

Abstract
Cruzain is the major cysteine protease of T. cruzi, which is the causative agent of Chagas’ disease and is a promising target for the development of new chemotherapy. With the goal of developing potent nonpeptidic inhibitors of cruzain, the Substrate Activity Screening (SAS) method was used to screen a library of protease substrates initially designed to target the homologous human protease cathepsin S. Structure-based design was next used to further improve substrate cleavage efficiency by introducing additional binding interactions in the S3 pocket of cruzain. The optimized substrates were then converted to inhibitors by the introduction of cysteine protease mechanism-based pharmacophores. Inhibitor 38 was determined to be reversible even though it incorporated the vinyl sulfone pharmacophore that is well documented to give irreversible cruzain inhibition for peptidic inhibitors. The previously unexplored β-chloro vinyl sulfone pharmacophore provided mechanistic insight that led to the development of potent irreversible acyl- and aryl-oxymethyl ketone cruzain inhibitors. For these inhibitors, potency did not solely depend on leaving group pKa, with 2,3,5,6-tetrafluorophenoxymethyl ketone 54 identified as one of the most potent inhibitors with a second order inactivation constant of 147,000 s−1M−1. This inhibitor completely eradicated the T. cruzi parasite from mammalian cell cultures and consequently has the potential to lead to new chemotherapeutics for Chagas’ disease.
Keywords: Cruzain, aryloxymethyl ketone, Chagas’ disease, protease inhibitor
Introduction
Chagas’ disease (American trypanosomiasis), caused by the parasitic protozoan Trypanosoma cruzi, is the leading cause of heart disease in Latin America. Today, at least 12 million people are infected with the parasite, resulting in more than 50,000 deaths each year.1 Chemotherapy for Chagas’ disease is unsatisfactory with current drugs, nifurtimox and benznidazole, having significant toxic side effects.2 Due to the toxicity of current chemotherapy and emerging drug resistance, there is an urgent need for developing an effective therapy against Chagas’ disease.
Cruzain, a cysteine protease of the papain family, is the primary cysteine protease of the T. cruzi parasite. It is involved in intracellular replication and differentiation and is essential at all stages of the parasite’s life cycle.3 Recently, it has been demonstrated that T. cruzi infection can be cured in cell, mouse, and dog models by treatment with irreversible inhibitors of cruzain.4,5 Parasite vulnerability to cruzain inhibition results from the lack of redundancy of this enzyme. Moreover, parasite localization provides a means for preferential inhibition of cruzain over the highly homologous human papain superfamily cysteine proteases cathepsins B, L, K, S, F and V because the parasite resides in the host cell cytoplasm whereas the cathepsins are located in the less accessible lysosomes.6 For these reasons, cruzain is a highly attractive therapeutic target for the treatment of Chagas’ disease.7
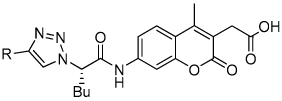
Dipeptidyl vinyl sulfone 1 is the most advanced inhibitor of cruzain and is currently in pre-clinical trials (Figure 1).8 Although this peptidic inhibitor has shown good efficacy with minimal toxicity, nonpeptidic inhibitors with improved oral bioavailability could prove even more effective. As only irreversible inhibitors of cruzain have been successful in curing parasitic infections, we sought to develop nonpeptidic irreversible inhibitors of cruzain.9
Figure 1.

Most advanced inhibitor of cruzain.
Recently, we developed Substrate Activity Screening (SAS) as a new method for the rapid identification of nonpeptidic enzyme inhibitors.10–14 The SAS method consists of the identification of nonpeptidic substrate fragments, substrate optimization, and then conversion of optimal substrates to inhibitors. Significantly, the SAS method has successfully been applied to the papain superfamily protease cathepsin S,10,12,13 which has high homology to cruzain.15 Using a focused substrate library developed for cathepsin S as a starting point, we report herein the development of a new class of nonpeptidic 2,3,5,6-tetrafluorophenoxymethyl ketone inhibitors that exhibit potent inhibitory activity against cruzain as well as complete eradication of T. cruzi parasites in cell culture. This class of compounds represents a promising and novel inhibitor class for the treatment of Chagas’ disease.
Results and Discussion
Initial Screening
High correlation between substrate cleavage efficiency and inhibitory activity was observed in the previous development of cathepsin S inhibitors.10,12 Substrate analogues were therefore first evaluated and optimized before conversion to inhibitors. A triazole-based substrate library consisting of more than 150 substrates was screened against cruzain. Substrate activity was measured by monitoring liberation of the 7-amino-4-methyl coumarin acetic acid (AMCA) fluorophore, which results from protease-catalyzed amide bond hydrolysis (Scheme 1).
Scheme 1.

