

Abstract
Trypanosoma brucei is one of the most ancient eukaryotes where RNA interference (RNAi) is operational and is the only single-cell pathogen where RNAi has been extensively studied and used as a tool for functional analyses. Here, we report that the T. brucei RNAi pathway, although relying on a single Argonaute protein (AGO1), is initiated by the activities of two distinct Dicer-like enzymes. Both TbDCL1, a mostly cytoplasmic protein, and the previously undescribed nuclear enzyme TbDCL2 contribute to the biogenesis of siRNAs from retroposons. However, TbDCL2 has a predominant role in generating siRNAs from chromosomal internal repeat transcripts that accumulate at the nucleolus in RNAi-deficient cells and in initiating the endogenous RNAi response against retroposons and repeats alike. Moreover, siRNAs generated by both TbDCL1 and TbDCL2 carry a 5′-monophosphate and a blocked 3′ terminus, suggesting that 3′ end modification is an ancient trait of siRNAs. We thus propose a model whereby TbDCL2 fuels the T. brucei nuclear RNAi pathway and TbDCL1 patrols the cytoplasm, posttranscriptionally silencing potentially harmful nucleic acid parasites that may access the cytoplasm. Nevertheless, we also provide evidence for cross-talk between the two Dicer-like enzymes, because TbDCL2 is implicated in the generation of 35- to 65-nucleotide intermediate transcripts that appear to be substrates for TbDCL1. Our finding that dcl2KO cells are more sensitive to RNAi triggers than wild-type cells has significant implications for reverse genetic analyses in this important human pathogen.
Keywords: repeat-derived siRNAs, retroposons, RNase III, siRNA modification
RNA-mediated silencing pathways through which double-stranded RNA (dsRNA) induces the inactivation of cognate sequences have been identified in a wide variety of eukaryotic organisms. One such pathway, named RNA interference (RNAi), is fueled by the activity of Dicer, which processes dsRNA substrates into 21- to 26-nucleotide (nt) small interfering RNAs (siRNAs), and by Argonaute (AGO), which directs the cleavage of complementary target RNAs (1). Despite high levels of functional conservation, the complexity of the RNAi machinery varies greatly between different organisms (1–3). Many metazoan organisms have multiple Argonautes, whereas single-celled organisms like the fission yeast Schizosaccharomyces pombe and the protozoan parasite Trypanosoma brucei encode only one Argonaute (4). Expansion of Dicer enzymes has also occurred: Arabidopsis thaliana has four Dicer-like proteins, whereas mammals and yeast encode a single Dicer sequence. The observed diversification of RNAi components raises the question of the composition of the ancestral RNAi machinery. It has been argued that the last common ancestor of eukaryotes likely had at least one Argonaute, one Dicer, and one RNA-dependent RNA polymerase (2) and that the role of these components was to provide a defense against genomic parasites, such as transposable elements and viruses. T. brucei represents one of the most ancient eukaryotes in which RNAi has been experimentally verified (5) and thus offers an opportunity to examine the pathway in an early divergent organism.
In addition to responding to exogenous dsRNA, the T. brucei RNAi machinery has been implicated in silencing retroposons at transcriptional and posttranscriptional levels (6, 7) and in maintaining genomic and possibly chromosome stability (8, 9). Whereas TbAGO1 was readily recognizable due to its well-conserved domain organization (6, 7), the Dicer-like protein involved in RNAi, TbDCL1, was described only a few years ago (10). The prototypical Dicer contains from N to C terminus a DExD/H-box helicase domain, a small domain of unknown function (DUF283), a PAZ domain, two tandem RNase III domains (RNase IIIa and RNase IIIb), and a dsRNA binding domain (dsRBD). Although Dicer-like proteins from animals, fungi, and plants maintain a similar overall domain organization, it appears that only the two RNase III motifs are conserved among all of the Dicers. Indeed, the only identifiable domains in TbDCL1 are the two RNase III domains (10). This highly divergent Dicer family member is predominantly located in the cytoplasm and RNAi knockdown of TbDCL1 inhibited the RNAi response, the accumulation of endogenous retroposon-derived siRNAs and cleavage of dsRNA in vitro (10). Here, we show that a second, mostly nuclear Dicer-like protein, TbDCL2, plays a major role in the production of siRNAs derived from satellite-like repeats and that DCL2 is essential for initiating the endogenous RNAi response. However, both DCL1 and DCL2 are involved in generating retrotransposon-derived siRNAs, suggesting that the two Dicer-like proteins have both distinct and redundant functions.
Results
A Class of Trypanosome siRNAs Derived from Satellite-Like Repeats.
We previously showed that the most abundant endogenous siRNAs in trypanosomes are derived from two retroposons, namely Ingi and SLACS (11). More recently, a survey of siRNAs from TbAGO1 immunoprecipitates (Figs. S1–S8) revealed the existence of a class of siRNAs originating from satellite-like repeats located in nontelomeric regions of T. brucei chromosomes 4, 5, and 8 (12). This family of repeats is comprised of 147-bp tandem units and was named CIR147 repeats (chromosome internal repeats). These repeats were independently identified as part of putative centromeric regions (12), which in S. pombe are under the control of the RNAi pathway (13).
