indicating that G6P can bind in either conformation (the more prevalent β-configuration was included in the crystallographic model).
Ligand binding affinities
The affinities of ligands that bind to C-CggR in our crystal structures were investigated by isothermal titration calorimetry (ITC). In our initial ITC experiments with C-CggR protein samples also used for X-ray analysis, the stoichiometry of FBP binding to C-CggR was determined to be about 1:2 (Fig. S3). This result supports the notion of partial preloading of effector-binding sites with DHAP acquired during protein expression, a fact that prevents rigorous ITC analysis. In order to produce ligand-free protein, we purified C-CggR from E. coli grown in a sugar-free medium with succinate as the main carbon source. Protein obtained in this way resulted in a stoichiometry for binding of FBP to C-CggR of 1:1, and was thus deemed to be ligand-free (apo). This apo-form was subsequently used for the determination of binding parameters of other ligands (ITC profiles are shown in Figs S4—S9). The ITC experiments confirm the highest affinity of C-CggR for FBP (Kd = 2.9 μM, Table 1), but also show similarly high affinity for DHAP (Kd = 3.3 μM, Table 1). Other ligands bind to C-CggR with lower affinity, with Kd values increasing in sequence G6P → G3P → GAP → F6P (Table 1).
Table 1.
Parameters of ligand binding to C-CggR determined by ITC.
| Ligand | Kd (μM) | stoichiometry |
|---|---|---|
| FBP | 2.87 ± 0.06 | 1.05 ± 0.004 |
| DHAP | 3.34 ± 0.15 | 1.03 ± 0.007 |
| GAP | 63.29 ± 3.1 | 2.86 ± 0.05 |
| G3P | 27.59 ± 2.4 | 1.97 ± 0.02 |
| G6P | 10.98 ± 1.2 | 0.97 ± 0.03 |
| F6P | 94.34 ± 3.1 | 0.98 ± 0.02 |
The observation that DHAP binds more strongly than GAP and G3P supports our interpretation of the electron density in the effector-binding site in crystals containing serendipitously bound ligand (discussed above).
Chemical cross-linking in the presence of ligands
It was shown previously that CggR can form oligomers and that oligomerization is disfavoured by FBP binding (Zorrilla et al., 2007). Here, we also investigated the effect of other ligands used in our crystallographic studies on the oligomerization of both full-length CggR and its C-terminal domain using chemical cross-linking with glutaraldehyde (GTA). In order to ensure sufficient binding, ligands were added at a concentration of 50 mM, a concentration that results in a more than 500-fold excess of protein-ligand complex over protein even for the ligand with the lowest affinity (F6P).
Chemical cross-linking of full-length CggR or its C-terminal domain without added ligand captured the oligomerization state, revealing the presence of monomers, dimers and, to a smaller extent, trimers and tetramers (Fig. 7A). Our results therefore indicate that oligomerization of CggR is mediated, at least in part, through its C-terminal effector-binding domain.
Fig. 7.
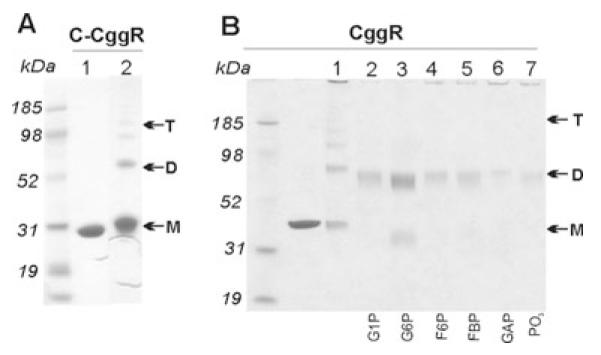
Glutaraldehyde (GTA) cross-linking SDS—PAGE analysis of C-CggR and full-length CggR cross-linked with 0.01% glutaraldehyde (GTA) in the presence of various ligands. Monomers, dimers and tetramers are indicated by M, D and T respectively.
A. In the absence of DNA: C-CggR was used at concentration of 1.6 μM and 1.4 μM respectively, and ligands were added at a concentration of 50 mM.
B. In the presence of DNA: full-length CggR was used at a concentration of 1.4 μM, ligands were used at a concentration of 50 mM, the DNA operator sequence was added at concentrations 0.45 μM in lane 3 and 1.4 μM in lanes 4–8 respectively.
