

Abstract
Aims
The presence of muscarinic receptors on bladder urothelial cells (BUC), suggests BUC may be a target for antimuscarinics. This study determined whether human BUC are responsive to a muscarinic agonist and if so, whether responses are altered in interstitial cystitis (IC) BUC.
Methods
Primary urothelial cell cultures were established from cystoscopic biopsies. Normal (NB) and IC BUC were studied using calcium imaging techniques as a means to monitor the response to muscarinic receptor activation with the agonist, carbachol (CCh). Changes in intracellular Ca2+ concentration ([Ca2+]i) were measured with fura-2 ratiometric microfluorimetry. Dose-response curves (CCh dose versus [Ca2+]i) were measured in IC and NB BUC. Tolterodine was used to confirm the specificity (muscarinic versus nicotinic) of CCh evoked increases in [Ca2+]i.
Results
CCh induced a dose-dependent increase in [Ca2+]i. Potency and efficacy of CCh was significantly greater in IC BUC. The maximal increase in [Ca2+]i was 136.3 ± 5.1% over baseline in 78 cells from 4 IC patients versus 92.4 ± 4.8% over baseline in 67 cells from 4 NB subjects (p<0.01). The EC50 of the evoked increase was 1.10 ± 0.14 μM vs 3.36 ± 0.72 μM (p<0.01) in BUC from IC and NB controls, respectively. Removal of extracellular calcium or application of tolterodine, abolished CCh evoked increase in [Ca2+]i in IC and NB BUC.
Conclusions
The greater sensitivity of IC BUC to CCh suggests that IC patholobiology may also include alterations muscarinic signaling. The physiologic sequelae of muscarinic activation in BUC need to be further investigated.
INTRODUCTION
There are currently two recognized bladder hypersensory syndromes – overactive bladder (OAB) and interstitial cystitis (IC). Both syndromes overlap in symptomatology, urinary urgency, frequency, and nocturia, except that IC is strongly associated with severe chronic idiopathic bladder pain while OAB is not. While the use of antimuscarinics have not been validated in a randomized placebo-controlled fashion for treatment of IC-symptoms, oral muscarinic receptor antagonists are the most common treatment for lower urinary tract symptoms. The presumed mode of action of these compounds is to block detrusor smooth muscle muscarinic receptors. However, these antagonists may exert their effects at other sites besides detrusor smooth muscle. .
The function of the bladder urothelium has traditionally been ascribed to that of a barrier, preventing urinary substances from accessing the underlying stromal compartment. Emerging evidence, however, suggests that the urothelium and suburothelium actively participates in the sensory signaling of the bladder. Bladder urothelial cells (BUC) have been found to express a range of receptors and release neurotransmitters in response to distension and receptor activation. Importantly, muscarinic receptors are present on BUC (2) and have been shown to mediate the release of adenosine triphosphate (ATP) and nitric oxide, which in turn may activate afferent nerves or feedback on the BUC themselves (3,4). That these receptors may normally contribute to the reflex arc underlying micturition is suggested by the present of dense parasympathetic innervation of the bladder with evidence of acetycholine containing nerves terminating near the base of the urothelium. Together, these observations raise the possibility that muscarinic receptors on BUC may contribute to the therapeutic efficacy of muscarinic antagonists in the treatment lower urinary tract symptoms. Consistent with this hypothesis, there is evidence that muscarinic receptor activation increases ATP release and intracellular calcium [Ca2+]i in rat BUC (5). However, the sensitivity of human BUC to muscarinic receptor activation remains to be determined. The present study was therefore designed to determine whether human BUC respond to muscarinic receptor activation and if so, whether this response is altered in BUC from patients with IC. Our results indicate that human BUC are not only responsive to muscarinic receptor activation, but that both the potency and efficacy of the muscarinic receptor agonist carbachol (CCh) is increased in BUC from patients with IC.
MATERIAL AND METHODS
All chemicals and buffers were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Bladder biopsies and urothelial cell culture
This study was approved by our Institutional Review Board. IC was defined according to the National Institute of Diabetes and Digestive and Kidney (NIDDK) Diseases criteria (6). Normal bladder (NB) controls were defined as those without bladder voiding symptoms by American Urological Association (AUA) symptoms questionnaire (score less than 7 out of 35), who had no bladder pain and who were to undergo other pelvic surgery.