To validate CIR147 small RNAs as siRNAs, we first analyzed their 5′ and 3′ end structures. Dicer cleavage of long dsRNA gives rise to siRNAs with a monophosphate at their 5′ end and siRNAs from plants (14), Drosophila (15), and Tetrahymena (16) carry a methyl group at the 3′ end. If the 5′ end of siRNAs has a terminal phosphate, treatment with calf intestine phosphatase (CIP) will result in a change in electrophoretic mobility, whereas treatment with Terminator exonuclease, which preferentially degrades substrates with a single 5′-phosphate, will lead to RNA degradation. However, resistance of RNA to treatment with periodate followed by β-elimination indicates a modified 3′ terminus (17). CIR147 siRNAs were susceptible to both CIP and Terminator enzymes (Fig. 1 A and C), but they were resistant to β-elimination (Fig. 1B lane 6). Control synthetic 25-nt RNA oligonucleotides with 5′-monophosphate or 3′-OH were sensitive to CIP and β-elimination, respectively (Fig. 1A lane 3, and C lane 2), and a 2′-O-methyl oligonucleotide was resistant to β-elimination (Fig. 1B lane 4). We thus deduced that CIR147 24- to 26-nt RNAs carry a single 5′-phosphate group and are blocked at their 3′ ends. A similar result was obtained for retroposon-derived siRNAs (Fig. 1 A and B). Thus, based on these biochemical criteria, the CIR147 small RNAs qualify as a class of endogenous siRNAs in trypanosomes.
Fig. 1.

Retroposon- and repeat-derived siRNAs have modified 5′ and 3′ termini. (A) A synthetic 30-nt RNA (lane 1) was sequentially treated with T4 polynucleotide kinase (PNK, lane 2) and calf intestinal alkaline phosphatase (CIP, lane 3), separated in a denaturing gel and visualized by hybridization with a complementary oligonucleotide. Small RNAs from wild-type cells were separated in a denaturing gel without (lanes 4 and 6) or with (lanes 5 and 7) CIP treatment and probed for CIR147 siRNAs (lanes 4 and 5) or SLACS siRNAs (lanes 6 and 7). (B) A synthetic 23-nt unmodified RNA (c, lanes 1 and 2), a synthetic 23-nt 2′-O-methyl oligonucleotide (2′-O, lanes 3 and 4), and small RNAs from wild-type cells (lanes 6 and 8) were subjected to periodate oxidation/β-elimination and probed for CIR147 siRNAs (lanes 5 and 6) or SLACS siRNAs (lanes 7 and 8). (C) Small RNAs from wild-type (wt) and dcl1KO cells (dcl1−/−) were treated with Terminator exonuclease and subjected to Northern analysis for the CIR147 siRNAs. 5S rRNA (5S), which harbors a 5′-triphosphate terminus, served as a control.
A Second Dicer-Like Enzyme, TbDCL2, Plays a Major Role in the Generation of CIR147 siRNAs.
To provide a direct link between CIR147 small RNAs and the RNAi pathway, we monitored siRNAs in cells where either AGO1 or the Dicer-like enzyme DCL1 were genetically ablated. Accumulation of CIR147 siRNAs was severely affected in ago1−/− cells (Fig. 2A lane 2), and the abundance was restored to wild-type levels in a complementation cell line (lane 3). Interestingly, CIR147-derived siRNAs showed little change in abundance in dcl1−/− cells (lane 4), indicating that DCL1 was not the main enzyme generating CIR147 siRNAs. However, ablation of DCL1 resulted in the accumulation of 35- to 65-nt-long transcripts (indicated by a bracket), whose identity and origin will be described below. Because our initial search for Dicer-like candidates in T. brucei identified a second RNase III-family polypeptide that was specific to the genomes of RNAi-positive trypanosomes (named Tb948 in reference 10 and herein referred to as TbDCL2), we generated a knock-out (KO) cell line of TbDCL2. In this genetic background, we could not detect CIR147 siRNAs (lane 6), and reintroduction of the Dcl2 gene in the dcl2−/− cell line restored accumulation of CIR147 siRNAs (lane 7). Thus, TbDCL2 appeared to have a prominent role in the accumulation of CIR147 siRNAs.
Fig. 2.

Expression pattern of siRNAs in different cell lines. (A and B) Northern blot hybridizations of low MW RNA isolated from the various cell lines, as indicated above each lane, and hybridized to a CIR147 (A) or SLACS (B) probe. tRNA hybridization served as loading control. Complementation cell lines are indicated by the letter c. The bracket indicates the 35- to 65-nt-long transcripts, and sizes are indicated in nucleotides.
Both DCL1 and DCL2 Participate in the Accumulation of Retroposon-Derived siRNAs.