When G1P, F6P, FBP, GAP/DHAP or phosphate was added to the cross-linking reaction, the CggR-oligomerization equilibrium shifted towards the dimer (Fig. 7B). By contrast, addition of G6P led to a shift towards the monomer. G6P, F6P, FBP and GAP/DHAP had the same effect on the C-terminal domain of CggR (data not shown), lending further support to the notion that the effector-binding domain mediates modulations of the CggR oligomeric state.
It was previously proposed that FBP is a conformational stabilizer of CggR, because it protected CggR from aggregation and proteolysis (Zorrilla et al., 2007). Our results show that similar stabilizing effects are also observed for other phosphorylated sugars.
Analysis of C-CggR crystal contacts
In an attempt to identify the interfaces mediating CggR oligomerization, we performed a detailed analysis of the intermolecular contacts observed in our C-CggR crystal structures. There are three major crystal-packing interfaces with areas over 500Å2, each (Table 2). Interface I comprises residues 183, 187 and 201–226 of one monomer and residues 179–188, 195, 201–224 and 252 of the second monomer and results in an almost symmetric dimer (Fig. 8). Strikingly, this interface involves protein regions that exhibit large structural changes upon ligand binding to the CggR effector-binding site, including the flap region. Minor structural differences in region 202–223 are also observed between the two molecules in the asymmetric unit when occupied by the same ligand (e.g. DHAP, G6P or F6P complex structures). Interface II involves the N-terminal region (residue 89–92) and residues 111–137, 159 and 330 of both monomers (Fig. 8). This interface also includes the three N-terminal residues representing the cloning artefact in the construct used in this study. In interface III, residues 96–107, 319, 337 and 338 of one monomer interact with residues 140–144, 202 and 225–233 of the second monomer (Fig. 8B). Both interfaces II and III result in asymmetric dimers.
Fig. 8.

C-CggR dimer interfaces observed in the crystal structures.
A. Assemblies formed through interfaces I and II. Transparent solvent-accessible surfaces are shown with underlying ribbon representations. Molecule A is coloured gray, and the two molecules interacting through interface I and II are coloured blue and yellow respectively. DHAP and FBP are shown as space-filling models.
B. Conservation of residues in C-CggR crystal interfaces. Residues conserved in at least 90% of the 58 closest homologues of CggR are coloured red. Residues of molecules A and B involved in interfaces are shown above and below the protein sequence respectively. Interfaces I, II and III are labelled 1, 2, 3 and coloured blue, yellow and green respectively.
The crystal interfaces and the stability of potential assemblies were evaluated using methods available through the Protein Interfaces, Surfaces and Assemblies (PISA) server (Krissinel and Henrick, 2005). The N-terminal residues representing the cloning artefact were omitted from the calculations (Table 2).
An assessment of the hydrophobicity of interface III shows suboptimal hydrophobic interactions, as reflected by a ΔiG P-values of 0.5 and higher. This interface is thus likely a crystal-packing artefact. By contrast, interfaces I and II are characterized by significantly larger hydrophobic contributions. It is therefore likely that these interfaces are involved in oligomer formation in solution. The results from our analysis did not allow us to deduce which of the two interfaces likely reflects the predominant dimeric interaction of C-CggR.
A potential solution can be obtained by examining residue conservation. The residues participating in interface I are more conserved throughout the SorC/DeoR family than those in interface II (Fig. 8B). Also, interface I involves residues in the vicinity of the effector-binding site. We conclude that interface I is likely responsible for dimer formation. The PISA analysis also suggests that interface stability varies depending on the ligands present in the effector-binding sites (Table 2). This result reflects the potential role of interface I in the modulation of CggR activity.
We cannot rule out an involvement of crystal interface II in CggR oligomerization in solution. Cooperative binding of two dimers on the DNA operator sequence was shown experimentally (Zorrilla et al., 2007). Because higher oligomers cannot be formed using interface I only, it is likely that two distinct interfaces are required for oligomer formation. It should be noted that interface II is near the N-terminus of C-CggR in close proximity to the DNA binding domain. Interface II, if engaged, might thus induce conformational changes in the DNA-binding domain. Further studies are required to investigate the structural basis of CggR oligomer formation.