All study patients were counseled and gave informed consent for this research study. Bladder biopsies were obtained using the cold-cup biopsy technique from patients with IC (n = 4, 2 females, 2 males mean age 42.5), and controls (n = 4, 2 female and 2 males, mean age 49.3) while the patients were under general or regional anesthesia. These specimens were transported in sterile saline to the laboratory within 30 minutes. They were then minced manually into 0.5 mm pieces. The samples were placed in uncoated plastic tissue culture plates (Falcon, Franklin Lakes, NJ) containing complete cell medium (MEM-COMP) composed of MEM plus L glutamine (Mediatech, Herndon, VA) supplemented with 1 unit per mL insulin, 10% heat inactivated FBS, 1.25μg/ml amphotericin B, 100 units per mL penicillin, and 100μg/ml streptomycin (Gibco BRL, Grand Island, NY). Samples were anchored to the bottom of the well with sterile cover glass slips, and incubated in 95% air/5% CO2 at 37 °C. The medium was changed twice weekly. Once cell growth could be seen and confluent monolayers were present, cells were harvested using 0.25% trypsin (Gibco BRL). After dissociation, the cell suspension was centrifuged at 2,000 g and resuspended in MEM-COMP. Cell counts were obtained and the suspension was titrated with MEM-COMP to give a cell count of 400 cells/20μl. 20μl of suspension was then pipetted onto individual 5mm round glass cover slips (Fisher, Fairlawn, NJ) arranged in a sterile Petri dish (Falcon) and incubated in 95% air/5% CO2 at 37°C overnight. This technique has been described and the cells derived from this technique have been shown to be epithelial in origin (7). Furthermore, routine quality control analyses of these cultured cells showed expression of cytokeratin (AE1/AE3) and lack of expression of α-smooth muscle actin.
For the experiments utilizing fibroblasts, the NIH/3T3 embryonic mouse fibroblast cell line (ATCC, Manassas, VA) was grown until confluent using standard ATCC protocol and plated onto round glass cover slips as described for intracellular calcium measurement.
Intracellular calcium measurement in BUC
Recordings of the intracellular free calcium concentration were performed in BUC within 20 to 36 hours after plating onto individual 5 mm cover slips. Individual cover slips were placed in a Petri dish and were loaded with 2.5 μM Fura-2-AM for 30 minutes. Cover slips were then transferred to the recording chamber mounted on an inverted microscope (Nikon, Tokyo Japan) and continuously perfused via gravity (1–3 ml/min) with a solution consisting of (in mM) 145 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES at pH 7.4 adjusted with Tris Base. Osmolality was adjusted with sucrose to 325 mOsm. Background corrected fluorescent images were acquired at 1 Hz with a slow scan CCD camera system (Princeton Instruments) through a 20x Nikon Fluor objective. Fura-2 was excited at 340 and 380 nm wavelengths (λ) using a high speed Lambda 10-2 filter wheel (Sutter Instruments, Novato, CA). Emitted light (510nm) was acquired with equal exposure time (200 ms) for the excitation wavelengths of 340 and 380 nm.
After the subtraction of background fluorescence, the ratio of fluorescence emission for 340/380 nm illumination was calculated by F340/F380 = R. Baseline Fura-2 ratios (Rbaseline) were characterized by the average baseline of the 30–45s prior to the application of test compounds. Test compounds were applied for 12 seconds and the change in R was assessed. The peak increase in Fura-2 ratios (Rpeak) was used to determine a percent change from baseline by the formula ((Rpeak −Rbaseline)/Rbaseline × 100). Data were analyzed and presented as a percent change in Fura-2 fluorescence ratio because this ratio is directly proportional to [Ca2+]i for concentrations of Ca2+ roughly an order of magnitude above and below the fura-2 Kd for Ca2+ (~250 nM). Based on a series of pilot studies with digitonin in the presence of normal bath solution (i.e., 2.5 mM Ca2+) and nominally Ca2+ free bath solution, resting and evoked Ca2+ transients within the BUC is neither close to the saturating concentration for Fura-2, nor below the threshold for which increase in [Ca2+]i may be detected (~5 nM in our system).
For application of chemicals, a fast 12 channel pressurized superfusion system with a common outlet manifold (ALA Scientific, Westbury, NY) was used. Pinch valves were controlled from a switch board operated by a windows application with external TTL triggering from the data collection and analysis software Metafluor (Molecular Devices, Sunnyvale, CA).
Over 80% of the BUC (both IC and NB) responded to CCh. However, because of the heterogeneity inherent in the culture system, we only analyzed data from cells that had >20% increase over baseline in fluorescence in response to any dose of CCh. This cutoff is based on a previous publication detailing similar responses measured in retinal non-pigmented epithelial (NPE) cells (8). In the BUC, the baseline values of [Ca2+]i was low, did not fluctuate, and was not significantly different between IC and NB BUC.
Tolterodine was obtained from Pfizer, Inc. The dose of tolterodine used was 70 nM based on the EC50 of tolterodine for muscarinic receptors. Tolterodine has equivalent antagonism to m1 through m5 receptor subtypes and no known activity at nicotinic receptors.