Fluorogenic substrate screening
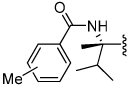
Shown in Table 1 is the structure activity relationship (SAR) for a subset of substrates from the triazole library that exemplifies cruzain’s substrate specificity requirements. The weakest substrate for which a signal could be detected was substrate 2 that incorporated a simple benzyl substituent on the triazole ring. A variety of more active hydroxyl substituted substrates were screened and the optimal aliphatic functionalities identified were the methyl and isopropyl substituents present in substrate 4. Replacement of the hydroxyl with a benzamide moiety in substrate 5 resulted in an increase in cleavage efficiency. The epimeric compounds 6 and 7 demonstrate that cruzain shows strong chiral recognition with epimer 7 being much more active. Substitutions on the benzamide moiety indicated that ortho substitutions were not tolerated by cruzain (substrates 8 and 11). In contrast, meta and para substituents resulted in increases in substrate activity (substrates 9, 10 and 12, 13) with p-methoxy substituted benzamide substrate 13 identified as the most efficient substrate from the screening of the initial triazole library.
Table 1.
Representative Substrates from the Initial 1,2,3-Triazole Library
 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Rel. kcat/Km | Cmpd | R | Rel. kcat/Km |
| 2 | 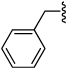 |
0.00047 | 6 | 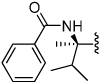 |
0.0074 |
| 3 | 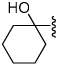 |
0.011 | 7 | 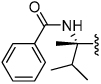 |
1.0 |
| 4 | 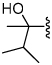 |
0.068 |
8 9 10 |
 |
o = 0.41 m = 1.8 p = 2.0 |
| 5 |  |
0.16 |
11 12 13 |
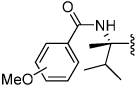 |
o = 0.22 m = 2.0 p = 2.8 |
Structure-guided Substrate Optimization
Further optimization of substrate binding to cruzain was accomplished using structure-based design. Recently, a crystal structure was obtained of chloromethyl ketone inhibitor 14 bound to cathepsin S.12 The amino acid sequences of cathepsin S and cruzain are 38% homologous and their active sites nearly identical (Figure 2).15 Taking into account the high homology between these two enzymes, molecular replacement was performed to model inhibitor binding to cruzain (Figure 3).
Figure 2.

Alignment of cathepsin S (PDB ID: 2H7J) and cruzain (PDB ID: 1F2C) amino acid sequences with areas around the catalytic triad highlighted in yellow. Identical residues are indicated with “*” and similar residues are indicated with “.”. The figure was produced using Swiss-Pdb Viewer (http://ca.expasy.org/spdbv/).
Figure 3.