DNA binding in the presence of ligands
We performed an electrophoretic mobility assay (EMSA) analysis to explore how the binding of C-CggR to its minimal 44 bp long DNA operator sequence is affected by the presence of the ligands used in our crystallographic studies (see Experimental procedures). In our experiment, the DNA duplex migrates with high mobility and forms a band at the bottom of the gel (lane 1 in Fig. 9A). Free CggR in the absence of DNA produces a smeared band towards the top of the gel (lane 10 in Fig. 9B). Upon addition of the DNA duplex, a CggR/DNA complex is formed. As a result, the mobility of the DNA is drastically reduced, and CggR is focused into a defined band (lanes 1–3 in Fig. 9A). There is only one band visible for the complex, which presumably represents a CggR tetramer bound to the DNA duplex. An excess of protein over DNA was used to ensure a maximal DNA shift. However, even at the high concentration of CggR, a weak band for the free DNA could still be observed (lane 3 in Fig. 9A), probably representing improperly paired DNA duplex incapable of binding to CggR.
Fig. 9.
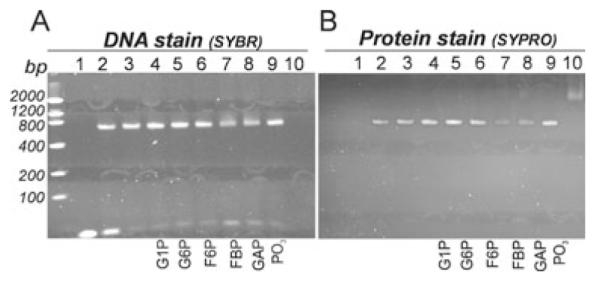
Electrophoretic mobility shift assay (EMSA) CggR/DNA/ligand-binding reaction analysed with 8% TBE polyacrylamide gel electrophoresis and stained with SYBR Green EMSA nucleic acid gel stain (A) and by the SYPRO Ruby EMSA protein gel stain (B). Lane 1: DNA operator (45 nM); lane 2: DNA operator (45 nM) + CggR (850 nM); lane 3: DNA operator (45 nM) + CggR (1.7 μM); lanes 4–9: DNA operator (45 nM) + CggR (1.7 μM) + ligand (1 mM); lane 10: CggR (1.7 μM).
In our assay, only FBP and GAP/DHAP had an effect on CggR-specific DNA binding. When FBP or GAP/DHAP were added to CggR and the DNA duplex, weaker and somewhat smeared bands for the DNA in the CggR/DNA complex as well as slightly more intense bands for free DNA were observed (lanes 7 and 8 in Fig. 9A). At the same time, significantly less intense bands for CggR in the CggR/DNA complex as well as faint smeared band for free CggR were observed (lanes 7 and 8 in Fig. 9B). We never observed full release of CggR from the DNA duplex, even for high concentrations of FBP or GAP/DHAP. It was recently shown that FBP destabilizes the CggR/DNA complex, but does not lead to irreversible dissociation (Zorrilla et al., 2007).
Discussion
Glycolysis, as one of the most general catabolic pathways, is subject to tight regulation depending on the presence of sugar as a carbon source. In B. subtilis, GapA transcription, as well as transcription of other enzymes of triose phosphate interconversion present on the gapA operon (Fig. 4), are repressed by the transcriptional regulator CggR (Doan and Aymerich, 2003). When glucose is available as an energy source CggR, dissociate from its operator sequence thereby abolishing transcriptional repression (Doan and Aymerich, 2003; Zorrilla et al., 2007).
CggR ligands
For CggR, we found that, in addition to the previously described effector molecule FBP (Doan and Aymerich, 2003; Zorrilla et al., 2007), several other intermediates of the glycolytic steps upstream of the GapA enzymatic step are able to bind to the same effector-binding site with a wide range of binding affinities (Kd from 3 to 94 μM, Table 1). These additional ligands feature a common phosphoryl group as well as a carbohydrate moiety.
Important insights into the characteristics of ligand binding to CggR can be gleaned from our crystallographic study. The ligand-binding site is largely preformed, as no significant structural adaptations are apparent upon ligand binding, except for the flap region (residues 174–184). Remarkably, the sugar portion of the ligands can either be glyceraldehyde, glycerol, dihydroxyacetone, D-fructose or D-glucose, and perhaps others. Although these sugars are structurally different, their polar groups establish similar interactions with surrounding residues. This type of promiscuity, and thus lack of specificity, usually occurs at the cost of binding energy. Conversely, strong binding usually results in pronounced specificity. Accordingly, specific sugar-binding proteins bind only one type of sugar. To accommodate different types, additional interactions are presumably necessary to guarantee efficient binding. In CggR, these additional interactions come from the phosphoryl group that is common to all ligands. This phosphoryl group binds into a pocked lined by residues Thr151, Thr152, Arg250 and Lys310, providing an anchor that appears to be strictly required. Binding of a phosphate ion into this pocket can also be observed crystallographically (data not shown). Accordingly, G1P, which does not have such a phosphoryl group, does not bind, although it has other features common to some of the ligands. The residues involved in phosphoryl binding are well conserved in CggR orthologues, indicating that these proteins might share a universal anion-binding pocket. Also, a phosphate ion is bound in this pocket in the homologous structure of the putative transcriptional regulator from E. faecalis (PDB code 2O0M).