The realization that a second Dicer-like enzyme participated in the T. brucei RNAi pathway prompted us to reevaluate the role of DCL1 and DCL2 in the accumulation of endogenous retroposon-derived siRNAs using our collection of KO and complemented cell lines. As reported (6), accumulation of SLACS (Fig. 2B lane 2) and Ingi (Fig. S1B) siRNAs was severely affected in ago1−/− cells. In contrast, although ablation of DCL1 decreased SLACS siRNAs noticeably (lane 4), the consequence was not comparable to that seen in ago1−/− cells (compare lanes 2 and 4), indicating that DCL1 was only in part responsible for the generation of retroposon-derived siRNAs. Indeed, the analysis of DCL2 KO cells revealed a similar result (lane 6), suggestive of an overlapping or redundant function of the two Dicer-like enzymes in the biogenesis of retroposon-derived siRNAs. As expected, in a genetic background devoid of both DCL1 and DCL2, retroposon siRNAs were below the detection level (Fig. 3B lane 1), but reappeared on complementation with Dcl2 (lane 3).
Fig. 3.

Processing and generation of intermediate-size transcripts. (A) Cells expressing an ectopic copy of Dcl1 under the control of a tet-inducible promoter were incubated with different tet concentrations (lanes 2 and 3) or without tet (lane 1). The expression of DCL1 was monitored by Western blotting (DCL1, Bottom). Low MW RNA was isolated and hybridized to a CIR147 probe. (B) The dcl1/2null cell line (lane 1) was complemented with dcl1 (dcl1c, lane 2) or dcl2 (dcl2c, lane 3), and low MW RNA isolated from the three cell lines was hybridized to a SLACS probe. Sizes are indicated in nucleotides.
A Role for DCL2 in Generating 35- to 65-nt Transcripts.
Our analysis of endogenous siRNAs in dcl1−/− cells revealed a population of transcripts, ranging in size from 35–65 nt in Northern blots for SLACS (Fig. 2B lane 4), Ingi (Fig. S1B lane 4), and CIR147 repeats (Fig. 2A lane 4). These longer transcripts were not seen in our previous experiments examining Ingi/SLACS siRNAs after RNAi-induced down-regulation of Dcl1 (10), most likely because the remaining levels of DCL1 were sufficient to prevent the accumulation of these transcripts. The 35- to 65-nt transcripts were detected by both sense and antisense probes (Fig. S1C), suggesting that they could form a dsRNA duplex in vivo. To characterize their structure and their relationship to siRNAs, 35- to 65-nt RNAs were gel-purified from total RNA isolated from dcl1−/− cells, converted to cDNA, and enriched for CIR147-specific sequences by hybrid selection with a biotinylated CIR147 RNA (11). The 87 analyzed sequences ranged in size from 23–63 nt with an average of 46 nt, and the two strands were represented almost equally (42 sense and 45 antisense), thus reflecting our hybridization results (Fig. S2). Both sense and antisense sequences were distributed relatively evenly along a CIR147 repeat, but no specific structural features, except their length, emerged from this analysis.
Because the 35- to 65-nt transcripts accumulated in dcl1−/− cells, we next asked whether they serve as a substrate for DCL1. Thus, we generated a cell line to conditionally express DCL1 under the control of a tet-inducible promoter in a genetic background, where both endogenous alleles of DCL1 were replaced with drug resistance markers. In the tet-off state, i.e., no detectable DCL1 by Western blotting (Fig. 3A), abundant CIR147 (Fig. 3A lane 1) and Ingi (Fig. S3A) 35- to 65-nt RNAs were detected. By titrating DCL1 expression with tet, the 35- to 65-nt transcripts progressively decreased in abundance, suggesting that these RNAs are DCL1 substrates. At the highest concentration of tet, there was a modest, but reproducible, enhancement of siRNA levels (1.3- to 1.5-fold accumulation relative to the uninduced cells). The observation that siRNA levels did not increase proportionately to the decrease in 35- to 65-nt transcripts might suggest that some of the molecules are degraded.
What enzymatic activity generates the 35- to 65-nt transcripts? A first clue came from the observation that these transcripts were sensitive to Terminator exonuclease (Fig. 1C lane 4) pointing to the presence of a monophosphate at the 5′ terminus. Although not conclusive, this result indicated that they could be primary products of digestion by a Dicer-like activity, i.e., DCL2. To test this hypothesis, we started with the dcl1/dcl2 double KO cell line (dcl1/2null) and reintroduced a copy of either Dcl1 or Dcl2 (Fig. 3B). Although the dcl1/2null cell line was depleted of siRNAs and 35- to 65-nt RNAs (lane 1), expression of DCL2, but not DCL1, restored the accumulation of 35- to 65-nt nt RNAs (compare lanes 2 and 3).
RNAi Components Control Steady-State Transcript Levels from Retroposons and CIR147 Repeats.