Statistical Analyses
The data are presented as the mean ± SEM. Data were analyzed by paired Student’s t tests and differences were considered significant at p< 0.05. Dose response curves were fitted with a modified Hill equation of the form EC50=(Rmax* xn)/(xn+Dn) where Rmax is the maximum effect (efficacy), and EC50 (potency) is the concentration of carbachol producing an effect that is 50% of the maximum response, n is the Hill coefficient and D is the concentration of carbachol. Curve fitting was performed with Sigmaplot (version 9.0, Systat, San Jose, CA). The sample sizes used in the statistical calculations were the numbers of subjects from which the experiments were performed, not the number of cells measured.
RESULTS
Carbachol induced dose-dependent increases in intracellular calcium in IC and NB BUC
Increasing concentrations of CCh (0.01μM – 1 mM) were applied to human BUC in order to determine the efficacy and potency of this muscarinic receptor agonist on these cells in isolation. The highest dose of CCh tested (1 mM) increased the concentration of intracellular Ca2+ ([Ca2+]i) in 94% of cells tested (Fig. 1). The response to CCh was concentration dependent. Increasing concentrations of CCh produced larger increases in [Ca2+]i (Fig. 2A, 2B). Dose response data from each cell was fitted with a modified Hill equation in order to determine the EC50 (potency) and maximal effect (efficacy). Pooled data fitted with a modified Hill equation revealed statistically significant differences between groups with respect to both efficacy and potency: these parameters were statistically significantly greater in IC compared to NB BUC (p < 0.05) (Table 1).
FIGURE 1. CCh evoked increases in [Ca2+]i.
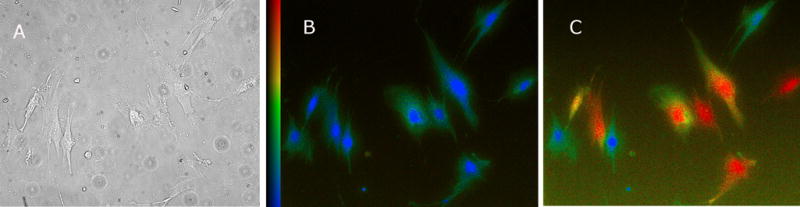
A: Brightfield image was taken prior to the application of CCh (single field of IC BUC).
B: Prior to the application of CCh, fura-2 fluorescence ratio was low (blue) (Scale bar on left is Fura-2 ratio (340/380)).
C: Twelve seconds after the application of CCh (100 μM), a significant increase in fura-2 fluorescence ratio is clearly seen (red).
FIGURE 2. Changes in [Ca2+]i in response to varying concentration of CCh (dose-response curves).
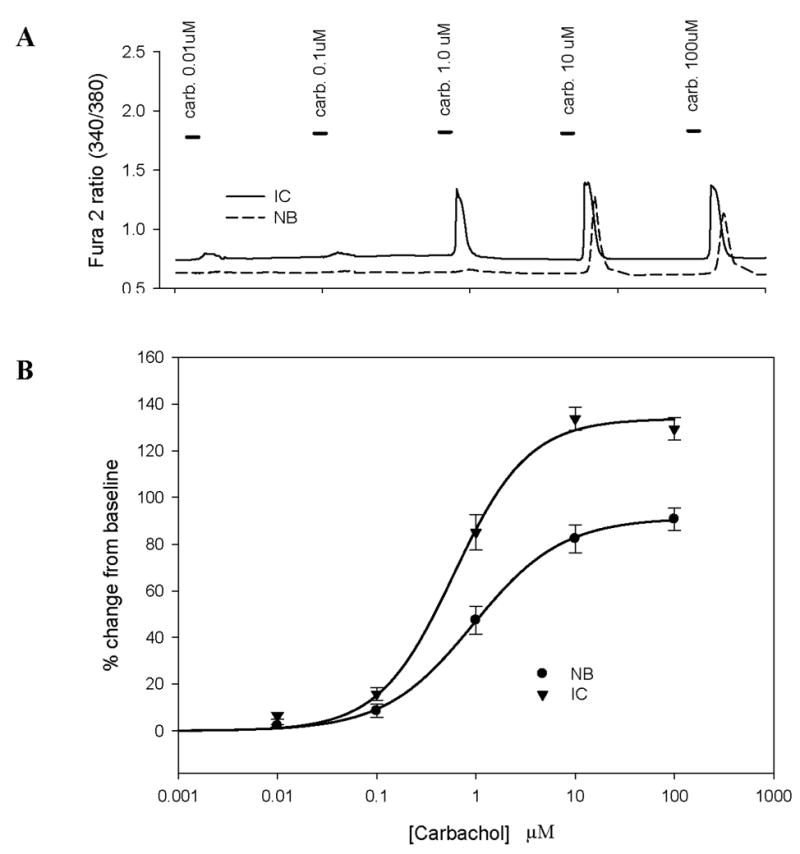
A: Representative raw ratiometric data from human interstitial cystitis (IC) and normal bladder (NB) bladder urothelial cells (BUC) used to generate dose response curves. Bars in the tracing represent the time duration of the CCh (carb) exposure.