Besides the interactions in monophosphorylated sugars, FBP engages in additional interactions with its second phosphoryl group. Based on the structural features for ligand binding to CggR, we surmise that other phosphorylated carbohydrates might be able to bind to the effector-binding site, for example, glucose-1,6-bisphosphate, which shares structural features with the ligands tested. Also binding of some triose phosphate intermediates downstream of GAP that possess a 3-phosphate group (Fig. 4), such as 1,3-bisphosphoglycerate and 3-phosphoglycerate, is possible and needs to be experimentally tested.
We have observed only one effector-binding site per C-CggR molecule. By contrast, binding isotherms based on fluorescence anisotropy experiments using a CggR/operator complex were interpreted as reflecting two distinct ligand-binding events (Zorrilla et al., 2007). Several possible rationalizations for these results exist. First, an additional binding event could occur in full-length CggR, but not when only the C-terminal domain is used. Second, our crystallization conditions might not be compatible with binding of a second ligand molecule. Third, the potential preloading of CggR binding sites with DHAP, serendipitously recruited during expression and retained throughout purification, may have affected the above-mentioned fluorescence anisotropy experiments and masked the true mechanism. It is evident that ligand binding to CggR is a complex phenomenon that requires a careful and thorough thermodynamic analysis to dissect its intricacies.
Ligand-bound states of C-CggR
Not all of the ligands observed to bind to CggR have been tested for their ability to affect CggR binding to its operator sequence and to repress transcription of gapA genes. Only FBP is known to have an effect on transcriptional regulation (Doan and Aymerich, 2003). This ligand is unique among the ones tested in that it induces significant structural changes near its distinctive second phosphoryl group attached to the C1 carbon of the fructose moiety. In the absence of any ligand, the region encompassing residues 174–184 (flap), assumes the extended, partially disordered U conformation. Binding of FBP uniquely induces a disorder-to-order transition in the flap region to adopt the L1 conformation. The U-to-L1 conversion does not result in large structural differences; free and FBP-bound C-CggR structures superimpose with an RMSD of 0.32 Å for all main-chain atoms, a value within the range observed for different crystal structures of identical proteins (Betts and Sternberg, 1999). However, in the L1 state, the flap region is significantly stabilized, as evidenced by the more pronounced electron density in our crystal structures and a reduced susceptibility to proteolytic cleavage in vitro (Zorrilla et al., 2007). In the presence of monophosphorylated carbohydrates (DHAP, G6P and F6P), the flap assumes the third, distinct L2 conformation. In this conformation, the absence of a second phosphoryl group allows the flap to close over the effector-binding site. This structural change is thus more pronounced than the U-to-L1 conversion.
The structurally related GlcN6P deaminase (Oliva et al., 1995; Rudino-Pinera et al., 2002) does not have a flap region equivalent to that in CggR. Instead, its active site is covered by a mobile lid provided by a different region (residues 163–182), for which, in turn, no equivalent structure is present in C-CggR (Fig. 2). The substrate of this enzyme (GlcN6P) is similar in structure to G6P, one of the ligands that are able to bind to CggR. Accordingly, the active site in GlcN6P deaminase shares much of its structural features with the effector-binding site in CggR. The acidic residue Asp72, which is crucial for the GlcN6P deaminase catalytic activity (Oliva et al., 1995), is in CggR replaced by Gln185.
CggR and GlcN6P deaminase thus represent an excellent example for proteins featuring a common fold that was adapted to carry out ligand-binding-dependent regulatory functions in one case and enzymatic activities in another. Other examples of structural similarity of transcriptional regulators with enzymes are provided by the GntR family regulator PhnF from E. coli and chorismate lyase UbiC (Gorelik et al., 2006), the negative transcriptional regulator of Aspergillus nidulans NmrA and UDP-galactose 4-epimerase (Holden et al., 2003) and yeast transcriptional regulators Gal80p and Gal3p and glucose-fructose oxidoreductase and galactosidase respectively (Bhat and Hopper, 1992; Thoden and Holden, 2003; Thoden et al., 2007).