One consequence of RNAi-deficiency in T. brucei, i.e., loss of AGO1, is the increased steady-state accumulation of long transcripts derived from retroposons (Fig. 4) (6). This scenario was reproduced for the CIR147 repeats. Whereas transcripts were below the detection level in wild-type cells (Fig. 4A lane 1), a heterogeneous collection of CIR147 RNAs ranging in size over several thousand nucleotides became evident in ago1−/− cells (Fig. 4A lane 2). At present, the extent of the various repeat arrays is not known, but CIR147 transcripts originated from both strands (Fig. S3B) and, based on α-amanitin sensitivity, were synthesized by RNA polymerase II.
Fig. 4.

Expression pattern of steady-state RNA. Northern blot of total RNA isolated from various cell lines as indicated above each lane. The membrane was hybridized to a CIR147-specific probe (A), an Ingi-specific probe (B), or a SLACS-specific probe (C), and α-tubulin (tub) served as a loading control. Approximate sizes in kb are indicated (Left).
We next examined whether loss of DCL1 or DCL2 also influenced the steady-state level of retroposon- and repeat-derived transcripts. Whereas ablation of DCL1 appeared to slightly increase Ingi transcript levels (Fig. 4B lane 4), there was no appreciable effect on CIR147 or SLACS transcripts, i.e., they were barely detectable and comparable to wild-type cells (Fig. 4 A and C lane 4). In contrast, the lack of DCL2 resulted in a noticeable increase of Ingi transcripts (Fig. 4B lane 6) and a striking accumulation of CIR147 (Fig. 4A lane 6) and SLACS transcripts (Fig. 4C lane 6). Complementation of the DCL2 null cell line restored transcript levels back to wild-type levels (Fig. 4 A–C lane 7). These observations suggested that DCL2 is the key Dicer in the endogenous RNAi pathway in T. brucei.
We took advantage of the significant accumulation of CIR147 transcripts in ago1−/− cells to probe their cellular localization by fluorescence in situ hybridization. This analysis revealed a predominant nuclear localization with one prominent hybridization signal colocalizing with the nucleolus and a few smaller foci, which varied in number among different nuclei (Fig. 5 and Fig. S4A). In contrast, in wild-type cells the CIR147 probe only revealed small hybridization foci (panel wt). The strong hybridization signals in ago1−/− nuclei disappeared upon RNase digestion, but the less intense signals in both ago1−/− and wild-type cells were not completely ablated by this treatment, suggesting that the small hybridization foci were, at least to some extent, the result of hybridization to the three known chromosomal CIR147 loci, whereas the strong hybridization signals were indicative of RNA transcripts residing in the nucleolus. RNase A resistance and RNase III sensitivity indicated that these transcripts are in part double-stranded (Fig. S4B), raising the question why they are not processed by DCL2. A possible explanation is that the DCL2-GFP fusion protein appeared to be excluded from the nucleolus (Fig. S8B).
Fig. 5.

FISH analysis of CIR147 transcripts. Wild-type and ago1−/− cells were fixed, processed for FISH, and DNA was stained with DAPI (red). A probe specific for the CIR147 repeats (green) was used for FISH. The DAPI and CIR147 signals were merged. (Bottom Left and Right) The slides were pretreated with ribonuclease A before processing for FISH. Exposure time is 150 ms for all slides.
Small SLACS Accumulation Depends on DCL2.
In addition to the accumulation of long SLACS transcripts, RNAi deficiency in T. brucei resulted in increased levels of short 450- to 550-nt SLACS transcripts, termed small SLACS (sSLACS), which are derived from the 3′ end of SLACS ORF1 (9). Although their mode of synthesis remains uncertain, the Terminator exonuclease assay revealed that <50% of the sSLACS were sensitive, thus exposing a mixed population with either a 5′-monophosphate terminus or a different 5′ terminus, possibly representing additional 5′-phosphate groups (Fig. S3C). The abundance of sSLACS increased in ago1−/− and dcl1−/− cells (Fig. 4C lanes 2 and 4), but they were not detectable in total RNA isolated from dcl2−/− cells (lane 6), implicating DCL2 in either the biogenesis or stabilization of sSLACS transcripts.
Functional Analysis of DCL2.
