

Abstract
Absence epilepsy is a neurological disorder that causes a recurrent loss of consciousness and generalized spike-and-wave discharges on an electroencephalogram (EEG). The role of metabotropic glutamate receptors (mGluRs) and associated scaffolding proteins in absence epilepsy has been unclear to date. We investigated a possible role for these proteins in absence epilepsy, focusing on the mGluR7a receptor and its PDZ-interacting protein, protein interacting with C kinase 1 (PICK1), in rats and mice. Injection of a cell-permeant dominant-negative peptide or targeted mutation of the mGluR7a C terminus, both of which disrupt the interaction between the receptor and PDZ proteins, caused behavioral symptoms and EEG discharges that are characteristic of absence epilepsy. Inactivation of the Pick1 gene also facilitated pharmacological induction of the absence epilepsy phenotype. The cortex and thalamus, which are known to participate in absence epilepsy, were involved, but the hippocampus was not. Our results indicate that disruption of the mGluR7a-PICK1 complex is sufficient to induce absence epilepsy—like seizures in rats and mice, thus providing, to the best of our knowledge, the first animal model of metabotropic glutamate receptor—PDZ protein interaction in absence epilepsy.
Typical absence epileptic seizures mainly affect children and are generalized nonconvulsive seizures that occur several times a day, usually during quiet wakefulness. They are characterized by brief unresponsiveness to environmental stimuli and cessation of activity. In EEGs, they are associated with bilateral, synchronous and regular low-frequency (3 Hz) spike-and-wave discharges (SWDs) that result from hypersynchronized rhythmicity of the thalamo-cortical circuitry. The pharmacological properties of absence seizures are unique, as they are suppressed by ethosuximide, which is ineffective in all other forms of epilepsies, and are aggravated by carbamazepine, which prevents partial, secondary generalized seizures1.
The metabotropic glutamate receptors mGluR7a/b are expressed on presynaptic terminals in different regions of the brain, including synapses of the thalamo-cortical circuitry that are involved in absence epilepsy2,3. Little is known about their pathophysiological functions in vivo. mGluR7a/b agonists show a controversial spectrum of actions, having either anti-convulsant4,5 or convulsant6 effects in vitro and in vivo. The mGluR7 knockout mouse shows hypersensitivity to proconvulsive agents, such as pentylenetetrazole and bicuculline, and develops convulsive seizures, but only under particular conditions7. These receptors downregulate neurotransmitter release through inhibition of voltage-dependent Ca2+ channels8 and prevent the neurotoxic effects of extracellular glutamate accumulation9. mGluR7a and mGluR7b are also involved in synaptic plasticity, particularly in long-term depression in hippocampal interneurons10. As a result of their low affinity for glutamate11, these receptors can only be activated under conditions of elevated synaptic activity.
A series of in vitro studies have highlighted the complexity of mGluR7a/b signaling12. The intracellular C-terminal domains of the mGluR7a and mGluR7b can potentially interact with several proteins, including Ca2+-calmodulin, MacMARCKS, G protein βγ subunits and the PDZ domain—containing proteins PICK1, glutamate receptor interacting protein (GRIP or ABP1) and syntenin13-16. The last three C-terminal amino acids of mGluR7a (LVI) constitute the recognition ligand motif of the PDZ interactions. Association with PICK1 has been shown to be necessary for the synaptic clustering and PKC-dependent phosphorylation of mGluR7a15,17, as well as for inhibition of voltage-dependent P/Q-type Ca2+ channels and EPSCs18. GRIP/ABP1 has a primarily postsynaptic localization in neurons19, making it unlikely to bind to the presynaptic mGluR7a, and syntenin binding to mGluR7a has not yet been demonstrated in neurons. Therefore, we focused our study on the interaction between mGluR7a and PICK1.
In the recent years, altered protein-protein interactions have been discussed as potentially causing various diseases. In particular, it is currently recognized that PDZ interactions are essential for membrane receptor signaling and therefore represent attractive therapeutic targets20. The aim of our study was to investigate whether an impaired interaction between mGluR7a and PICK1 could be involved in the development of some form of epilepsy. We found that disruption of the mGluR7a-PICK1 complex was able to trigger absence-like seizures in the mouse and rat.
RESULTS
TAT-R7-LVI peptide triggers absence-like seizures in vivo
We designed a peptide encompassing the last nine amino acids of the mGluR7a C terminus, which included the PDZ-ligand motif LVI, conjugated to the cell-membrane transduction domain of the HIV-1 Tat protein (TAT—R7-LVI; Fig. 1a). TAT-conjugated peptides can cross the blood-brain barrier and plasma membrane21, thus making them well suited for in vivo applications. We first validated the efficacy of the TAT—R7-LVI peptide in blocking both binding of PICK1 to the mGluR7a C terminus and PICK1-dependent mGluR7a/b signaling in cultured mouse cerebellar neurons. A similar peptide bearing three alanines instead of the C-terminal LVI served as a control (TAT—R7-AAA; Fig. 1a). We carried out peptide-affinity chromatography with an immobilized peptide comprising the last nine amino acids of the mGluR7a C terminus (Fig. 1b). We found that the TAT—R7-LVI, but not TAT—R7-AAA, peptide inhibited the interaction of mGluR7a with PICK1 in a dose-dependent manner. Similarly, co-immunoprecipitation experiments performed on mouse brain lysates showed a decrease in PICK1 binding to mGluR7a/b when TAT—R7-LVI was added to the lysate (Fig. 1c).
Figure 1.

in vitro validation of the uncoupling between mGluR7a and PICK1 by the TAT—R7-LVI peptide. (a) Schematic representation of the dominantnegative action of the TAT—R7-LVI, but not TAT—R7-AAA, peptide on PDZ interaction with the mGluR7a C terminus. (b) Pull-down experiment carried out using mouse brain extracts supplemented with no TAT peptide, 100 μM TAT—R7-AAA peptide or 1, 10 or 100 μM TAT—R7-LVI peptide. The right lane (empty beads) was obtained from beads only, with no covalently bound C-terminal peptide. (c) Co-immunoprecipitation of endogenous mGluR7a (antibody to mGluR7a/b) and PICK1 from mouse brain lysates with either no TAT peptide, TAT—R7-LVI or TAT—R7-AAA peptide (10 μM each). (d) Light (upper) and fluorescence (lower) microscopy images obtained from the same culture of cerebellar granule cells, following 10-min incubation with 10 μM fluorescein-tagged TAT—R7-LVI peptide. Scale bar represents 10 μm. (e) Effect of TAT peptides on whole-cell patch-clamp recordings of Ba2+ currents obtained from cultured cerebellar granule cells. Top, typical current traces obtained at 0 mV in the absence (control) and presence of 600 μM D,L-AP4. Scale bars indicate 50 ms and 50 pA. Bottom, percentage inhibition of Ba2+ current induced by D,L-AP4, in the absence and presence of the indicated TAT peptides (n = 10 each, **P < 0.001, Student’s t test compared with control). Error bars represent mean ± s.e.m.