Ligand-modulated transcriptional repression by CggR
The fact that C-CggR is able to bind different intermediates of the glycolytic steps preceding the step catalysed by the GapA enzyme raises the question whether these ligands play a physiological role. Some intermediates were previously tested for their ability to inhibit CggR binding to its DNA operator in vitro using DNase I footprinting (Doan and Aymerich, 2003) and recently. Only FBP was shown to have such an activity. By contrast, F6P, fructose-2,6-bisphosphate and GAP/DHAP did not show any effect. It may be necessary to revisit these studies, given our observations that CggR is capable of procuring DHAP from the expression host. A preloading of the effector-binding sites would likely mask the true effects of ligands.
Fructose-1,6-bisphosphate has dual effects on the CggR structure and regulatory function (Zorrilla et al., 2007). At millimolar concentrations, FBP destabilizes the CggR/DNA-operator complex. At micromolar concentrations, FBP also acts as a conformational stabilizer protecting CggR against extensive aggregation and proteolytic cleavage. Our cross-linking experiments revealed that CggR aggregation (self-association) is mediated through the C-terminal effector-binding domain and is modulated not only by FBP but also by other phosphorylated sugars (Fig. 8). These sugars likely exert stabilizing effects similar to those described for FBP.
On the other hand, the negative effect on specific DNA binding could only be observed for FBP and GAP/DHAP, but not for other tested glycolytic intermediates. FBP and GAP/DHAP do not lead to an irreversible dissociation of the CggR/DNA complex but only to a destabilization. The two sugars that have an effect on CggR function also have similarly high binding affinities, with Kd values of 2.9 μM and 3.3 μM for FBP and DHAP respectively. Also, the fact that DHAP was procured as a ligand from the E. coli cytoplasm and could not be displaced even by 90 mM FBP during crystallization suggests a physiological relevance of DHAP binding to CggR. Our crystal structures revealed some dimers where each monomer adopted a different state, e.g. one monomer was bound to DHAP, and the other monomer was free or bound to FBP. These observations raise the possibility that asymmetrically occupied dimers might be of physiological importance. This notion is particularly intriguing, given the fact that CggR activity depends on its oligomeric status and considering that differential ligand binding may have specific effects on CggR oligomer formation through at least two distinct interfaces.
Although the physiological role of ligands that bind CggR with lower affinity and have an effect on its oligomeric state but not on its DNA binding function remains unclear, the regulation of CggR appears to be more complex than previously assumed. If the different glycolytic metabolites have different effects on CggR stability and DNA binding activity, CggR might be able to capture a rather complex snapshot of the glycolytic conditions and perhaps the general metabolic state.
Experimental procedures
Protein expression and purification
The gene for the effector-binding domain of CggR (residues 89–340, referred to as C-CggR) was cloned into the pMCSG7 vector using ligase-independent cloning (Stols et al., 2002). The gene of full-length CggR was cloned into the pET151/D-TOPO vector using the Gateway Cloning Technology (Invitrogen). Both vectors included a sequence encoding an N-terminal His6 affinity tag followed by a sequence encoding the tobacco etch virus (TEV) protease recognition site.
Proteins were overexpressed in E. coli BL21 (DE3). For production of selenomethionyl-substituted C-CggR protein for SAD analysis, E. coli cells were grown in enriched M9 medium supplemented with 0.8% (v/v) glycerol and 20 μg ml−1 ampicillin in the presence of selenomethionine, conditions known to inhibit methionine biosynthesis (Doublie, 1997). For the production of native C-CggR and CggR proteins, Terrific Broth II medium (Q-Biogene) supplemented with 0.8% (v/v) glycerol and 20 μg ml−1 ampicillin was used. Cells were grown at 37°C, and protein expression was induced with 1 mM isopropyl-β-D-thiogalactopyranoside at an OD600 of about 1 for a total of 12 h at 20°C. Cells were harvested by centrifugation, resuspended in 5 vols of lysis buffer [50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM imidazole, 0.02% (v/v) β-mercaptoethanol and 5% (v/v) glycerol], and lysed by sonication after the addition of protease inhibitors (1 mM PMSF, 1 mM benzamide). The lysate was clarified by centrifugation and subjected to affinity chromatography using Ni2+-nitrilotriacetic acid (Ni-NTA). Superflow Resin (Quiagen) equilibrated with buffer A [50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM imidazole, 5% (v/v) glycerol]. The His6-tagged protein was eluted with 250 mM imidazole in buffer A.