B: Data from IC and NB BUC were analyzed as a percent (%) change from baseline and pooled. BUC were obtained from n=4 IC (78 cells total measured) and n=4 NB (n=67 cells total measured) subjects. Each BUC was exposed to exponential doses of CCh and the response fitted with a modified Hill equation to obtain maximum effect (efficacy) and EC50 (potency). There was significant increase in both efficacy and EC50 of CCh in IC compared to normal BUC (See Table 1).
TABLE 1. Efficacy and Potency of IC and normal BUC.
Efficacy and Potency of CCh on increasing [Ca2+]i in IC and NB BUC
| # of cells measured | # of pts | Efficacy (% rise [Ca2+]i) | Potency (μM) | |
|---|---|---|---|---|
| IC | 78 | 4 | 136.3 ± 5.1* | 1.10 ± 0.14* |
| NB | 67 | 4 | 92.4 ± 4.8 | 3.36 ± 0.72 |
p<0.05 comparing IC vs NB
Tolterodine blocked CCh induced increases of [Ca2+]i
CCh (1 mM) evoked increase in [Ca2+]i were blocked by the muscarinic antagonist tolterodine (70 nM) in a reversible manner in IC (Fig. 3A) and NB BUC cells (Fig. 3B). Pooled data indicate that there was no difference between IC BUC (10 cells) and NB BUC (10 cells) with respect to either the magnitude of tolterodine induced block or the degree of recovery from block (compare Figs. 3A and 3B). Tolterodine alone, had no detectable influence on [Ca2+]i (data not shown)
FIGURE 3. Effect of tolterodine on maximal CCh evoked increases in [Ca2+]i.
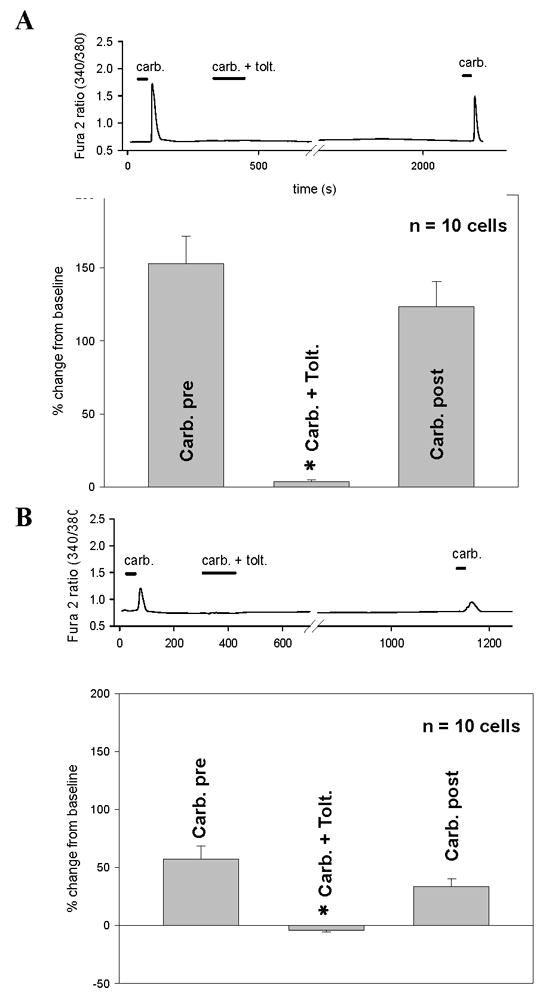
A: Tracing depicts CCh (carb, 1 mM) evoked increase in [Ca2+]i in IC BUC which then was reversibly blocked by tolterodine (carb. + tolt). This effect was reversible with a subsequent exposure to 1 mM CCh represented by the last peak in the tracing. Bars in tracing represent duration of exposure. The graph depicts pooled data from n=10 cells showing that tolterodine completely abrogated CCh evoked responses (p<0.01) and that this effect was completely reversible (Carb. post).
B: Same as panel A, except performed in NB BUC. Tolterodine had similar effects on NB BUC and these effects were reversible.
Absence of extracellular Ca2+ ([Ca2+]o) eliminates CCh evoked increases in [Ca2+]i
CCh evoked increases in [Ca2+]i in both IC and NB BUC were eliminated in Ca2+ free extracellular solution (Fig. 4A and 4B). CCh (1000μM) was applied NB BUC three times. The first application was in the presence of 2.5mM bath calcium ([Ca2+]o). The second time was in the presence of 0 mm [Ca2+]o with 1mM EGTA present. The third time was after restoration of the conditions used for the first application (i.e., 2.5mM [Ca2+]o with no EGTA). The CCh evoked increase in [Ca2+]i was completely eliminated in the absence of extracellular Ca2+. Pooled data indicate that there was no difference between BUC from IC (5 cells) and NB controls (5 cells) with respect to either the dependence of CCh evoked response on Ca2+ influx or the degree of recovery after restoring extracellular Ca2+ levels (compare Figs. 4A and 4B).