A fluorescein-tagged version of the TAT—R7-LVI peptide applied to cultured cerebellar granule cells was rapidly (<10 min) taken up by almost all of the neurons (Fig. 1d), and in an indiscriminating manner by soma, dendrites and axons. This confirmed the efficiency of TAT-mediated peptide incorporation into neurons. As has been previously shown8 in cerebellar granule cells transfected with mGluR7a, the group-III mGluR agonist, D,L-2-amino-4-phosphonobutyrate (D,L-AP4), inhibited whole-cell voltage-dependent Ba2+ currents by 36 ± 2% (mean ± s.e.m., n = 10; Fig. 1e). This inhibition is strictly dependent on PICK1 interaction with mGluR7a18. Cultured neurons pretreated with the TAT—R7-LVI, but not TAT—R7-AAA, peptide (10 μM each) showed reduced D,L-AP4—mediated inhibition of voltage-dependent Ca2+ channels (3 ± 2% inhibition with TAT—R7-LVI, 32 ± 3% inhibition with TAT—R7-AAA, n = 10 each; Fig. 1e). These results confirmed the efficiency of the TAT—R7-LVI peptide for blocking mGluR7a signaling in neurons, which was consistent with its ability to disrupt the mGluR7a-PICK1 complex.
We then tested the effect of the TAT—R7-LVI peptide in vivo. Our animal models of choice were rats and mice, each for their specific advantages. Rats provide precise stereotaxy for deep electrode and cannula implantations, whereas mice provide the necessary models for studying genetic modifications (knockout and knock-in mice, see below). Whenever possible, we carried out similar experiments on both rats and mice, with comparable results. Adult Sprague-Dawley rats were intravenously injected with 1 μmol of either TAT—R7-LVI or TAT—R7-AAA peptide. We examined the brain distribution of the peptides at 1, 2 and 3 h after injection. All of the brain areas that we examined 1 h after injection contained the injected peptides (Fig. 2a,b). Only a nucleolar peptide accumulation persisted 2 h after injection, whereas the peptide was no longer detected in the brain 3 h after injection (Supplementary Fig. 1 online). These results confirmed that the peptide was able to cross the blood-brain barrier, reaching all of the regions of the brain that we examined, and penetrated vessels and a majority of the neurons.
Figure 2.

Brain diffusion and mGluR7-PICK1 dissociating effect of in vivo—administered TAT peptides. (a) Left, biotin-conjugated TAT—R7-LVI peptide visualized with FITC-conjugated avidin in the rat cortex (Cx), hippocampus (Hi) and ventrobasal (Vb) and reticular (Rt) thalamic nuclei 1 h after intravenous injection. Right, mGluR7a immunolabeling and counterstaining with haematoxylin, in the same regions. Scale bar represents 50 μmin a and b.(b) Control experiments carried out in a rat injected with non-biotinylated TAT—R7-LVI peptide (left) and immunoreactivity obtained using only the secondary antibody for mGluR7a staining in the Vb (middle) and Rt nuclei (right). Data were collected from different rats. (c) Co-immunoprecipitation experiment from peptide-injected mouse brain lysate using an antibody to PICK1 for precipitation and an antibody to mGluR7a/b for western blotting. Input lanes are 1% of the lysate used for co-immunoprecipitation. This blot is representative of three other separate experiments.
Immunocytochemistry confirmed the widespread distribution of mGluR7a throughout the brain, including neocortex, hippocampus, ventrobasal and reticular thalamic nuclei (Fig. 2a,b), as previously described2,3. The TAT—R7-LVI, but not TAT—R7-AAA, peptide reduced the amount of mGluR7a/b-PICK1 complex that could be co-immunoprecipitated from the brains of intravenously injected rats by 49 ± 15% (n = 3; Fig. 2c). This result confirmed the ability of the TAT—R7-LVI peptide to disrupt the interaction between mGluR7a/b and PICK1 in vivo.
What are the consequences of this peptide on brain activity? Intravenous injection of TAT—R7-LVI in rats induced a series of paroxystic, bilateral concomitant discharges (Fig. 3a) that were accompanied by an arrest of locomotor activity, facial myoclonus and vibrissal twitching (Supplementary Movie 1 online). The first discharges appeared with a latency of 16 ± 2 min after peptide administration and once the animal had calmed down after injection. The number and duration of paroxystic EEG events progressively increased with time, reaching a maximum of 19 ± 3 events per 20-min period and a duration of 2.8 ± 0.2 s per seizure (n = 5 rats, 94 discharges) at 40–60 min after injection (Supplementary Fig. 2 online). The discharges had pattern characteristics close to those of SWDs that are observed in the GAERS and WAG/Rij rats, two well-characterized absence epilepsy models22,23, although their duration was shorter. Fourier analysis of the discharges revealed a main intrinsic frequency of 6.9 ± 0.5 Hz with harmonics at 13 and 22 Hz (n = 5 rats, n = 30 discharges; Fig. 3b). The short-time Fourier spectrogram showed that the core frequency was stable throughout the discharge (Fig. 3b). The phenotype disappeared 2 h after the TAT—R7-LVI peptide injection, corresponding to the elimination of the peptide. The control TAT—R7-AAA peptide had no detectable effect on the EEG or on animal behavior (Fig. 3a,d and Supplementary Fig. 3 online).
Figure 3.

Concomitant bilateral cortical discharges induced by disruption of the interaction between mGluR7a and PDZ domain proteins by TAT—R7-LVI in vivo.(a) Left and right cortical EEG recordings were obtained from a rat 30 min after intravenous injection of TAT—R7-LVI peptide (1 μmol, round and square symbols). Scale bars indicate 5 s and 50 μV. (b) Relative power and spectrogram obtained from Fourier analysis of the EEG discharge shown in the insert (same discharge as in a, squaresymbol).(c) EEG cortical recordings obtained from two different mice 30 min after injection of either TAT—R7-LVI (top) or TAT—R7-AAA (bottom) peptides. Dots indicate concomitant epileptiform discharges from the left and right cortex. Scale bars represent 1 s and 100 μV. (d) Mean intrinsic frequency of TAT—R7-LVI peptide—induced EEG discharges in rat and mouse (fast Fourier analysis, n = 5 rats and n = 12 mice). Error bars represent mean ± s.e.m.