The affinity tag was removed from C-CggR by incubation with recombinant His6-tagged TEV protease overnight at 25°C while dialysing against buffer A. The sample was subjected to a second Ni-NTA metal-affinity chromatography to remove the TEV protease and cleaved His6 tag, extensively dialysed against 20 mM Tris-HCl, pH 7.5, 125 mM NaCl, and concentrated to using Vivaspin Concentrators (ISC BioExpress). Proteins that were > 95% pure as judged by SDS-PAGE were flash-frozen in liquid nitrogen and stored at −80°C until further use.
Protein crystallization
Initial C-CggR crystals were obtained by the vapour diffusion method in sitting drop mode using Index (Hampton Research) and Wizard I and II (Emerald Biostructures) crystallization screens carried out with a HoneyBee crystallization workstation (Cartesian Technologies). Crystals appeared after several days in various conditions containing PEG 3350 as the precipitant. Further optimization was done manually and involved changing to the hanging drop mode. Optimal crystals for C-CggR selenomethionyl-substituted protein were obtained by mixing 1 μl of protein (25 mg ml−1 in 10 mM Tris-HCl, pH 7.5, 125 mM NaCl) with 1 μl of reservoir solution [16% (w/v) PEG 3350, 0.1 M MES, pH 6.5, 0.1 M MgCl2, 0.1 M 6-aminohexanoic acid] and equilibration at 20°C over 1 ml reservoir solution. Crystals for C-CggR complexes were obtained in a similar way using native protein supplemented with 90 mM FBP, 10 mM GAP, 10 mM G6P or 10 mM F6P. The reservoir solutions were: 0.2 M ammonium formate pH 6.6, 20% (w/v) PEG 3350 for the C-CggR/DHAP complex, 0.2 M sodium fluoride, 20% (w/v) PEG 3350 for the C-CggR/FBP complex, 0.2 M sodium formate, 20% (w/v) PEG 3350 for the C-CggR/G6P complex and 0.2 M sodium thiocyanate, 20% (w/v) PEG 3350 for the C-CggR/F6P complex. Generally, rod-shaped crystals appeared within a day, and grew to a final size of 0.2 × 0.1 × 0.1 mm3 within 1 week. For cryoprotection, the crystals were transferred into reservoir solution supplemented with 10% (v/v) ethylene glycol, flash-cooled by plunging into liquid nitrogen and stored in liquid nitrogen until used for X-ray diffraction experiments.
Data collection
Diffraction data were collected at 100°K at beamlines 19-ID and 19-BM of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory, Argonne, IL, USA as well as on an in-house FR-E rotating-anode X-ray source equipped with an R-AXIS IV image-plate detector (Rigaku). All diffraction data were processed using the HKL-3000 suite of programs (Minor et al., 2006). Crystals exhibited the symmetry of space group P212121 and contained two molecules in the asymmetric unit with a solvent content of ~40%. Crystal parameters and data collection statistics are given in Table 3.
Structure determination, refinement and analysis
The C-CggR crystal structure was determined by single-anomalous dispersion (SAD) utilizing the anomalous signal from selenium atoms. All procedures for phasing and phase improvement by density modification were carried out using the HKL-3000 software package (Schneider and Sheldrick, 2002; Terwilliger, 2003; Minor et al., 2006). Phasing statistics are given in Table 4. The initial model, containing 95% of all residues, was constructed automatically by ARP/wARP (Perrakis et al., 1999) using electron density maps after density modification with the program DM (Emsley and Cowtan, 2004). Model refinement was carried out using the program REFMAC 5.2 (Murshudov et al., 1997) from the CCP4 package (CCP4, 1994), interspersed with manual adjustments using Coot (Emsley and Cowtan, 2004). The final steps included TLS (translation, libration, screw) refinement (Winn et al., 2001).
Structures of C-CggR complexes with ligands were determined by the difference-Fourier method or, in cases where crystals were not isomorphous, by molecular replacement using the program Molrep (Vagin and Teplyakov, 2000) with the structure of the C-CggR monomer as the search model. In all cases, coordinates for the ligands were added after complete refinement of the C-CggR and solvent model. The quality of the final models was validated with Molprobity (Lovell et al., 2003). Final refinement statistics for all structures is given in Table 4. All figures showing structural representations were prepared with the program PyMOL (DeLano, 2002). The following services were used to analyse the structures: PISA server (Krissinel and Henrick, 2005), Protein—protein interaction server (Jones and Thornton, 1996) and Consurf (Landau et al., 2005).