(a) Crystal structure of cathepsin S (PDB ID: 2H7J) and (b) molecular replacement model of cruzain (PDB ID: 1F2C) with chloromethyl ketone inhibitor 14. The atoms are shaded according to element: protein carbons are green, inhibitor carbons are grey, nitrogens are blue, and oxygens are red. The figure was produced using PyMOL (www.pymol.org).
Due to the similarities in the active sites, the inhibitors are predicted to bind in a similar fashion with the n-butyl group in the S1 pocket, the methyl and isopropyl groups in the S2 pocket, and the benzamide moiety in the S3 pocket. The majority of prior inhibitor development for cruzain has focused on the S1’, S1, and S2 pockets. The S3 pocket of cruzain is largely unexplored with no previous reports of significant binding interactions in this pocket. Upon closer inspection of our molecular replacement model, key differences in the S3 pockets were noted. The S3 pocket of cathepsin S is small and well-defined whereas that of cruzain is large and open-ended. To take advantage of cruzain’s larger S3 pocket, a focused library of substrate analogues incorporating planar heterocycles in place of the phenyl ring of the benzamide moiety was designed. Heterocycles were chosen with potential for hydrophobic interactions with the hydrophobic side of the pocket and with potential for hydrogen bonding interactions with the serine residue in the S3 pocket.
The synthesis of the 1,4-disubstituted-1,2,3-triazole substrates containing the AMCA fluorophore was accomplished on solid support (Scheme 2). We were pleased to find that all but one of the substrates of the focused library were more active than the unsubstituted benzamide substrate 7 (Table 2). The most potent substrates identified were quinoline 30 and benzothiazole 31 with 7–9 fold increases in cleavage efficiency. These substrates both contained a nitrogen atom para to the amide bond, which could be interacting with the serine residue in the S3 pocket (Figure 3b). When comparing naphthyl 20 and quinoline 30, a 4-fold increase in activity was observed presumably due to this polar interaction. Moreover, the 4-fold increase in cleavage efficiency of indole 29 relative to benzotriazole 21 demonstrated that hydrophobic interactions also contribute to binding. Substrate 18 incorporating a morpholine moiety was prepared because this substituent has led to potent vinyl sulfone inhibitors of cruzain.16 However, this substituent resulted in a decrease in substrate cleavage efficiency.
Scheme 2.

General Synthesis of Focused Library 1,2,3-Triazole Substratesa
a Reagents: (a) CuI, i-Pr2EtN, THF, rt; (b) R’CO2H, triphosgene, THF, rt; (c) R’COH, NaBH(OAc)3, AcOH, THF, rt; (d) CF3CO2H, CH2Cl2, rt.
Table 2.
Cleavage Efficiencies of 1,2,3-Triazole Amide Substrates against Cruzain
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | R | Rel. kcat/Km | Cmpd | R | Rel. kcat/Km | Cmpd | R | Rel. kcat/Km |
| 18 | 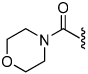 |
0.6 | 22 | 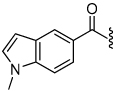 |
2.5 | 27 | 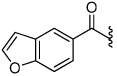 |
3.6 |
| 7 | 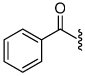 |
1.0 | 23 | 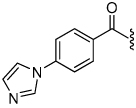 |
2.8 | 28 | 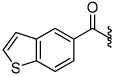 |
5.3 |
| 19 |  |
1.4 | 24 | 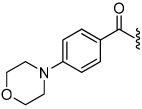 |
3.0 | 29 | 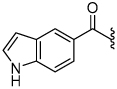 |
6.5 |
| 20 |  |
1.6 | 25 | 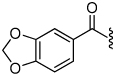 |
3.1 | 30 | 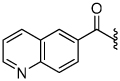 |
7.4 |
| 21 | 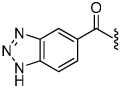 |
1.7 | 26 | 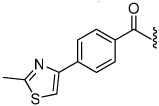 |
3.3 | 31 | 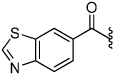 |
9.0 |
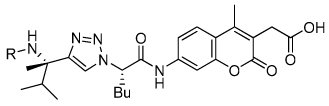
A notable feature in the inhibitor binding model is the nonessential nature of the benzamide carbonyl (Figure 3b). Therefore, amine 33 corresponding to benzamide substrate 7 was prepared resulting in a 3-fold increase in cleavage efficiency (Table 3). To determine if amine substrate SAR correlated with the SAR trends observed for the corresponding amide substrates, additional amine analogues were synthesized and evaluated. High correlation was observed between the SAR for the amide and amine substrate series, resulting in the identification of quinoline amine substrate 36 and benzothiazole amine substrate 37 with 19-fold greater cleavage efficiency than unsubstituted benzamide 7.
Table 3.
Comparison of 1,2,3-Triazole Amide and Amine Substrate Activity against Cruzain
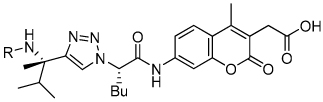 | |||||
|---|---|---|---|---|---|
| Cmpd | R | Rel. kcat/Km | Cmpd | R | Rel. kcat/Km |
| 7 | 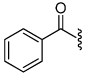 |
1.0 | 33 | 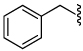 |
3.5 |
| 32 | 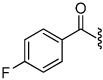 |
1.3 | 34 | 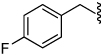 |
4.0 |
| 13 | 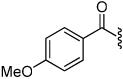 |
2.8 | 35 | 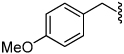 |
6.5 |
| 30 | 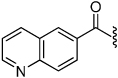 |
7.4 | 36 | 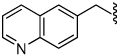 |
19 |
| 31 | 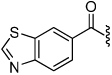 |
9.0 | 37 | 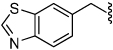 |
19 |
Conversion of Substrates into Inhibitors
A key advantage of the SAS method is that the aminocoumarin group has to be precisely oriented in the active site for amide bond hydrolysis to occur and can therefore be directly replaced with mechanism-based pharmacophores. The optimal quinoline amine substrate 36 was first converted to inhibitors to evaluate the effectiveness of different cysteine protease mechanism-based pharmacophores.
We initially chose to investigate the vinyl sulfone pharmacophore because it has been incorporated in potent inhibitors of cruzain that have proven effective at eradicating Chagas’ disease in both cell culture and animal models.4,5,16,17 Vinyl sulfone inhibitor 38 was prepared via a Horner-Wadsworth-Emmons olefination (Scheme 3). Kinetic analysis of the vinyl sulfone inhibitor, surprisingly, indicated no time dependence and was consistent with competitive reversible inhibition (Figure 4a).18
Scheme 3.