We obtained analogous results in C57/Bl6 mice that we intravenously injected with the TAT peptides (400 nmol per mouse). The TAT—R7-LVI peptide induced EEG discharges with a mean frequency of 6.4 ± 0.4 Hz and a duration of 1.5 ± 0.1 s (n = 12 mice, 102 EEG discharges; Fig. 3c,d and Supplementary Fig. 4 online). The number of discharges increased over time and we measured a maximum of 34 ± 3 discharges per 20-min period 40–60 min after intravenous injection of the peptide. As in rats, these EEG activities were concomitant on right and left cortex and were accompanied by locomotor arrest and facial myocloniae. No effect was observed with the TAT—R7-AAA peptide. We found similar results as those obtained in the C57/Bl6 mice in the Swiss mouse strain (data not shown). The differences in discharge characteristics (namely the duration) that we observed in mice versus rats are also present in other rat and mouse models of absence epilepsy1, and may therefore be specific to animal species.
No convulsive seizure was detected on TAT—R7-LVI peptide injection, both in rat and mouse. Instead, the uniform discharges throughout the cortical EEG and the concomitant behavior clearly resembled symptoms seen in nonconvulsive generalized absence epilepsy1. Our results strongly suggest that TAT—R7-LVI peptide induces an absencelike epileptic phenotype in rodents.
TAT—R7-LVI peptide activates absence-related brain regions
The minimal criteria for absence epilepsy in an animal model are as follows: (i) the EEG should display generalized SWDs, (ii) the animal should show locomotor arrest, with or without facial myoclonus, (iii) these symptoms should be concomitant to paroxystic activity of the thalamo-cortical loop, and (iv) they should respond to anti- and pro-absence—specific drugs. Such a phenotype is found in established animal models of absence epilepsy, such as the Hcn2 knockout mouse and spontaneous mouse (C3H/He, ducky, lethargic, mocha2, stargazer and tottering) and rat (GAERS and WAG/Rij) mutant strains1,24,25.
In this study, the first two criteria were met by the results described above. To check for the remaining criteria and to disclose the anatomical substrates of TAT—R7-LVI peptide—dependent generation of SWD-like events, we analyzed c-fos expression as a marker for neuronal activation induced by seizures in mice treated with either TAT—R7-LVI or TAT—R7-AAA peptide (Fig. 4a). An increase in the intensity of c-fos immunolabeling was evident in specific brain regions 1 h after injection of TAT—R7-LVI peptide, but not TAT—R7-AAA peptide. These regions were thalamic nuclei, including the reticular and ventrobasal thalamic nuclei, the somatosensory cortex, the paraventricular area and the ventromedial hypothalamus. Other regions, such as the hippocampus, did not show any increase in c-fos immunostaining. These results suggest that the TAT—R7-LVI peptide preferentially activates thalamo-cortical regions. Notably, those regions have been implicated in absence-like seizures and represent the socket for SWD generation26.
Figure 4.

Region-specific absence seizures induced by disruption of mGluR7a-PDZ ligand interactions. (a) Immunolabeling of c-fos in brain sections from rats that were intravenously injected with either TAT—R7-LVI or TAT—R7-AAA peptide. Scale bar represents 100 μm. (b) Cortical and thalamic EEG recordings in a rat 30 min after intravenous injection with the TAT—R7-LVI peptide. Dots show concomitant discharges in the two structures. (c) Cortical and hippocampal EEG recordings in a rat 30 min after intravenous injection of TAT—R7-LVI peptide. Dots show EEG discharges in the cortex. Scale bars represent 1 s and 100 μVin b and c.
To functionally map the sites of peptide activation, we carried out deep EEG recordings in the thalamic ventrobasal nucleus and in the hippocampus, in parallel with cortical recording, in the rat. Systemic injection of TAT—R7-LVI peptide elicited repetitive discharges on the cortex that were concomitant to thalamic discharges (Fig. 4b and Supplementary Fig. 5 online), with the animal stopping locomotor activity for the duration of the epileptiform EEG activity period. The thalamic discharges had a mean intrinsic frequency of 6.9 ± 0.5 Hz and a meanduration of2.8 ± 0.2s (n = 40 discharges from eight rats) and occurred at an average rate of 15 ± 2 discharges per 20-min period. We observed no EEG modification in the hippocampus (Fig. 4c). Intravenous injection of the control TAT—R7-AAA peptide did not modify any of the recorded deep EEG activities. These results confirmed that the TAT—R7-LVI peptide functionally activated the thalamo-cortical, but not the hippocampal, circuitry.
Local injections of the TAT—R7-LVI or TAT—R7-AAA peptide into the cortex, ventrobasal thalamus or hippocampus were also carried out. The TAT—R7-LVI peptide was readily taken up by neurons at the site of injection 10 min after injection (14 pmol in 0.7 μl), as shown by co-immunolabeling with the neuronal marker NeuN (Fig. 5a—d). The peptide was found in the controlateral structure 30 min after injection in the thalamus (Fig. 5a). We obtained similar results with the TAT—R7-AAA peptide (data not shown). Consistent with the restricted increase of c-fos expression in the thalamus, intra-thalamic injection of TAT—R7-LVI peptide induced paroxystic EEG activity, which spread to the entire cortex concomitantly to the thalamic epileptiform activity (Fig. 5e). The EEG discharges occurred at a frequency of 26 ± 2 per 20-min period (n = 5 rats), and had a mean intrinsic frequency of 6.8 ± 0.4 Hz and a mean duration of 3.8 ± 0.4 s (n = 5 rats, 75 discharges). The EEG seizures were accompanied by the arrest of locomotor activity, as it was the case after intravenous injection of the peptide. Similar effects were obtained when injection was carried out in the somatosensory cortex (Fig. 5f). The discharges elicited from this region had a mean intrinsic frequency of 6.4 ± 0.2 Hz and a duration of 2.8 ± 0.2 s (n = 5 rats, 40 discharges). The control TAT—R7-AAA peptide did not elicit a seizure, regardless of whether it was injected in the thalamus or in the cortex. Notably, intra-hippocampal injection of the active peptide had no detectable effect on the EEG in this structure, nor did it produce any behavioral abnormality (data not shown). These results further demonstrated that (i) EEG discharges induced by the TAT-R7-LVI peptide involved the thalamus and cortex, (ii) they had similar frequencies (although shorter duration) and cortical spreading as generalized SWDs of GAERS and WAG/Rij rats and (iii) were correlated with the typical behavioral arrest of the animal. These characteristics are reminiscent of the generalized nonconvulsive absence epilepsy seizures.
Figure 5.

Absence-like seizures induced by local injection of TAT—R7-LVI peptide in either cortex or thalamus in the rat. (a,b) Avidin-peroxidase labeling of biotin-conjugated TAT—R7-LVI peptide in Vb and Rt thalamic nuclei 10 and 30 min after unilateral intra-thalamic (a) or intra-cortical (b) injection of the TAT—R7-LVI peptide (arrowhead in a, cannula track). (c) Endogenous biotin detection in a non-injected rat (control experiment). (d) FITC-avidin detection of the biotinylated TAT—R7-LVI peptide (green) in neurons labeled with NeuN (red) from rat Vb nucleus, Rt nucleus and cortex 30 min after injection of the peptide. No FITC-avidin was detected in neurons in a non-injected rat (without peptide). Scale bar represents 50μmin a—d.(e,f) Left, position of the cannula and recording electrodes. Right, EEG recordings obtained 30 min after injection of TAT—R7-LVI peptide in the Vb nucleus and cortex. Scale bars represent 3 s and 100 μV.