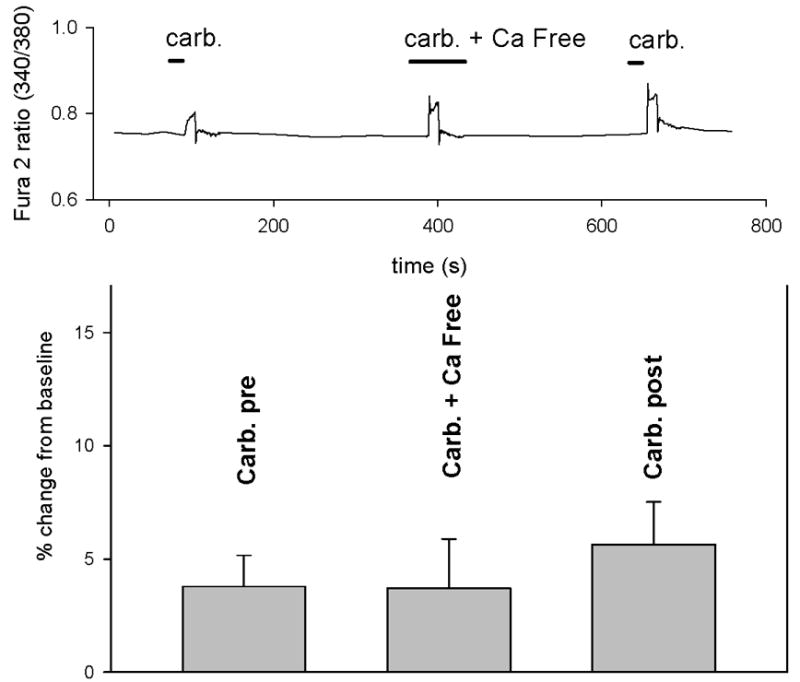
FIGURE 4. Effect of removal of extracellular calcium on CCh-evoked changes in [Ca2+]i.
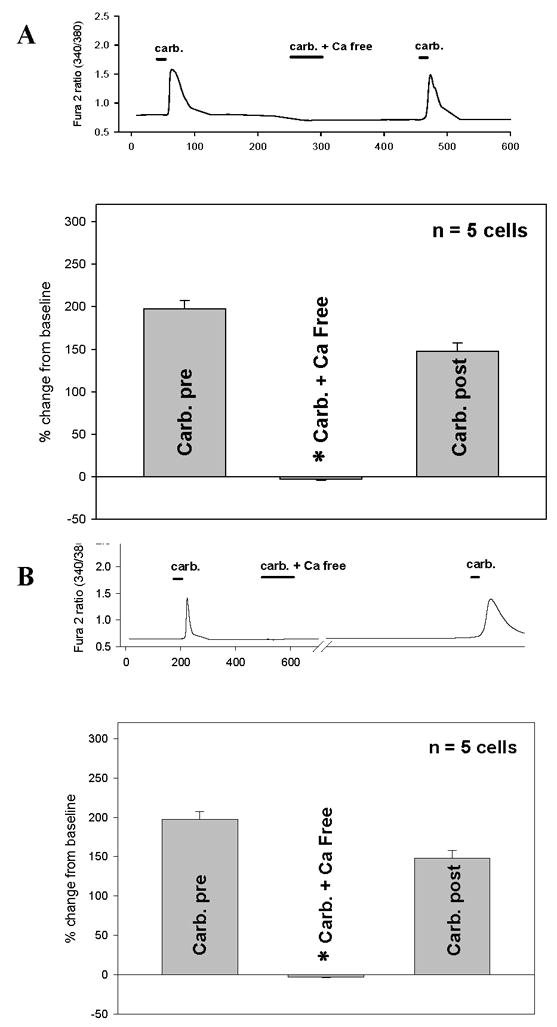
A: Tracing depicts CCh (carb, 1 mM) evoked increase in [Ca2+]i in IC BUC which then was reversibly blocked by removal of extracellular calcium (carb. + Ca free). This effect was reversible with a subsequent exposure to 1 mM CCh represented by the last peak in the tracing. Bars in the tracing represent duration of exposure. The graph depicts pooled data from n=5 cells showing that removal of extracellular calcium (carb. + Ca free) completely abrogated CCh evoked responses (p<0.01) and that this effect was completely reversible (Carb. post).
B: Same as panel A, except performed in NB BUC. Removal and restitution of extracellular calcium had identical effects on NB BUC.