In our initial analysis, TbDCL2 had only one recognizable RNase III domain at the carboxy terminus, and a GFP fusion protein was enriched in the nucleus (10). However, using less stringent criteria for domain searches, a second RNase III domain close to the amino terminus of TbDCL2 became apparent (Fig. S5A). No other feature(s) associated with Dicer or Dicer-like enzymes were evident. The TbDCL2 RNase IIIa and IIIb domains could be aligned with Aquifex aeolicus RNase III (Fig. S6) and mouse RNase IIIb (Fig. S7), respectively, which allowed us to generate three-dimensional structure models (Fig. S5 B and C). However, because both RNase III domains appeared rather divergent at both the primary and predicted tertiary structural level, we deemed it necessary to provide experimental evidence that TbDCL2 is an active RNase III enzyme. To this end, we engineered cell lines that expressed TbDCL2 with a dual TY/FLAG epitope tag at the amino terminus, either in the wild-type form or carrying substitutions in the RNase IIIb domain. Specifically, the glutamic acid residue at position 885 that we predicted to be part of the four invariant catalytic carboxylates (18–20) and the lysine residue at position 878 that we deduced to correspond to a recently identified catalytic residue in the mouse Dicer (21) were mutated to alanine. Wild-type and mutant proteins were immunopurified from cell extracts and, without removal of the tag, were incubated with an 83-nt dsRNA substrate in the presence of ATP and Mg2+ for 15, 30, and 60 min (Fig. 6). The expression level and size of the mutant proteins was comparable to that of the wild-type enzyme (Fig. S8A). At the earliest time point, wild-type TbDCL2 gave rise to two sets of fragments, a ladder of ≈20- to 24-nt small fragments, and a doublet of ≈65 nt, most likely representing dsRNA molecules that have been cleaved at one end. Both sets of products were Mg2+-dependent and increased in intensity at later time points. We also noticed the appearance of intermediate size fragments, but their origin was not investigated further. Importantly, mutation of either K878 or E885 significantly inhibited the in vitro cleavage of dsRNA by DCL2 (Fig. 6 lanes 3, 4, 7, 8, 11, and 12), indicating that these two residues were critical for TbDCL2 activity.
Fig. 6.
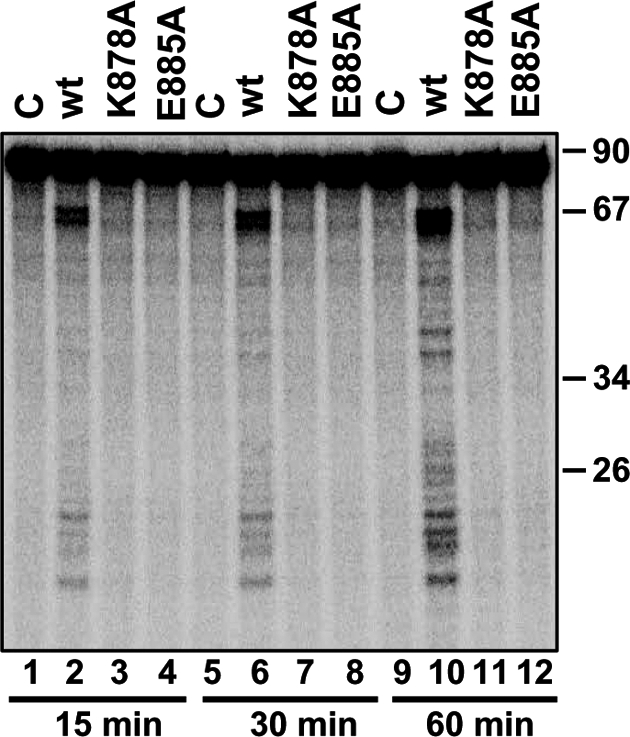
Functional analysis of TbDCL2. Dicing was assayed with affinity-purified DCL2 from dcl2−/− cells complemented with wild-type DCL2 (wt), DCL2 containing the K878A mutation, and DCL2 containing the E885A mutation.
Ablation of DCL2 Enhanced the RNAi Response.
Next, we addressed the role of the two Dicer-like enzymes in response to transfection of exogenous dsRNA. We challenged dcl1KO and dcl2KO cells by electroporation with α-tubulin dsRNA that, after degradation of α-tubulin mRNA, leads to the formation of FAT cells, i.e., cells that are blocked in cytokinesis and accumulate multiple nuclei, flagella, and basal bodies (5). By this assay, ablation of DCL1 resulted in a >10-fold reduction of the RNAi response (Table 1), as gauged by transfection of 1 or 2 μg of α-tubulin dsRNA. Nevertheless, dcl1KO cells were still able to partially respond to dsRNA when 5 μg of dsRNA were used (Table 1). The RNAi response was restored, albeit not to the full wild-type level, when one copy of the dcl1 gene was reintroduced into the dcl1KO cell line (dcl1c). This result indicated that DCL1 is the major player in processing transfected dsRNA. Surprisingly, dcl2KO cells had a 2-fold enhanced RNAi response to dsRNA, as compared to wild-type cells, and this phenotype returned to wild-type levels in the complemented cell line (Table 1). We reasoned it was unlikely that the observed effect was due to an increase in the expression level and thus activity of DCL1, because we have shown that DCL1 overexpression did not affect the magnitude of the RNAi response to transfected dsRNA (10). However, it was possible that the second step of RNAi, including loading of siRNAs into AGO1, was more efficient in dcl2−/− cells. Thus, as a means to bypass the DCL1 cleavage step, dcl2−/− cells were transfected with synthetic α-tubulin siRNA-315, which was shown to induce 85% FAT cells using 60 μg per transfection (22). We found that dcl2−/− cells were exquisitely sensitive to siRNA-315; whereas 5 μg of siRNA-315 gave rise to ≈80% FAT cells in dcl2−/− cells, the same siRNA concentration induced only 10% FAT cells in the wild-type background. We observed a similar, albeit less pronounced enhanced response in dcl1−/− cells transfected with siRNA-315. Thus, this analysis exposed a rather unexpected outcome, namely that ablation of DCL2 resulted in cells with an enhanced response to transfection of long dsRNA or synthetic siRNAs.