Pharmacology of the TAT—R7-LVI—induced phenotype
We then characterized the pharmacological properties of the TAT—R7-LVI peptide—induced phenotype in mice, using drugs that promote or reduce human absence epilepsy seizures. The drugs were injected intraperitoneally 30 min before the peptide intravenous injection. The specific anti—absence epilepsy drug ethosuximide (250 mg per kg of body weight) significantly reduced the number (P < 0.001; Fig. 6) and duration of EEG discharges (1.0 ± 0.1 s, n = 5 mice and n = 41 discharges, measured 20 to 40 min after peptide injection).
Figure 6.
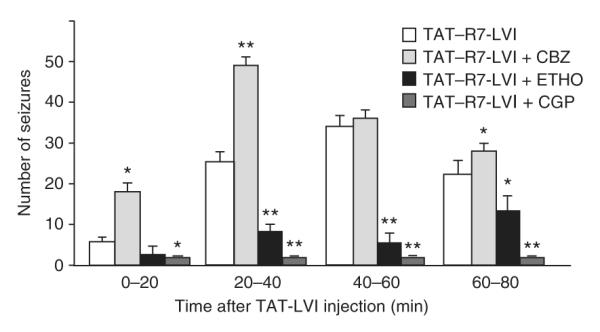
Pharmacology of TAT—R7-LVI—induced absence-like discharges. Time course of EEG seizures per 20-min interval after injection of TAT—R7-LVI peptide and effects of the indicated drugs (CBZ, carbamazepine; ETHO, ethosuximide; CGP, CGP 35348; *P < 0.01; **P < 0.001, Student’s t test compared with TAT—R7-LVI alone, n = 5 mice for each condition). Error bars represent mean ± s.e.m.
The anti-convulsant carbamazepine, which promoted absence epilepsy seizure, (20 mg per kg) increased the number and duration of EEG discharges induced by the TAT—R7-LVI peptide (n = 5 mice and n = 70 discharges measured 20–40 min after peptide injection; Fig. 6). An explicit means of distinguishing absence epilepsy discharges from other paroxystic EEG activity unrelated to absence epilepsy is to test their sensitivity to GABA-B receptor blockade27. The GABA-B receptor antagonist CGP 35348 (300 mg per kg) readily abolished the TAT—R7-LVI peptide—induced EEG discharges, confirming their absence-like pharmacological profile (Fig. 6).
Absence-like phenotype of the mGluR7AAA/AAA KI mouse
Several proteins, such as GluR2, GluR5 and GluR6 AMPA receptor subunits, PKCα, the dopamine transporter DAT1 and ASIC potassium channels, have PDZ ligand motifs that are analogous to that of mGluR7a and bind PICK1 (ref. 28). Therefore, the TAT—R7-LVI peptide could act in a dominant-negative fashion on any of these PDZ protein interactions, as it does at the mGluR7a-PICK1 complex. To demonstrate that the absence seizures triggered by the TAT—R7-LVI peptide were the result of specific disruption of PDZ protein interactions with mGluR7a, we studied a knock-in mouse bearing a constitutive mutation of the mGluR7a PDZ ligand motif (mGluR7aAAA/AAA, C.Z., unpublished observations). This mouse expresses three alanines instead of the LVI C-terminal motif in the mGluR7a carboxyl tail, a mutation that has previously been demonstrated to abolish the binding of PICK1 to the receptor29.
The mGluR7aAAA/AAA mouse showed spontaneous absence-like discharges with characteristics that were similar to those elicited by injection of the TAT—R7-LVI peptide in wild-type mice (Fig. 7a and Supplementary Fig. 6 online). The discharges had a mean duration of 1.4 ± 0.1 s and an intrinsic frequency of 7.7 ± 0.6 Hz and occurred 28 ± 5 times over a 20-min period once the animal had adjusted to the quiet recording environment. The discharges were accompanied by behavioral arrest, facial myoclonus and vibrissal twitching (Supplementary Movie 2 online), symptoms that were similar to those induced by the TAT—R7-LVI peptide. As expected, injection of the TAT—R7-LVI peptide in mGluR7aAAA/AAA mice did not significantly alter the number or duration of these events (P > 0.05; Fig. 7b). Moreover, these seizures responded to the same drugs as those induced by TAT—R7-LVI peptide in wild-type animals, being aggravated by carbamazepine and blocked by ethosuximide and CGP 35348 (Fig. 7b). These results were consistent with disruption of the mGluR7a C terminus, rather than any other PDZ ligand protein interaction, by the TAT—R7-LVI peptide. These experiments were carried out in animals older than 8 weeks. Notably, the spontaneous absence-like behavior and EEG changes were absent in knock-in mice that were younger than 5 weeks (Fig. 7c). In young animals (both wild-type and mGluR7aAAA/AAA mice), the TAT—R7-LVI peptide did not alter the cortical EEG, nor did it induce any behavioral change.
Figure 7.
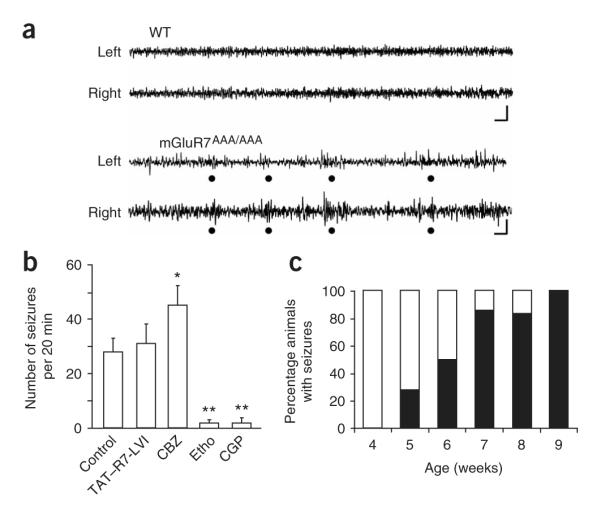
Spontaneous discharges recorded from the mGluR7aAAA/AAA mouse. (a) EEG recordings obtained from left and right cortex, in a wild-type (WT) and an mGluR7aAAA/AAA mouse. Dots indicate simultaneous discharges in both hemispheres in the mGluR7aAAA/AAA mouse. Scale bar represents 1 s and 100 μV. (b) Time course of EEG seizures per 20-min interval in mGluR7aAAA/AAA mice 40 min after intravenous injection of vehicle (control) or TAT—R7-LVI, or after intraperitoneal treatment with the indicated drugs (same legend as in Fig. 6; n = 5–7 mice for each condition, *P < 0.01 or **P ≤ 0.001, Student’s t test compared with control). (c) Age-related development of the absence-like phenotype in the mGluR7aAAA/AAA mouse (n = 6–7 animals per age group). Error bars represent mean ± s.e.m.