Synthesis of Vinyl Sulfone Inhibitor 38a
a Reagents: (a) CuI, i-Pr2EtN, THF, rt; (b) diazomethane, THF, rt; (c) DIBAL-H, Et2O, −78 °C; (d) (OEt)2P(O)CH2SO2Ph, LiBr, NEt3, CH3CN, −40 °C.
Figure 4.

Time-dependence of (a) vinyl sulfone 38 and (b) β-chloro vinyl sulfone 43.
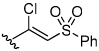
To gain further insight, we decided to investigate the reversible nature of vinyl sulfone inhibitor 38. Vinyl sulfones are thought to irreversibly alkylate cysteine proteases via a Michael addition followed by protonation of the α-carbon by the active site histidine to form a covalent thioether adduct (Figure 5a).19 Two potential reasons for the reversibility of vinyl sulfone 38 are therefore either that the active site cysteine is not adding into the vinyl sulfone or that the active site histidine is not properly oriented for protonating the resulting anion. We postulated that a β–chloro vinyl sulfone could distinguish between these possibilities because cysteine protease inactivation could be accomplished via Michael addition followed by β-elimination of chloride ion thereby eliminating the need for anion protonation (Figure 5b). Although this particular pharmacophore has never before been investigated, it is analogous to previously characterized β–chloro α,β–unsaturated ester inhibitors.20
Figure 5.

Mechanism of inhibition of cysteine proteases by (a) vinyl sulfones and (b) β-chloro vinyl sulfones.
β-Chloro vinyl sulfone inhibitor 43 was prepared according to the route depicted in Scheme 4 with the key step being conversion of ketosulfone 45 to vinyl chloride 46 via the vinyl triflate. Gratifyingly, time-dependence analysis and dilution experiments indicated that β-chloro vinyl sulfone inhibitor 43 was an irreversible inhibitor of cruzain (Figure 4b). This result suggests that the active site cysteine is adding into vinyl sulfone 38 and that the lack of a protonation event resulted in a reversible inhibitor. The β-chloro sulfone inhibitor 43 had a modest second order rate of inactivation constant of 805 s−1M−1 (Table 4). Encouraged by this result, we decided to explore other pharmacophores that irreversibly inactivate cysteine proteases according to mechanisms that do not utilize a protonation step.
Scheme 4.

Synthesis of β-Chloro Vinyl Sulfone Inhibitor 43a
a Reagents: (a) diazomethane, THF, rt; (b) methylphenylsulfone, n-BuLi, THF, 0 °C to −78 °C; (c) Tf2O, i-Pr2EtN, THF, −20 °C to rt; (d) TBACl, THF, rt; (e) Na ascorbate, CuSO4, 1:1 H2O:t-BuOH, 39, rt.
Table 4.
Second-Order Inactivation Rates of Cruzain Inhibitors with Varying Pharmacophores
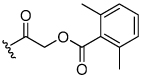
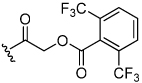
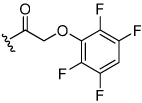
Acyloxymethyl ketone inhibitors were designed by Krantz as more stable halomethyl ketone analogues.21,22 This pharmacophore has led to potent time-dependent inhibitors of cathepsins B, L, and S.19 Hence, we next incorporated the acyloxymethyl ketone pharmacophore. The synthesis of the acyloxymethyl ketone inhibitors required the preparation of azide intermediates 47a and 47b (Scheme 5). Beginning with L-norleucine azido acid 40, the common bromomethyl ketone precursor 48 was obtained in three steps. Displacement of the bromide followed by cyclization with propargyl amine 39 then afforded the acyloxymethyl ketone inhibitors 49 and 50 as mixtures of diastereomers.
Scheme 5.