Table 1.
Summary of dsRNA and siRNA transfections
| Cell line | α-Tubulin dsRNA, μg |
siRNA-315, μg |
||||||
|---|---|---|---|---|---|---|---|---|
| 0.25 | 0.5 | 1.0 | 2.0 | 5.0 | 1.0 | 2.0 | 5.0 | |
| Wild type | 14 | 42 | 57 | 85 | 100 | <5 | <5 | 8 |
| dcl1KO | nd | nd | <5 | <5 | 20 | <5 | 12 | 30 |
| dcl1C | nd | 34 | 50 | 80 | nd | nd | nd | nd |
| dcl2KO | 50 | 87 | 90 | 95 | nd | 25 | 63 | 82 |
| dcl2C | 14 | 35 | 50 | 70 | nd | nd | nd | nd |
Cell lines were transfected with the indicated amounts of dsRNA or siRNA and the FAT cell phenotype was scored 16 hr post-transfection and is represented as % FAT cells. nd, not determined. Percentage values for each cell line are raw numbers and were not adjusted relative to the wild-type control, which is shown as an example. For each cell line the experiments were repeated three times, and the average value is shown.
Discussion
The RNAi pathway in the early divergent parasitic protozoan T. brucei relies on a single member of the Argonaute family of proteins (6, 7) and is initiated by two distinct Dicer-like enzymes, namely TbDCL1, a previously characterized Dicer-like enzyme that is mostly found in the cytoplasm (10), and TbDCL2, characterized here, which is primarily localized in the nucleus (Fig. S8B) (10). Our biochemical and genetic analyses further indicated that DCL2 and DCL1 are the major and, most likely, the only enzymes of this class that drive the RNAi pathway in T. brucei (Fig. 3B and Fig. S3E).
Both DCL1 and DCL2 are required for the generation of siRNAs from retroposon transcripts. In contrast, accumulation of CIR147 repeat-derived siRNAs was only slightly affected by the absence of DCL1, but required DCL2 activity. Thus, DCL1 and DCL2 have overlapping and distinct functions. Once generated, siRNAs of all classes are loaded into AGO1 (ref. 6 and this article). Our investigations of siRNA termini revealed that the vast majority of both retroposon- and repeat-derived siRNAs in T. brucei possess a 5′-monophosphate. One hypothesis is that the ancestral eukaryotic cell also contained an RNA-directed RNA polymerase (RdRP) presumably involved in the amplification of the RNAi trigger to generate “secondary siRNAs,” which are characterized by 5′-triphosphate termini (23). At present, our biochemical studies do not indicate that such siRNAs play a prominent role in the T. brucei RNAi pathway, and bioinformatics analysis, using blast, psi-blast, and conserved domain searches, did not reveal a canonical RdRP in the genome. However, our data are consistent with a blocked 3′ end, which is suggestive of a 2′-O-methyl modification. Thus, 3′ end modification of siRNAs has ancient roots in the eukaryotic lineage.
Our results further suggest that there is cross-talk between DCL2 and DCL1, because DCL2 is implicated in the generation of 35- to 65-nt transcripts that appear to be substrates for DCL1. In addition, the biogenesis of sSLACS requires DCL2, suggesting that they may also feed into the DCL1 pathway. The accumulation of 35- to 65-nt RNAs was observed in dcl1KO cells not only for the endogenous retroposon and repeat-derived substrates, but also for dsRNA expressed from a transgene equipped with opposing T7 RNA polymerase promoters (Fig. S3D), indicating that their biogenesis is not dependent on the identity of the RNA polymerase transcribing the substrate. Although our results suggested that a portion of these RNAs are converted into siRNAs by the action of DCL1 (Fig. 3A), the potential role and processing of the 35- to 65-nt transcripts will require further investigation.
The presence of abundant nucleolar CIR147 transcripts in ago1−/− cells and the observation that CIR147 siRNA biogenesis depended on DCL2 further supports the hypothesis that the RNAi pathway in T. brucei is operational both in the nucleus and in the cytoplasm. Additional evidence for the functional compartmentalization of the RNAi pathway comes from the phenotypic differences between dcl1−/− and dcl2−/− cells. First, up-regulation of retroposon and repeat transcript levels only occurs when DCL2 is ablated, whereas DCL1 elimination is of little consequence for this phenotype. Second, dcl1KO cells are deficient in the response to transfected dsRNA, indicating that DCL2 is not able to access/use dsRNA transfected by electroporation, a method that mostly accesses the cytoplasm. On the other hand, dcl2−/− cells are significantly more responsive to dsRNA or siRNA transfections than dcl1−/− or wild-type cells. At first glance, this result appears somewhat paradoxical, in that the RNAi response is actually more efficient in the absence of an important RNAi protein. Past experiments have shown that nascent AGO1 is needed for a full RNAi response in T. brucei, suggesting that siRNAs are preferentially loaded on synthesized AGO1 protein (24). We therefore hypothesize that in the absence of DCL2, and consequently the absence of many endogenous siRNAs, there could be less competition by endogenous siRNAs for loading of synthesized AGO1. Thus, dcl2KO cells may be primed to accept exogenous RNAi triggers, i.e., siRNAs that are transfected directly or are derived from transfected dsRNAs. The fact that Dicer-deficient cells are more responsive to RNAi triggers could have exciting implications for the trypanosomatid field at large by enabling the generation of cell lines with a more robust RNAi response that would facilitate analysis of gene function.