PICK1 uncoupling from mGluR7a causes absence-like seizures
The above results did not reveal the identity of the PDZ domain—containing protein that was uncoupled from mGluR7a to confer the absence-like phenotype. We therefore used a TAT fusion peptide (TAT—GluR2-EVKI) that was specifically able to block PICK1 PDZ interactions. This peptide mimics the last 11 amino acids of the C-terminal tail of the AMPA receptor subunit GluR2 and has previously been shown to bind PICK1, but not GRIP/ABP1 and syntenin30 (Fig. 8a). We reasoned that because of this specific interaction, the TAT—GluR2-EVKI peptide should compete with binding of PICK1, but not GRIP/ABP1 or syntenin, on any PDZ ligand protein, including mGluR7a. Intravenous injection of TAT—GluR2-EVKI in mice induced cortical discharges with characteristics that were similar to those produced by the TAT—R7-LVI peptide (Supplementary Fig. 7 online). Their number (Fig. 8b) and duration increased over time, peaked 40 min after injection (23 ± 4 discharges, n = 5 mice) and disappeared 2 h after injection. These results suggest that blocking PICK1, rather than other PDZ protein interactions, causes absence-like EEG discharges.
Figure 8.

Absence-like seizures induced by disruption of PICK1-PDZ ligand interaction. (a) Schematic representation of the dominant-negative effect of the TAT—GluR2-EVKI peptide on the GluR2 C-terminal PDZ ligand interaction with PICK1, but not GRIP/ABP1. (b)Time course of EEG seizures per 20-min interval after intravenous injection of the TAT—GluR2-EVKI peptide (n = 5 mice). (c) Time course of EEG seizures after intravenous injection of CBZ in WT and Pick1-/- mice (n = 5 mice for each condition, *P < 0.01 or **P ≤ 0.001, Student’s t test compared with wild type). Error bars represent mean ± s.e.m.
To further confirm this interpretation, we used a knockout mouse with an inactivated Pick1 gene (Pick1-/-)31. This mouse has a complex phenotype, as expected from the lack of a ubiquitously interacting protein. However, an absence-like phenotype, characterized by episodes of locomotor arrest concomitant to generalized paroxystic EEG events, was unmasked in the Pick1-/- mouse by carbamazepine (CBZ, 20 mg per kg injected intraperitoneally). This effect was specific to the loss of PICK1 protein, as wild-type mice similarly treated with CBZ showed virtually no EEG modification (Fig. 8c). The discharges had an average duration of 2.1 ± 0.3 s, a frequency of 6.7 ± 0.3 Hz and appeared at a rate of 11 ± 1 per 20-min recording period at 40 min after injection of the drug (n = 5 mice, 48 discharges analyzed). Together these results indicated that the loss of PICK1 interaction was sufficient to promote an absence-like phenotype.
DISCUSSION
We found that disruption of PDZ interaction between mGluR7a and PICK1 can induce absence-like seizures, providing evidence that a single PDZ protein-protein interaction can result in a specific neurological disorder. Existing genetic murine models of absence epilepsy are either of multigenic origin (GAERS and WAG/Rij rats) or, when monogenic, have symptoms that are often associated with other severe neurological disorders, such as ataxia1. Our study describes a more genetically restricted model of absence epilepsy.
A number of studies exist in the literature that show that TAT-conjugated dominant-negative peptides can be used to study the pathophysiological role of neuronal protein-protein interactions in vivo21,32. For example, a TAT-conjugated peptide that specifically inhibits the interaction between the NR2B subunit of NMDA receptors and the postsynaptic PDZ protein PSD-95 can protect rat brain from ischemic damage33. We employed a similar approach to study the role of the interaction between the PDZ protein, PICK1 and mGluR7a receptor in absence epilepsy. We found that disruption of the PDZ interaction with mGluR7a is sufficient to induce an absence epilepsy—like phenotype in mice and rats.
Theoretically, the TAT—R7-LVI peptide that we employed could not only interfere with PICK1, but also with other mGluR7a PDZ partners, such as GRIP/ABP1 and syntenin. We found that deletion of the Pick1 gene, or injection of the specific PICK1 dominant-negative peptide TAT—GluR2-EVKI, induced absence epilepsy—like EEG discharges and behavior. This strongly suggests that disruption of PICK1, rather than other PDZ protein interactions, is sufficient to generate the absence epilepsy phenotype. This conclusion does not exclude the possibility that the TAT—R7-LVI peptide antagonized protein-protein interactions other than mGluR7a-PICK1 binding. However, these effects of the peptide were not sufficient to generate absence-like seizures. We therefore conclude that disruption of PDZ interactions, different from the mGluR7a-PICK1 interaction, is not required to induce absence-like seizures. Consistent with this hypothesis, no absence epilepsy symptoms have been described in knockout mice lacking the PICK1 interactors GluR2, GluR5, GluR6, ASIC1, ASIC2 and DAT1. Moreover, it appears that the Gria2-/- (Glur2) mouse is less sensitive to the absence epilepsy—promoting drug γ-hydroxybutyric acid and that AMPA receptor agonists generally promote, rather than decrease, SWDs in various animal models of absence epilepsy34-36. These results further support the hypothesis that disrupted mGluR7a-PICK1 interaction is sufficient to induce an absence epilepsy phenotype.
Mutation of stargazin, a protein that binds to PSD-95 and functionally targets AMPA glutamate receptors at the postsynaptic membrane37 results in an absence epilepsy phenotype with accompanying ataxia, the stargazer mouse38. High levels of stargazin transcripts are found in cerebellum and thalamus39. Thus, defective functional expression of AMPA receptors in cerebellar granule cells may contribute to the stargazer ataxic phenotype. Whether depletion of stargazin in the thalamus contributes to SWDs and absence epilepsy phenotype in this mutant remains uncertain. The promoting effect of AMPA receptor agonists on absence epilepsy and the Gria2-/- phenotype described above argue against this hypothesis. Indeed, high voltage—activated Ca2+ currents, which include the P/Q type, are increased in stargazer thalamo-cortical slices, causing SWDs40. We also found that disruption of the mGluR7-PICK1 complex reduced inhibition of voltage-dependent P/Q-type Ca2+ currents in cultured cerebellar granule neurons. Therefore, we anticipate that absence epilepsy results from facilitated voltage-dependent Ca2+ channels, rather than decreased AMPA receptor activation, in both animal models.
In contrast to rodents injected with the TAT—R7-LVI peptide and the mGluR7AAA/AAA mouse, the Glur7-/- mouse has not been reported to have absence epilepsy seizures, but is rather hypersensitive to convulsant drugs7. This suggests a specific pathophysiological function for the mGluR7a-PICK1 interaction over the global mGluR7 signaling.