CCh has minimal effect on [Ca2+]i in NIH/3T3 fibroblasts
As an additional control to assure that the BUC cultures were not differentiated into fibroblasts, responses of NIH/3T3 fibroblast cells to CCh were measured. These cells were exposed to CCh (1 mM) before and after substituting normal bath solution (2.5mM Ca2+) with Ca2+ free bath solution (Fig 5). Pooled data from NIH/3T3 cells (n = 5) demonstrated minimal CCh effect on [Ca2+]i and that this “response” was unaffected with the removal of extracellular Ca2+ (p > 0.05).
FIGURE 5. Effect of CCh on [Ca2+]i in NIH/3T3 fibroblasts.
Tracing shows typical response of NIH/3T3 cell to CCh (carb., 1 mM). There was minimal response to CCh with and without extracellular calcium. Note that the y-axis on the tracing is of a smaller scale. Graph depicts pooled data from n=5 NIH/3T3 cells. Fibroblasts responded minimally to carbachol. Removal of extracellular calcium had minimal effect.
DISCUSSION
Our study of BUC in vitro yielded two important observations. First, BUC are responsive to CCh, demonstrating dose-dependent increases in [Ca2+]i. Second, differences between BUC from IC and NB persist in vitro and are manifest as an increase in the potency and efficacy of CCh evoked increases in [Ca2+]i.
The sensitivity of human BUC to CCh are similar to what has been previously shown in primary cultured rat bladder urothelial cells (5). In this previous study, only muscarinic agonists (oxotremorine and muscarine) were employed, so the response to nicotinic receptor activation was not determined. CCh was used in the present study because it is the prototypical muscarinic receptor agonist. However, because this agonist is not as selective as oxotremorine and muscarine, with minimal efficacy at nitotinic receptors, it is possible that at least some of the CCh evoked responses were due to nicotinic receptor activation, particularly since these receptors have been found in human urothelium (9). However, the fact that tolterodine, a muscarinic antagonist with no known activity at nicotinic receptors, completely blocked CCh-evoked rise in [Ca2+]i, it is likely that CCh mediated its actions through muscarinic, not nicotinic, receptors.
The muscarinic receptor subtype(s) underlying the actions of CCh on human BUC remain to be determined. Five genes, m1-m5 encoding muscarinic receptors have been identified. In rat BUC, it was found that m1, m2 and m3 all contributed to the rise in [Ca2+]i, in response to oxotremorine (5). mRNA for all 5 receptor subtypes are detectable in human BUC with m2 expressed at highest levels and m4 and m1 present at lowest levels. Immunohistochemical data suggests protein for all 5 subunits are present in BUC as well, although the receptor subtypes appear to be differentially distributed (10). Despite evidence from rat BUC to the contrary, only m1, m3 and m5 receptor subtypes are typically thought to be coupled to a G-protein, Gq, generally associated with increases in [Ca2+]i (11). Nevertheless, indirect mechanisms have been described whereby both Gs and Gi (12, 13) coupled receptors are able to drive an increase in [Ca2+]i.
The observation that CCh evoked increases in [Ca2+]i were eliminated in the absence of extracellular Ca2+ suggests that evoked Ca2+ transients were due to Ca2+ influx rather than release from intracellular stores. This was unexpected given evidence from other systems that muscarinic receptor mediated increase in [Ca2+]i reflect the release of Ca2+ from internal stores following activation of phospholypase C (PLC) and the subsequent release of inositol trisphosphate (IP3). There are at least two potential explanations for this observation. One is that muscarinic receptors are not coupled to a PLC dependent pathway in BUC. If this was the case, Ca2+ influx would occur secondary to the activation of Gs or Gi coupled pathways, for example, as a result of a Gs mediated increase in cAMP resulting in the activation of a cation permeable cyclic nucleotide gated channel. Alternatively, CCh may still act through a PLC dependent pathway, where the increase in [Ca2+]i is dependent on influx rather than release. There is evidence for PLC mediated activation of the Ca2+ permeable channel TRPV1 (14). This channel appears to be constitutively inhibited by PIP2 such that PLC mediated cleavage of PIP2, results in an increase in channel activity. Importantly, there is evidence that TRPV1 is present and functional in BUC (15). An implication of this latter possibility, however, is that Ca2+ stores in BUC are maintained at such low levels that the liberation of IP3 results in an indictable increase in [Ca2+]i, as has been demonstrated in other cell types.
More striking than the presence of functional muscarinic receptors on BUC was the observation that differences between IC and NB BUC persist in vitro. While a simple increase in receptor expression could account for the increase in efficacy, such a change, alone, should not produce an increase in potency as well. Thus, there are at least 3 explanations for the increase in both potency and efficacy of CCh in BUC from IC patients. These include: 1) a change in the properties of muscarinic receptors (i.e., a shift in the relative proportion of M2 and M3 receptor subtypes), 2) a change in the coupling of receptor activation to Ca2+ influx, and/or 3) a decrease in outward currents that counter Ca2+ influx mechanism. We have recently obtained evidence in support of the latter possibility (16). Additional studies will be needed to assess the relative contribution of the first two possibilities.