Synthesis of Aryl- and Acyl-oxymethyl Ketone Inhibitorsa
a Reagents: (a) isobutyl chloroformate, N-methylmorpholine, THF, −40 °C; (b) diazomethane, THF, 0 °C; (c) HBr, THF, 0 °C; (d) KF, DMF, 0 °C; (e) Na ascorbate, CuSO4, 1:1 H2O:t-BuOH, rt.
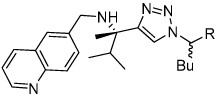
We initially investigated the 2,6-dimethyl acyloxymethyl ketone inhibitor 49 and observed that it was an irreversible inhibitor of cruzain with a second-order rate constant of 2,690 s−1M−1 (Table 4). Acyloxymethyl ketone inhibitors of the cathepsins have shown a strong correlation between the leaving group pKa and the rate of inactivation.23 Accordingly, we prepared the 2,6-bis-trifluoromethyl acyloxymethyl ketone inhibitor 50 and were delighted to find that inhibitor 50 was 58-fold more potent than inhibitor 49 with a second-order rate constant of 157,000 s−1M−1 (Table 4). Inhibitors incorporating this pharmacophore were subsequently prepared corresponding to both the amides and amines of the benzothiazole and quinoline substrates (Scheme 5). There was good correlation between substrate activity and inhibitor potency with the amine inhibitors 50 and 51 being more potent than the amide inhibitors 52 and 53 (Table 5).
Table 5.
Second-Order Inactivation Rates of 1,2,3-Triazole Cruzain Inhibitorsa
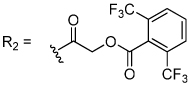
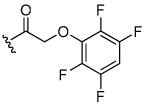
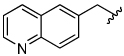
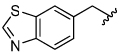
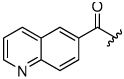
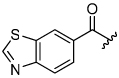
The aryloxymethyl ketone pharmacophore has the same mechanism of inhibition as the acyloxymethyl ketone pharmacophore. It is more attractive, however, because it should be less prone to nucleophilic attack, cannot undergo hydrolysis, and has a lower molecular weight. This pharmacophore has proven to be particularly effective for caspase inhibition.25,26 In particular, Idun pharmaceuticals used 2,3,5,6-tetrafluorophenol as the leaving group in an aryloxymethyl ketone pan-caspase inhibitor that has progressed to Phase II clinical trials.27 In contrast, there has only been one report of aryloxymethyl ketone inhibitors of a member of the papain superfamily, and only modest inhibition was observed.21 Nevertheless, we prepared 2,3,5,6-tetrafluorophenol aryloxymethyl ketone inhibitor 54 (Scheme 5). Unexpectedly, the aryloxymethyl ketone inhibitor 54 was equipotent to the acyloxymethyl ketone inhibitors with a second-order inactivation constant of 147,000 s−1M−1 despite the >105 difference in the acidity of the corresponding leaving groups (pKa of 2,3,5,6-tetrafluorophenol = >5.5, pKa of 2,6-bis-trifluoromethyl benzoic acid = 0.58) (Table 4). Inhibitor potency is clearly not solely dependent on the pKa values of the leaving group with leaving group binding and/or orientation also playing a significant role.25
The preparation of both the acyl- and aryl-oxymethyl ketone inhibitors resulted in epimerization alpha to the pharmacophore carbonyl. To investigate the configurational stability of the inhibitors in vivo, an alternative sequence for the preparation of diastereomerically pure aryloxymethyl ketone inhibitor 54 was developed (Scheme 6). Specifically, racemization through enolization was prevented by reducing the acyloxymethyl ketone 47c prior to the cycloaddition step. Alcohol 57 was then oxidized back to the acyloxymethyl ketone. The diastereomerically pure inhibitor 58 was then subjected to the assay buffer conditions at 37 °C for several hours (Scheme 7). This resulted in complete racemization of the inhibitor, suggesting that in vivo the inhibitor will be able to funnel through the active diastereomer.
Scheme 6.