Consistent with our functional studies, we found that the thalamocortical loop was preferentially affected by the TAT—R7-LVI peptide. We also found strong c-fos expression in paraventricular and hypothalamic nuclei, which have previously been reported to be activated by chemically induced absence-like seizures41. This result does not exclude the possibility that other structures of the brain may be affected, as we detected the injected TAT—R7-LVI peptide in virtually all of the brain regions that were examined. Nonetheless, it appears that in these other regions, namely the hippocampus, substantial neuronal discharges did not occur.
The question of which synapses are functionally altered by disruption of the mGluR7a-PICK1 complex to induce absence epilepsy remains open. We detected mGluR7a/b immunoreactivity in brain structures that are known to be involved in absence epilepsy. These include the ventrobasal and reticular thalamic nuclei, as well as the somatosensory cortex. Previous studies have shown the expression of the receptor at the release zones of glutamatergic and GABAergic terminals in these regions3,42. In particular, the mGluR7a/b immunoreactivity is observed on axon terminals of GABAergic neurones of the reticular thalamic nucleus that innervate the ventrobasal thalamic neurones43. As a consequence, the TAT—R7-LVI peptide could affect a range of synapses in these regions. Given the role of the GABAergic and glutamatergic systems in physiological oscillations and SWDs in the thalamo-cortical loop, we tentatively suggest that disruption of the mGluR7-PICK1 complex may alter glutamate release in the ventrobasal thalamic nucleus and/or on thalamo-cortical afferents, and/or affect GABA release in the reticular nucleus. Pharmacological studies are consistent with this interpretation44,45.
We found the age of the rodents at the onset of absence seizures to be interesting. In the mGluR7AAA/AAA mouse, seizures appear at an age of 5 weeks or older. Similarly, all other animal models of absence epilepsy (rat and mouse) develop seizures at a pre-adult or adult age. In contrast, typical absence epilepsy in humans develops during childhood and disappears during adolescence1. This apparent discrepancy could be ascribed to differences in corticalization between primates and rodents.
Absence epilepsy is a human genetic disease with a poorly defined origin. This is also true for some animal models of absence epilepsy, such as the GAERS and WAG/Rij rats1. At present, it is not known whether or not the Glur7 and/or Pick1 genes are altered in these animals and humans. Our results provide a solid background to encourage future search for such genetic alterations. All three groups of mGluRs are present in the thalamo-cortical loop. Pharmacological and knockout approaches have suggested that mGluR1/5, mGluR2/3 and mGluR4 are involved in absence epilepsy46,47, but specific studies supporting mGluR7 as a possible therapeutic target for the suppression of SWDs are not available. Our results hint in this direction, but also imply that therapeutic efforts should be targeted to the mGluR7a-PICK1 interaction, rather than to the ligand-binding site of the receptor. To the best of our knowledge, approaches for a therapeutic modification of protein-protein interactions have not been developed, but might provide a promising strategy for treating this and other types of diseases. Until such therapeutic tools become available, it would be worthwhile to screen patients with absence epilepsy for mutations of the PDZ ligand domain of mGluR7. Lack of mutations in this domain would still not exclude participation of the mGluR7-PICK1 complex in absence epilepsy. Indeed, one should bear in mind that altered mGluR7 functional targeting (for example, as a result of post-translational modifications) could result in similar receptor dysfunction and absence seizures in patients. In conclusion, our study presents a new animal model of absence-like seizures and shows that a single PDZ protein interaction can be directly linked to this disease.
METHODS
Neuronal cultures
Cerebellar granule cell cultures were prepared and transfected as previously described13. Whole-cell patch-clamp recording of Ba2+ currents were carried out on mature (9–11 d in vitro) cultures13 (Supplementary Methods online).
Peptide-affinity chromatography
The R7BEAD peptide (see Materials) was coupled via its N-terminal extremity to activated CH-sepharose 4B (GE-Healthcare) according to the manufacturer’s instructions. Brains from Swiss mice (Janvier) were washed in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4mM KH2PO4, pH 7.4), homogenized with a polytron and centrifuged at 200g for 3 min. Pellets were resuspended in CHAPS extraction buffer (Roche) for 3 h under continuous rotation at 1 rpm at 4 °C. Samples were then centrifuged for 1 h at 10,000 g. Solubilized proteins (10 mg per condition) were incubated overnight at 4 °C with 10 μg of immobilized peptide and the TAT-conjugated peptide of interest. Samples were washed twice with 1 ml of extraction buffer and then four times with phosphate-buffered saline before elution in SDS sample buffer.
Co-immunoprecipitation experiments
Swiss mouse brain lysates were incubated with 2 mg of rabbit antibody to mGluR7 (generous gift from R. Shigemoto, National Institute for Physiological Sciences, Japan) and the indicated peptides (10 μM final concentration) overnight at 4 °C and then with protein A—sepharose beads for 1 h at room temperature (20–22 °C). Beads were washed and bound protein was eluted with SDS sample buffer. In separate experiments, brain lysates were prepared from mice intravenously injected with either TAT—R7-LVI or TAT—R7-AAA peptide (400 nmol) 40 min previous to brain extraction. We then co-immunoprecipitated 5 mg of protein incubated with 4 μg of goat antibody to PICK1 (Santa Cruz Biotechnology) overnight at 4 °C and then with protein A—sepharose beads for 1 h at 20–22 °C. After washing of the beads, bound proteins were eluted with SDS sample buffer.
Western blotting
Proteins obtained from peptide-affinity chromatography or co-immunoprecipitation experiments were analyzed by western blotting using goat antibody to PICK1 (1:500 in blocking buffer, Upstate) or rabbit antibody to mGluR7 (1:500 in blocking buffer) as primary antibodies and horseradish peroxidase—conjugated rabbit antibody to goat (1:3,000, Calbiochem) or goat antibody to rabbit (1:4,000, Amersham) as secondary antibodies (Supplementary Methods).
Mutant mice
The generation and characterization of the mutant mice are described elsewhere (mGluR7aAAA/AAA mouse, C.Z., unpublished observations, and Supplementary Methods; PICK1-/- mouse)48.
Surgery, injections and EEG recording
All animal procedures were conducted in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) and approved by the French Ministry for Agriculture. Male Sprague-Dawley rats (Janvier) were anaesthetized using 3 ml per kg of body weight equithesin, and C57/Bl6 or Swiss mice with 3 ml per kg of a saline solution containing 40% ketamin and 20% xylazine (vol/vol). Animals were then placed in a David Kopf stereotaxic apparatus. Gold screws (rat) or silver wires (mouse) were extradurally inserted in the skull, two on each frontal and parietal bone for unilateral fronto-parietal bipolar recording and one on the occipital bone for ground. For deep EEG recording, bipolar electrodes (100-μm diameter nickel-chrome—insulated twisted wires) or cannulae (length 12 mm, 26 G) were implanted according to the rat brain atlas49 (right hippocampus: posterior 3.5, lateral. 2.5, inferior 3 to bregma; right ventrobasal thalamus: posterior 3, lateral 3, inferior 6 to bregma; right frontal cortex: anterior 1.5, lateral 1.5, inferior 1 to bregma). In these animals, only the left side of the skull and the occipital bone were implanted with extradural screws. Deep electrodes and screws were linked to a microconnector fixed to the skull with acrylic cement. After surgery, rats and mice were individually housed and allowed to recover for at least 7 d before experiments.