Materials and Methods
Trypanosome Cell Lines.
T. brucei rhodesiense Ytat 1.1 cells were used as parental cell line unless otherwise indicated. Construction of the ago1−/− cell line has been described in ref. 6. KO cell lines were made by sequentially replacing the two alleles with drug resistance markers via homologous recombination (25). Complementation cell lines were made by replacing one drug resistance marker with a full-length, untagged version of the wild-type gene in the original locus. Cell lines expressing epitope-tagged versions of a protein were constructed with PCR-assembled cassettes (25).
RNA Manipulations.
Northern blot analysis of SLACS, Ingi, and CIR147 transcripts was performed using total RNA extracted with either the TRIzol (Invitrogen) or RNeasy (Qiagen) method and processed for Northern blot analysis as described in ref. 6. For detection of siRNAs, low molecular weight RNAs were enriched by passage through a Centricon-100 column (Millipore) and separated on a 15% denaturing polyacrylamide gel for subsequent Northern blot analysis as described in ref. 11.
Dicer Activity Assays.
Double-stranded RNA substrate was prepared by annealing a gel-purified 83-nt transcript synthesized with SP6 RNA polymerase in the presence of [α-32P]CTP to ≈20-fold excess of a cold 109-nt T7 RNA polymerase transcript (see SI Materials and Methods). Dicer activity assays were performed in 10-μL reactions, containing 5-μL extract, 2.5-μL transcription buffer, 1 mM ATP, 20 U Protector RNase Inhibitor (Roche Diagnostics), 10,000 cpm (≈20 fmol) dsRNA substrate, in the presence or absence of 10 mM EDTA. Reactions were incubated for 75 min at 24 °C. The assays with the purified Dcl2 proteins contained 2-μL eluted protein, 5.5-μL transcription buffer, and the rest of the above components and proceeded for the indicated times. RNA was extracted with phenol, precipitated with ethanol, separated on denaturing 15% polyacrylamide gels, and analyzed by PhosphorImager.
Enzymatic Assays of Small RNAs.
Small RNAs were size-selected (11) and treated with a variety of enzymes under manufacturer-recommended conditions: Calf intestinal alkaline phosphatase (CIP; Amersham) and T4 polynucleotide kinase (T4 PNK; New England Biolabs) assays were carried out for 1 h at 37 °C, and Terminator exonuclease (Epicentre) was added for 1 h at 30 °C. Periodate oxidation/β-elimination chemical reactions were done as described in ref. 15.
RNA in Situ Hybridization.
Plasmid pCR2.1Topo-CIR3, containing three 147-bp CIR147 repeat units, was linearized and in vitro-transcribed with SP6 or T7 RNA polymerase using a DIG RNA labeling kit (Roche). RNA was hydrolyzed with 0.2 M sodium carbonate buffer, pH 10.2, to an average fragment size of 100 bases, ethanol-precipitated in the presence of 0.1 mg/mL herring sperm DNA and yeast tRNA and dissolved in hybridization buffer (50% deionized formamide, 2× SSC, 10% dextran sulfate) at 6 ng/μL.
Trypanosomes were fixed in 4% formaldehyde in PBS for 20 min, attached to silanized slides, and permeabilized with 0.2 M HCl for 10 min. Cells were either treated with 100 μg/mL boiled RNase in PBS for 60 min at 20 °C or mock-treated with RNase-free PBS and prehybridized in hybridization buffer, including 10 μg/mL herring sperm DNA and yeast tRNA, for 30 min at room temperature. Hybridization was done for 16 h at 50 °C. Slides were washed with 50% formamide, 2× SSC at 50 °C for 5 min, 0.1× SSC at 50 °C for 30 min, 4× SSC at 20 °C for 5 min, incubated with 0.5 μg/mL sheep anti-digoxigenin antibody (Roche) in PBS, 0.1 mg/mL BSA, for 45 min, washed with PBS, 0.05% Tween 20, and developed with 10 μg/mL FITC anti-sheep antibody in PBS/Tween (Vector Laboratories). Cells were embedded in Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) and examined using a Nikon Eclipse 80i epifluorescence microscope, equipped with a CoolSNAP ES CCD camera (Photometrics) controlled by MetaVue software (Molecular Devices). Images were assembled and pseudocolored in Adobe Photoshop CS3.
Supplementary Material
Acknowledgments.