Synthesis of Diastereomerically Pure Aryloxymethyl Ketone Inhibitor 58a
a Reagents: (a) NaBH4, 95:5 THF:H2O, 0 °C to rt; (b) Na ascorbate, CuSO4, 1:1 H2O:t-BuOH, rt; (c) Dess-Martin periodonane, CH2Cl2, rt.
Scheme 7.

Configurational Lability of Aryloxymethyl Ketone Inhibitor 54
Cell Culture Assays
Acyloxymethyl ketone inhibitors 50–53 and aryloxymethyl ketone inhibitors 54–55 were tested for their effectiveness in eliminating T. cruzi infection in irradiated (9000 rad) J744 macrophages. Host cells died of T. cruzi infection after 5 days without treatment (Table 6). Notably, all of the inhibitors significantly delayed T. cruzi intracellular replication at concentrations of 5 or 10 µM. The T. cruzi infected cells were initially treated with all of the inhibitors at 10 µM concentrations. The cultures treated with each different inhibitor were compared daily by contrast phase microscopy to uninfected macrophage controls. Inhibitors 50, 51, 52, and 55 showed toxicity as evidenced by cells that rounded up or detached from the wells, condensed, died, or became granular. For this reason, the concentration of inhibitors 50, 51, 52, and 55 was lowered to 5 µM. The 2,6-bis-trifluoromethyl acyloxymethyl ketone inhibitors 50, 51, and 52 remained toxic at 5 µM. As a result, it was necessary to stop treatment by day 14 after which point the cells died. Acyloxymethyl ketone inhibitor 53 was quite effective at 10 µM in delaying T. cruzi replication, however, by day 23 the cell monolayer had been destroyed by the infection.
Table 6.
Effect of inhibitors on survival of T. cruzi infected J744 macrophages
| cmpd | survival of T. cruzi infected cells (days)a | last day of treatment (day) | type of inhibition | cell toxicitye |
|---|---|---|---|---|
| none | 5 | − | − | − |
| 1 | >40 | 27c | trypanocidal | nontoxic |
| 50b | 30 | 14d | trypanostatic | toxic |
| 51b | 26 | 14d | trypanostatic | toxic |
| 52b | 19 | 14d | trypanostatic | toxic |
| 53 | 23 | 23 | trypanostatic | nontoxic |
| 54 | >40 | 27c | trypanocidal | nontoxic |
| 55b | 33 | 27c | trypanostatic | nontoxic |
Effect of inhibitors on survival of J744 macrophages infected with T. cruzi trypomastigotes, treated daily with a solution of inhibitor (10 µM). Survival time is defined as the time before the cell monolayer is destroyed by the infection.4
The concentration was lowered to 5 µM after 9 (50), 7 (51), and 12 (52, 55) days.
Treatment was ended to distinguish between trypanostatic and trypanocidal inhibitors.
Treatment was ended due to compound toxicity.
Determined by contrast phase microscopy comparison to uninfected macrophage controls.
To distinguish between trypanostatic compounds that only delay parasite replication and trypanocidal compounds that effectively kill T. cruzi, treatment for the remaining aryloxymethyl ketone inhibitors was ended on day 27 and the cells were monitored for two more weeks. Benzothiazole aryloxymethyl ketone inhibitor 55 proved to be trypanostatic at 5 µM against T. cruzi because parasites destroyed the cell monolayer by day 33 (6 days after ending the treatment). Most significantly, the quinoline aryloxymethyl ketone inhibitor 54 was trypanocidal at 10 µM and had completely eradicated the T. cruzi parasite with no parasites observed at day 40 post-infection. The performance of inhibitor 54 was comparable to vinyl sulfone 1, which is the most advanced inhibitor of cruzain to date.
Plasma stability
Due to the excellent cell culture activity of inhibitor 54, mouse plasma stability studies were performed by ADMETRx as a prelude to the inhibitor’s evaluation in animal models of the Chagas’ disease. The inhibitor was incubated at 37°C in mouse plasma for 0, 5, 10, 30, or 60 minutes and analyzed by LC/MS for remaining inhibitor. Incubation of the inhibitor with mouse plasma showed no disappearance of the inhibitor with time, indicating it to be 100% stable to mouse plasma under these conditions.