Freely moving animals were placed into individual Plexiglas boxes and their microconnectors were plugged to an EEG preamplifier. The EEG was filtered (0.5–60 Hz) and acquired with a computer-based system (Micromed). After 20 min of habituation, a 1-h period of baseline recording was carried out before injections under short anesthesia (5 min) by isoflurane inhalation. We intravenously administered TAT peptides in the tail (rat: 1 μmol in 140 μl of saline per 300 g of body weight; mouse: 400 nmol in 70 μl of saline per 30 g of body weight). Intracerebral injection of TAT peptides in the rat consisted of a bolus of 0.7 μl at 20 μM. Anaesthesia was stopped and the animals were replaced in their boxes and reconnected to the EEG apparatus. The EEG recording period (≥80 min) was performed in parallel with observation of the animal behavior, which was monitored with a digital video camera (Sony). Carbamazepine (20 mg per kg), ethosuximide (250 mg per kg) or CGP35348 (300 mg per kg) were intraperitoneally injected after 30 min of control EEG recording and before TAT peptide administration.
The EEG signals were analyzed using IGOR software (Wavemetrics) implemented with fast- and short-time Fourier transformation analysis routines. EEG discharges were identified as previously described50. Only signals with a voltage amplitude that was twice that of the background signal and with a minimum duration of 0.7 s were included in the analysis. Data were analyzed by two different investigators blinded to drug treatments and genotypes.
Histological detection of TAT conjugates and immunohistochemistry
We killed the rats and mice 10 min, 30 min, 1 h, 2 h and 3 h after intracerebral or intravenous injection of the indicated peptides, and detection of biotinylated peptides was carried out with either peroxidase-conjugated avidin or fluorescent avidin. Double immunostaining was performed to identify the TAT peptide—positive cells using a mouse monoclonal antibody to NeuN (1:500, Chemicon). Expression of c-fos and mGluR7 was detected using primary rabbit polyclonal antibodies to mGluR7 (1:1000) or c-fos (1:2,000, Santa Cruz Biotechnology). See Supplementary Methods for details on immunohistochemistry.
Materials
Synthetic peptides (>95% purity) were purchased from Millagen. The peptide sequence was YGRKKRRQRRRYVSYNNLVI for TAT—R7-LVI, YGRKKRRQRRRYVSYNNAAA for TAT—R7-AAA, YGRKKRRQRRRYNVY GIEEVKI for TAT—GluR2-EVKI, and CYVSYNNLVI for R7BEAD, for and N-terminal fluorescein- and biotin-conjugated versions of the TAT-R7-LVI.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Cohen-Solal for animal handling, M.-C. Rousset and M. Gien-Asari for assistance with immunohistochemistry and movie preparation, and A. Depaulis, B. Chanrion and C. Sharpe for helpful discussion. This work was supported by the Agence Nationale de la Recherche (ANR-05-NEURO-035 and ANR-06-NEURO-035), the Max-Planck-Gesellschaft, European Community (QLG3-CT-2001-00929), the Fonds der Chemischen Industrie and Ligue Française Contre l’Epilepsie (F.B.).
Footnotes
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat. Rev. Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- 2.Bradley SR, Rees HD, Yi H, Levey AI, Conn PJ. Distribution and developmental regulation of metabotropic glutamate receptor 7a in rat brain. J. Neurochem. 1998;71:636–645. doi: 10.1046/j.1471-4159.1998.71020636.x. [DOI] [PubMed] [Google Scholar]
- 3.Kinoshita A, Shigemoto R, Ohishi H, van der Putten H, Mizuno N. Immuno-histochemical localization of metabotropic glutamate receptors, mGluR7a and mGluR7b, in the central nervous system of the adult rat and mouse: a light and electron microscopic study. J. Comp. Neurol. 1998;393:332–352. [PubMed] [Google Scholar]
- 4.Folbergrova J, Haugvicova R, Mares P. Seizures induced by homocysteic acid in immature rats are prevented by group III metabotropic glutamate receptor agonist (R,S)-4-phosphonophenylglycine. Exp. Neurol. 2003;180:46–54. doi: 10.1016/s0014-4886(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 5.Gasparini F. (R,S)-4-phosphonophenylglycine, a potent and selective group III metabotropic glutamate receptor agonist, is anticonvulsive and neuroprotective in vivo. J. Pharmacol. Exp. Ther. 1999;289:1678–1687. [PubMed] [Google Scholar]
- 6.Ghauri M, Chapman AG, Meldrum BS. Convulsant and anticonvulsant actions of agonists and antagonists of group III mGluRs. Neuroreport. 1996;7:1469–1474. doi: 10.1097/00001756-199606170-00005. [DOI] [PubMed] [Google Scholar]
- 7.Sansig G, et al. Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J. Neurosci. 2001;21:8734–8745. doi: 10.1523/JNEUROSCI.21-22-08734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perroy J, et al. Selective blockade of P/Q-type calcium channels by the metabotropic glutamate receptor type 7 involves a phospholipase C pathway in neurons. J. Neurosci. 2000;20:7896–7904. doi: 10.1523/JNEUROSCI.20-21-07896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lafon-Cazal M, et al. mGluR7-like metabotropic glutamate receptors inhibit NMDA-mediated excitotoxicity in cultured mouse cerebellar granule neurons. Eur. J. Neurosci. 1999a;11:663–672. doi: 10.1046/j.1460-9568.1999.00475.x. [DOI] [PubMed] [Google Scholar]
- 10.Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto N, et al. Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J. Biol. Chem. 1994;269:1231–1236. [PubMed] [Google Scholar]
- 12.El Far O, Betz H. G-protein-coupled receptors for neurotransmitter amino acids: C-terminal tails, crowded signalosomes. Biochem. J. 2002;365:329–336. doi: 10.1042/BJ20020481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertaso F, et al. MacMARCKS interacts with the metabotropic glutamate receptor type 7 and modulates G protein—mediated constitutive inhibition of calcium channels. J. Neurochem. 2006;99:288–298. doi: 10.1111/j.1471-4159.2006.04121.x. [DOI] [PubMed] [Google Scholar]
- 14.O’Connor V, et al. Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science. 1999;286:1180–1184. doi: 10.1126/science.286.5442.1180. [DOI] [PubMed] [Google Scholar]
- 15.Dev KK, et al. PICK1 interacts with and regulates PKC phosphorylation of mGluR7. J. Neurosci. 2000;20:7252–7257. doi: 10.1523/JNEUROSCI.20-19-07252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirbec H, et al. The PDZ proteins PICK1, GRIP, and syntenin bind multiple glutamate receptor subtypes. Analysis of PDZ binding motifs. J. Biol. Chem. 2002;277:15221–15224. doi: 10.1074/jbc.C200112200. [DOI] [PubMed] [Google Scholar]