Antimuscarinics, such as tolterodine, are widely used clinically to control overactive bladder symptoms such as urinary frequency, urgency, and nocturia, despite the fact that efficacy of tolterodine in IC has not been demonstrated in randomized placebo controlled trials. In fact, clinical experience suggests that antimuscarinics may be of limited therapeutic efficacy. This may reflect the complexity of muscarinic signaling in the bladder, with receptors coupled to both excitatory and inhibitory processes that are present on both afferent and efferent fibers innervating this structure as well as multiple cell types within the structure. Our present finding add to this complexity and suggest that in vitro urothelial muscarinic signaling alterations also may be involved in the pathobiology of IC.
The mechanism(s) underlying persistent differences between IC and NB BUC in vitro remain to be determined. It is certainly possible that these differences are an artifact as a result of cell culture given multiple lines of evidence of phenotypic changes in vitro. Two lines of evidence argue against this possibility, however. First, responsiveness to CCh is not an artifact of cell culture as suggested by data from the fibroblast cell line (NIH/3T3 cells), indicating that these cells, cultured under identical conditions, do not respond to CCh like human BUC. We also have immunostained our cultured cells with smooth muscle α-actin antibodies and have not found any expression of this smooth muscle specific protein (data not shown). Second, cells from NB and IC were consistently different arguing that even if there are changes in culture, there is a persistent interaction between the properties of the cells when introduced into culture and the culture conditions. Importantly, phenotypic differences between IC and NB BUC in culture have been noted by others who have described differences cytokine expression and purinergic signaling (17–22). A more intriguing possibility, however, is that the persistent differences between NB and IC reflect epigenetic changes in BUC from IC. This would provide a mechanism for the persistent “memory” of the IC BUC properties, even after repeated passages in vitro. Such a mechanism would account for the persistent of IC symptoms, despite temporary relief provided by a number of interventions. Such a mechanism would also suggest that an approach for treating IC may be to reverse epigenetic changes.
CONCLUSIONS
IC BUC, when compared to control BUC, were more sensitive to CCh as measured by efficacy and potency of CCh in increasing [Ca2+]i. This was due to muscarinic rather than nicotinic activation as tolterodine, a non-specific muscarinic blocker without nicotinic effects, totally blocked CCh’s ability to increase [Ca2+]i. Furthermore, CCh’s effect on [Ca2+]i. depended on presence of extracellular Ca2+. The greater sensitivity of IC BUC to CCh suggests that IC pathophysiology may include alterations in urothelial cellular muscarinic signaling. The downstream effects of muscarinic receptor activation in BUC are unknown and should be further investigated.
Acknowledgments
Supported by NIH R01-DK075728 and Pfizer (TCC) and NIH-R01- NS 44992 (MSG)
References
- 1.Apodaca G, Balestreire E, Birder LA. The Uroepithelial-associated sensory web. Kidney Int. 2007;72:1057–1064. doi: 10.1038/sj.ki.5002439. [DOI] [PubMed] [Google Scholar]
- 2.Tyagi S, Tyagi P, Van-le S, Yoshimura N, Chancellor MB, de Miguel F. Qualitative and quantitative expression profile of muscarinic receptors in human urothelium and detrusor. J Urol. 2006;176:1673–1678. doi: 10.1016/j.juro.2006.06.088. [DOI] [PubMed] [Google Scholar]
- 3.Kullmann FA, Artim DE, Birder LA, de Groat WC. Activation of muscarinic receptors in rat bladder sensory pathways alters reflex bladder activity. J Neurosci. 2008;28:1977–1987. doi: 10.1523/JNEUROSCI.4694-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersson MC, Tobin G, Giglio D. Cholinergic nitric oxide release from the urinary bladder mucosa in cyclophosphamide-induced cystitis of the anaesthetized rat. Br J Pharmacol. 2008;153:1438–1444. doi: 10.1038/bjp.2008.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kullmann FA, Artim D, Beckel J, Barrick S, de Groat WC, Birder LA. Heterogeneity of muscarinic receptor-mediated Ca2+ responses in cultured urothelial cells from rat. Am J Physiol Renal Physiol. 2008;294:F971–F981. doi: 10.1152/ajprenal.00313.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillenwater JY, Wein AJ. Summary of the National Institute of Arthritis, Diabetes, Digestive and Kidney Diseases Workshop on Interstitial Cystitis, National Institutes of Health, Bethesda, Maryland, August 28–29, 1987. J Urol. 1988;140:203–206. doi: 10.1016/s0022-5347(17)41529-1. [DOI] [PubMed] [Google Scholar]