This work was funded by Public Health Service Grants AI28798 and AI56333 (to E.U) and AI43594 (to C.T.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0907766106/DCSupplemental.
References
- 1.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 2.Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: From protists to man. Curr Genet. 2006;50:81–99. doi: 10.1007/s00294-006-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutvagner G, Simard MJ. Argonaute proteins: Key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 4.Siomi H, Siomi MC. On the road to reading the RNA-interference code. Nature. 2009;457:396–404. doi: 10.1038/nature07754. [DOI] [PubMed] [Google Scholar]
- 5.Ngo H, Tschudi C, Gull K, Ullu E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc Natl Acad Sci USA. 1998;95:14687–14692. doi: 10.1073/pnas.95.25.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi H, Djikeng A, Tschudi C, Ullu E. Argonaute protein in the early divergent eukaryote Trypanosoma brucei: Control of small interfering RNA accumulation and retroposon transcript abundance. Mol Cell Biol. 2004;24:420–427. doi: 10.1128/MCB.24.1.420-427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durand-Dubief M, Bastin P. TbAGO1, an Argonaute protein required for RNA interference is involved in mitosis and chromosome segregation in Trypanosoma brucei. BMC Biol. 2003;1:2. doi: 10.1186/1741-7007-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durand-Dubief M, et al. The Argonaute protein TbAGO1 contributes to large and mini-chromosome segregation and is required for control of RIME retroposons and RHS pseudogene-associated transcripts. Mol Biochem Parasitol. 2007;156:144–153. doi: 10.1016/j.molbiopara.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 9.Patrick KL, et al. Genomic rearrangements and transcriptional analysis of the spliced leader-associated retrotransposon in RNA interference-deficient Trypanosoma brucei. Mol Microbiol. 2008;67:435–447. doi: 10.1111/j.1365-2958.2007.06057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi H, Tschudi C, Ullu E. An unusual Dicer-like1 protein fuels the RNA interference pathway in Trypanosoma brucei. RNA. 2006;12:2063–2072. doi: 10.1261/rna.246906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djikeng A, Shi H, Tschudi C, Ullu E. RNA interference in Trypanosoma brucei: Cloning of small interfering RNAs provides evidence for retroposon-derived 24–26-nucleotide RNAs. RNA. 2001;7:1522–1530. [PMC free article] [PubMed] [Google Scholar]
- 12.Obado SO, et al. Functional mapping of a trypanosome centromere by chromosome fragmentation identifies a 16-kb GC-rich transcriptional “strand-switch” domain as a major feature. Genome Res. 2005;15:36–43. doi: 10.1101/gr.2895105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White SA, Allshire RC. RNAi-mediated chromatin silencing in fission yeast. Curr Top Microbiol Immunol. 2008;320:157–183. doi: 10.1007/978-3-540-75157-1_8. [DOI] [PubMed] [Google Scholar]
- 14.Yang Z, Ebright YW, Yu B, Chen X. HEN1 recognizes 21–24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 3′ terminal nucleotide. Nucleic Acids Res. 2006;34:667–675. doi: 10.1093/nar/gkj474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horwich MD, et al. The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr Biol. 2007;17:1265–1272. doi: 10.1016/j.cub.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 16.Kurth HM, Mochizuki K. 2′-O-methylation stabilizes Piwi-associated small RNAs and ensures DNA elimination in Tetrahymena. RNA. 2009;15:675–685. doi: 10.1261/rna.1455509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neu HC, Heppel LA. Nucleotide sequence analysis of polyribonucleotides by means of periodate oxidation followed by cleavage with an amine. J Biol Chem. 1964;239:2927–2934. [PubMed] [Google Scholar]
- 18.Zhang H, et al. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 19.Blaszczyk J, et al. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure. 2001;9:1225–1236. doi: 10.1016/s0969-2126(01)00685-2. [DOI] [PubMed] [Google Scholar]
- 20.Macrae IJ, et al. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 21.Du Z, et al. Structural and biochemical insights into the dicing mechanism of mouse Dicer: A conserved lysine is critical for dsRNA cleavage. Proc Natl Acad Sci USA. 2008;105:2391–2396. doi: 10.1073/pnas.0711506105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Best A, Handoko L, Schluter E, Göringer HU. In vitro synthesized small interfering RNAs elicit RNA interference in African trypanosomes: An in vitro and in vivo analysis. J Biol Chem. 2005;280:20573–20579. doi: 10.1074/jbc.M414534200. [DOI] [PubMed] [Google Scholar]
- 23.Pak J, Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315:241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- 24.Shi H, Tschudi C, Ullu E. Depletion of newly synthesized Argonaute1 impairs the RNAi response in Trypanosoma brucei. RNA. 2007;13:1132–1139. doi: 10.1261/rna.474707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arhin GK, Shen S, Ullu E, Tschudi C. A PCR-based method for gene deletion and protein tagging in Trypanosoma brucei. Methods Mol Biol. 2004;270:277–286. doi: 10.1385/1-59259-793-9:277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.