- 17.Boudin H, et al. Presynaptic clustering of mGluR7a requires the PICK1 PDZ domain binding site. Neuron. 2000;28:485–497. doi: 10.1016/s0896-6273(00)00127-6. [DOI] [PubMed] [Google Scholar]
- 18.Perroy J, et al. PICK1 is required for the control of synaptic transmission by the mGluR7 receptor. EMBO J. 2002;21:2990–2999. doi: 10.1093/emboj/cdf313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burette A, Wyszynski M, Valtschanoff JG, Sheng M, Weinberg RJ. Characterization of glutamate receptor interacting protein-immunopositive neurons in cerebellum and cerebral cortex of the albino rat. J. Comp. Neurol. 1999;411:601–612. [PubMed] [Google Scholar]
- 20.Dev KK. Making protein interactions druggable: targeting PDZ domains. Nat. Rev. Drug Discov. 2004;3:1047–1056. doi: 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]
- 21.Dietz GP, Bahr M. Peptide-enhanced cellular internalization of proteins in neuroscience. Brain Res. Bull. 2005;68:103–114. doi: 10.1016/j.brainresbull.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog. Neurobiol. 1998;55:27–57. doi: 10.1016/s0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- 23.van Luijtelaar EL, Coenen AM. Two types of electrocortical paroxysms in an inbred strain of rats. Neurosci. Lett. 1986;70:393–397. doi: 10.1016/0304-3940(86)90586-0. [DOI] [PubMed] [Google Scholar]
- 24.Frankel WN, et al. Development of a new genetic model for absence epilepsy: spike-wave seizures in C3H/He and backcross mice. J. Neurosci. 2005;25:3452–3458. doi: 10.1523/JNEUROSCI.0231-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pietrobon D. Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr. Opin. Neurobiol. 2005;15:257–265. doi: 10.1016/j.conb.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Meeren H, van Luijtelaar G, da Silva F. Lopes, Coenen A. Evolving concepts on the pathophysiology of absence seizures: the cortical focus theory. Arch. Neurol. 2005;62:371–376. doi: 10.1001/archneur.62.3.371. [DOI] [PubMed] [Google Scholar]
- 27.von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, Xia J. Structure and function of PICK1. Neurosignals. 2006;15:190–201. doi: 10.1159/000098482. [DOI] [PubMed] [Google Scholar]
- 29.El Far O, et al. Interaction of the C-terminal tail region of the metabotropic glutamate receptor 7 with the protein kinase C substrate PICK1. Eur. J. Neurosci. 2000;12:4215–4221. doi: 10.1046/j.1460-9568.2000.01309.x. [DOI] [PubMed] [Google Scholar]
- 30.Daw MI, et al. PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 31.Gardner SM, et al. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 32.Cao G, et al. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aarts M, et al. Treatment of ischemic brain damage by perturbing NMDA receptor—PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 34.Hu RQ, et al. γ-hydroxybutyric acid-induced absence seizures in GluR2 null mutant mice. Brain Res. 2001;897:27–35. doi: 10.1016/s0006-8993(01)02076-5. [DOI] [PubMed] [Google Scholar]
- 35.Avanzini G, de Curtis M, Franceschetti S, Sancini G, Spreafico R. Cortical versus thalamic mechanisms underlying spike and wave discharges in GAERS. Epilepsy Res. 1996;26:37–44. doi: 10.1016/s0920-1211(96)00037-x. [DOI] [PubMed] [Google Scholar]
- 36.Ramakers GM, Peeters BW, Vossen JM, Coenen AM. CNQX, a new non-NMDA receptor antagonist, reduces spike wave discharges in the WAG/Rij rat model of absence epilepsy. Epilepsy Res. 1991;9:127–131. doi: 10.1016/0920-1211(91)90023-9. [DOI] [PubMed] [Google Scholar]
- 37.Chen L, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 38.Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990;7:129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- 39.Letts VA, et al. The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nat. Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Mori M, Burgess DL, Noebels JL. Mutations in high voltage—activated calcium channel genes stimulate low voltage—activated currents in mouse thalamic relay neurons. J. Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andre V, Pineau N, Motte JE, Marescaux C, Nehlig A. Mapping of neuronal networks underlying generalized seizures induced by increasing doses of pentylenetetrazol in the immature and adult rat: a c-Fos immunohistochemical study. Eur. J. Neurosci. 1998;10:2094–2106. doi: 10.1046/j.1460-9568.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- 42.Dalezios Y, Lujan R, Shigemoto R, Roberts JD, Somogyi P. Enrichment of mGluR7a in the presynaptic active zones of GABAergic and non-GABAergic terminals on interneurons in the rat somatosensory cortex. Cereb. Cortex. 2002;12:961–974. doi: 10.1093/cercor/12.9.961. [DOI] [PubMed] [Google Scholar]
- 43.Somogyi P, et al. High level of mGluR7 in the presynaptic active zones of select populations of GABAergic terminals innervating interneurons in the rat hippocampus. Eur. J. Neurosci. 2003;17:2503–2520. doi: 10.1046/j.1460-9568.2003.02697.x. [DOI] [PubMed] [Google Scholar]
- 44.Wan H, Cahusac PM. The effects of L-AP4 and L-serine-O-phosphate on inhibition in primary somatosensory cortex of the adult rat in vivo. Neuropharmacology. 1995;34:1053–1062. doi: 10.1016/0028-3908(95)00091-j. [DOI] [PubMed] [Google Scholar]
- 45.Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J. Physiol. (Lond.) 1999;519:481–491. doi: 10.1111/j.1469-7793.1999.0481m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexander GM, Godwin DW. Metabotropic glutamate receptors as a strategic target for the treatment of epilepsy. Epilepsy Res. 2006;71:1–22. doi: 10.1016/j.eplepsyres.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 47.Snead OC, III, Banerjee PK, Burnham M, Hampson D. Modulation of absence seizures by the GABA(A) receptor: a critical role for metabotropic glutamate receptor 4 (mGluR4) J. Neurosci. 2000;20:6218–6224. doi: 10.1523/JNEUROSCI.20-16-06218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steinberg JP, et al. Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron. 2006;49:845–860. doi: 10.1016/j.neuron.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 49.Paxinos G, Watson C, Pennisi M, Topple A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J. Neurosci. Methods. 1985;13:139–143. doi: 10.1016/0165-0270(85)90026-3. [DOI] [PubMed] [Google Scholar]
- 50.Kim D, et al. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α(1G) T-type Ca2+ channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.