- 7.Trifillis AL, Cui X, Jacobs S, Warren JW. Culture of bladder epithelium from cystoscopic biopsies of patients with interstitial cystitis. J Urol. 1995;153:243–248. doi: 10.1097/00005392-199501000-00085. [DOI] [PubMed] [Google Scholar]
- 8.Ohuchi T, Yoshimura N, Tanihara H, Kuriyama S, Ito S, Honda Y. Ca2+ mobilization in nontransformed ciliary nonpigmented epithelial cells. Invest Ophthalmol Vis Sci. 1992;33:1696–1705. [PubMed] [Google Scholar]
- 9.Bschleipfer T, Schukowski K, Weidner W, Grando SA, Schwantes U, Kummer W, Lips KS. Expression and distribution of cholinergic receptors in the human urothelium. Life Sci. 2007;80:2303–2307. doi: 10.1016/j.lfs.2007.01.053. [DOI] [PubMed] [Google Scholar]
- 10.Mansfield KJ, Liu L, Mitchelson FJ, Moore KH, Millard RJ, Burcher E. Muscarinic receptor subtypes in human bladder detrusor and mucosa, studied by radioligand binding and quantitative competitive RT-PCR: changes in ageing. Br J Pharmacol. 2005;144:1089–1099. doi: 10.1038/sj.bjp.0706147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishii M, Kurachi Y. Muscarinic acetylcholine receptors. Curr Pharm Des. 2006;12(28):3573–81. doi: 10.2174/138161206778522056. [DOI] [PubMed] [Google Scholar]
- 12.Thomas RL, Mistry R, Langmead CJ, Wood MD, Challiss RA. G protein coupling and signaling pathway activation by m1 muscarinic acetylcholine receptor orthosteric and allosteric agonists. J Pharmacol Exp Ther. 2008 Nov;327(2):365–74. doi: 10.1124/jpet.108.141788. [DOI] [PubMed] [Google Scholar]
- 13.Michal P, El-Fakahany EE, Dolezal V. Muscarinic M2 receptors directly activate Gq/11 and Gs G-proteins. J Pharmacol Exp Ther. 2007 Feb;320(2):607–14. doi: 10.1124/jpet.106.114314. [DOI] [PubMed] [Google Scholar]
- 14.Rohacs T, Thyagarajan B, Lukacs V. Phospholipase C mediated modulation of TRPV1 channels. Mol Neurobiol. 2008 Apr-Jun;37(2–3):153–63. doi: 10.1007/s12035-008-8027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, Wang E, Ruiz G, De Groat WC, Apodaca G, Watkins S, Caterina MJ. Altered urinary bladder function in mice lacking the vanilloid receptor TRPV1. Nat Neurosci. 2002 Sep;5(9):856–60. doi: 10.1038/nn902. [DOI] [PubMed] [Google Scholar]
- 16.Gupta GN, Li M, Sun Y, Gold MS, Simard JM, Chai TC. Increased efficacy and potency of carbachol in inducing increases in intracellular calcium ([Ca2+])i and outward potassium currents (Io) in interstitial cystitis (IC) bladder urothelial cells (BUC): evidence of persistent phenotypic alteration of culture. Abstract #117 presented at 2007 Annual AUA Meeting. [Google Scholar]
- 17.Keay S, Kleinberg M, Zhang CO, Hise MK, Warren JW. Bladder epithelial cells from patients with interstitial cystitis produce an inhibitor of heparin-binding epidermal growth factor-like growth factor production. J Urol. 2000;164:2112–2118. [PubMed] [Google Scholar]
- 18.Keay S, Zhang CO, Shoenfelt JL, Chai TC. Decreased in vitro proliferation of bladder epithelial cells from patients with interstitial cystitis. Urology. 2003;61:1278–1284. doi: 10.1016/s0090-4295(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Chen M, Lowentritt B, Van Zijl PS, Koch KR, Keay S, Simard JM, Chai TC. EGF and HB-EGF modulate inward potassium current in human bladder urothelial cells from normal and interstitial cystitis patients. Am J Physiol Cell Physiol. 2007;292:C106–C114. doi: 10.1152/ajpcell.00209.2006. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Chai TC. Up-regulation of P2X3 receptor during stretch of bladder urothelial cells from patients with interstitial cystitis. J Urol. 2004;171:448–452. doi: 10.1097/01.ju.0000099660.46774.3c. [DOI] [PubMed] [Google Scholar]
- 21.Sun Y, Chai TC. Augmented extracellular ATP signaling in bladder urothelial cells from patients with interstitial cystitis. Am J Physiol Cell Physiol. 2006;290:C27–C34. doi: 10.1152/ajpcell.00552.2004. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Keay S, De Deyne PG, Chai TC. Augmented stretch activated adenosine triphosphate release from bladder uroepithelial cells in patients with interstitial cystitis. J/Urol. 2001 Nov;166(5):1951–6. [PubMed] [Google Scholar]