Atomic coordinates and experimental structure factors have been deposited with the Protein Data Bank with the codes 2OKG, 3BXF, 3BXE, 3BXG and 3BXH (Table 4).
Isothermal titration calorimetry
Thermodynamic analysis by ITC was performed using a VP-ITC microcalorimeter (Microcal) at 25°C in ITC buffer (20 mM Tris/HCl, pH 8.0, 150 mM NaCl, 0.02% (v/v) β-mercaptoethanol). C-CggR at a concentration of 30 μM was titrated with ligand in ITC buffer at concentrations of 11–14 times that of protein. At each injection (a total of 35), 8.5 μl of ligand was added to the sample cell. Each experiment was accompanied by a corresponding control experiment, in which ligand was injected into buffer alone. Protein concentration was determined by amino acid analysis. Association constants and thermodynamic parameters were estimated using the program Origin (version 7.0, Microcal). The data reported are the average of two measurements.
Chemical cross-linking
Glurataldehyde cross-linking reactions were carried out in 10 mM sodium phosphate, pH 7.5, 100 mM NaCl. Protein (1.6 and 1.4 μM for C-CggR and full-length CggR respectively) was mixed with 50 mM ligand and 0.1% GTA. Ligands were dissolved in the reaction buffer, and the pH was adjusted to 7.5. A large excess of ligand over CggR was used to ensure maximum binding. The cross-linking reaction was allowed to proceed for 3 h on ice before quenching by the addition of 200 mM lysine. Reactions were then mixed with 2 × SDS sample buffer (with final concentrations for SDS and DTT of 2.5% and 0.25 M, respectively), and the samples were boiled for 2 min before being subjected to SDS—PAGE. Protein bands were visualized through Colloidal Coomassie G-250 staining (Invitrogen).
Electrophoretic mobility shift assay
A non-radioactive EMSA (Invitrogen) was used. This assay uses SYBR Green EMSA nucleic acid gel stain for DNA detection and SYPRO Ruby EMSA protein gel stain for protein detection. A DNA duplex containing 44 bp, representing the minimal DNA regulatory sequence of CggR (Doan and Aymerich, 2003), was used: CGGGACGTTTTTTGTCAT AGCGGGACA TATAATGTCCAGC.
In the binding reaction, 45 nM DNA was mixed with 0.85–1.7 μM CggR protein in buffer (10 mM sodium phosphate, pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT). Ligands at concentrations of 10 mM were added, and the binding reaction was let to proceed for 20 min. The reaction mixture was then analysed on 8% TBE polyacrylamide gels. Gel staining was carried out according to the manufacturer’s instructions. Imaging of was carried out using a 300 nm transilluminator.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health Grant GM074942 and in part by projects Nos. AV0Z50520514 and AV0Z40550506 awarded by the Academy of Sciences of the Czech Republic and Grants 1M0508 and ME08016 from the Ministry of Education of the Czech Republic. The authors wish to thank Min Zhou and other members of the Structural Biology Center at Argonne National Laboratory for their help with protein purification and conducting data collection at 19-ID and 19-BM beamlines, and Dominika Borek for her help with diffraction data collection. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Footnotes
Supplementary material This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1365-2958.2008.06318.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing is not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Betts MJ, Sternberg MJ. An analysis of conformational changes on protein—protein association: implications for predictive docking. Protein Eng. 1999;12:271–283. doi: 10.1093/protein/12.4.271. [DOI] [PubMed] [Google Scholar]
- Bhat PJ, Hopper JE. Overproduction of the GAL1 or GAL3 protein causes galactose-independent activation of the GAL4 protein: evidence for a new model of induction for the yeast GAL/MEL regulon. Mol Cell Biol. 1992;12:2701–2707. doi: 10.1128/mcb.12.6.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA, USA: 2002. http://www.pymol.org [Google Scholar]
- Deutscher J, Kuster E, Bergstedt U, Charrier V, Hillen W. Protein kinase-dependent HPr/CcpA interaction links glycolytic activity to carbon catabolite repression in gram-positive bacteria. Mol Microbiol. 1995;15:1049–1053. doi: 10.1111/j.1365-2958.1995.tb02280.x. [DOI] [PubMed] [Google Scholar]
- Doan T, Aymerich S. Regulation of the central glycolytic genes in Bacillus subtilis: binding of the repressor CggR to its single DNA target sequence is modulated by fructose-1,6-bisphosphate. Mol Microbiol. 2003;47:1709–1721. doi: 10.1046/j.1365-2958.2003.03404.x. [DOI] [PubMed] [Google Scholar]
- Doan T, Martin L, Zorrilla S, Chaix D, Aymerich S, Labesse G, Declerck N. A phospho-sugar binding domain homologous to NagB enzymes regulates the activity of the Central glycolytic genes Repressor. Proteins. 2008;71:2038–2050. doi: 10.1002/prot.21883. [DOI] [PubMed] [Google Scholar]
- Doublie S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gorelik M, Lunin VV, Skarina T, Savchenko A. Structural characterization of GntR/HutC family signaling domain. Protein Sci. 2006;15:1506–1511. doi: 10.1110/ps.062146906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkin TM. The role of CcpA transcriptional regulator in carbon metabolism in Bacillus subtilis. FEMS Microbiol Lett. 1996;135:9–15. doi: 10.1111/j.1574-6968.1996.tb07959.x. [DOI] [PubMed] [Google Scholar]
- Holden HM, Rayment I, Thoden JB. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem. 2003;278:43885–43888. doi: 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Searching protein structure databases has come of age. Proteins. 1994;19:165–173. doi: 10.1002/prot.340190302. [DOI] [PubMed] [Google Scholar]
- Jones S, Thornton JM. Principles of protein—protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Detection of protein assemblies in crystals. In: Istrail S, Pevzner P, Waterman M, editors. CompLife 2005, LNBI 3695. Springer-Verlag; Berlin, Heidelberg: 2005. pp. 163–174. [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Ludwig H, Homuth G, Schmalisch M, Dyka FM, Hecker M, Stulke J. Transcription of glycolytic genes and operons in Bacillus subtilis: evidence for the presence of multiple levels of control of the gapA operon. Mol Microbiol. 2001;41:409–422. doi: 10.1046/j.1365-2958.2001.02523.x. [DOI] [PubMed] [Google Scholar]
- Ludwig H, Rebhan N, Blencke HM, Merzbacher M, Stulke J. Control of the glycolytic gapA operon by the Catabolite control protein A in Bacillus subtilis: a novel mechanism of CcpA-mediated regulation. Mol Microbiol. 2002;45:543–553. doi: 10.1046/j.1365-2958.2002.03034.x. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution — from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Oliva G, Fontes MR, Garratt RC, Altamirano MM, Calcagno ML, Horjales E. Structure and catalytic mechanism of glucosamine 6-phosphate deaminase from Escherichia coli at 2.1 A resolution. Structure. 1995;3:1323–1332. doi: 10.1016/s0969-2126(01)00270-2. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Rudino-Pinera E, Morales-Arrieta S, Rojas-Trejo SP, Horjales E. Structural flexibility, an essential component of the allosteric activation in Escherichia coli glucosamine-6-phosphate deaminase. Acta Crystallogr D Biol Crystallogr. 2002;58:10–20. doi: 10.1107/s0907444901016699. [DOI] [PubMed] [Google Scholar]
- Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- Stulke J, Hillen W. Regulation of carbon catabolism in Bacillus species. Annu Rev Microbiol. 2000;54:849–880. doi: 10.1146/annurev.micro.54.1.849. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Improving macromolecular atomic models at moderate resolution by automated iterative model building, statistical density modification and refinement. Acta Crystallogr D Biol Crystallogr. 2003;59:1174–1182. doi: 10.1107/S0907444903009922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden JB, Holden HM. Molecular structure of galactokinase. J Biol Chem. 2003;278:33305–33311. doi: 10.1074/jbc.M304789200. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Sellick CA, Reece RJ, Holden HM. Understanding a transcriptional paradigm at the molecular level. The structure of yeast Gal80p. J Biol Chem. 2007;282:1534–1538. doi: 10.1074/jbc.C600285200. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. An approach to multicopy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Zorrilla S, Doan T, Alfonso C, Margeat E, Ortega A, Rivas G, et al. Inducer-modulated cooperative binding of the tetrameric CggR repressor to operator DNA. Biophys J. 2007;92:3215–3227. doi: 10.1529/biophysj.106.095109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.