Conclusion
A potent irreversible 2,3,5,6-tetrafluorophenoxymethyl ketone inhibitor 54 was developed that completely eradicates T. cruzi parasites in cell culture. A substrate library containing more than 150 triazole-based substrates developed for cathepsin S, was first evaluated against cruzain to define important structural features for efficient substrate cleavage. Subsequent optimization of the substrate scaffold in the S3 pocket was guided by structure-based design and led to nonpeptidic substrates with even greater cleavage efficiency.
Vinyl sulfone, β-chloro vinyl sulfone, acyl- and aryloxymethyl ketone pharmacophores were then explored in the conversion of the most efficient substrates to inhibitors. The β-chloro vinyl sulfone pharmacophore, which had not previously been reported, led to key mechanistic insight and ultimately resulted in the development of potent irreversible aryloxymethyl ketone inhibitors, a pharmacophore class that had previously been little explored against the papain superfamily.
Evaluation of 2,3,5,6-tetrafluorophenoxymethyl ketone inhibitor 54 in animal models of the disease is currently underway. The nonpeptidic nature of this potent class of inhibitors, coupled with their potent cell-based activity and plasma stability, makes these compounds very promising starting points for the development of chemotherapy for Chagas’ disease.
Supplementary Material
Acknowledgment
J.A.E. acknowledges support from the NIH (GM54051) and J.H.M. acknowledges support from the NIH (AI35707) and the Sandler Family Supporting Foundation.
Footnotes
Supporting Information Available: Complete experimental details, including cell culture assay and plasma stability procedures, spectral data for all compounds, and full author list for ref 27. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
James H. McKerrow, Email: jmck@cgl.ucsf.edu.
Jonathan A. Ellman, Email: jellman@uclink.berkeley.edu.
References
- 1. www.who.int/tdr/diseases/chagas/direction.htm.
- 2.de Castro SL. Acta tropica. 1993;53:83–98. doi: 10.1016/0001-706x(93)90021-3. [DOI] [PubMed] [Google Scholar]
- 3.Harth G, Andrews N, Mills AA, Engel JC, Smith R, McKerrow JH. Mol. Biochem. Parasitol. 1993;58:17–24. doi: 10.1016/0166-6851(93)90086-d. [DOI] [PubMed] [Google Scholar]
- 4.Engel JC, Doyle PS, Hsieh I, McKerrow JH. J. Exp. Med. 1998;188:725–734. doi: 10.1084/jem.188.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barr SC, Warner KL, Kornreic BG, Piscitelli J, Wolfe A, Benet L, McKerrow JH. Antimicrob. Agents Chemother. 2005;49:5160–5161. doi: 10.1128/AAC.49.12.5160-5161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKerrow JH, Engel JC, Caffrey CR. Biorg. Med. Chem. 1999;7:639–644. doi: 10.1016/s0968-0896(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 7.McKerrow JH, McGrath ME, Engel JC. Parasitology Today. 1995;11:279–282. doi: 10.1016/0169-4758(95)80039-5. [DOI] [PubMed] [Google Scholar]
- 8.Jacobsen W, Christians U, Benet LZ. Drug Metab. Dispos. 2000;28:1343–1351. [PubMed] [Google Scholar]
- 9.For the only other report of nonpeptidic cruzain inhibitors see: Du X, Guo C, Hansell E, Doyle PS, Caffrey CR, Holler TP, McKerrow JH, Cohen FE. J. Med. Chem. 2002;45:2695–2707. doi: 10.1021/jm010459j.
- 10.Wood WJL, Patterson AW, Tsuruoka H, Jain RK, Ellman JA. J. Am. Chem. Soc. 2005;127:15521–15527. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]
- 11.Salisbury CM, Ellman JA. Chem Bio Chem. 2006;7:1034–1037. doi: 10.1002/cbic.200600081. [DOI] [PubMed] [Google Scholar]
- 12.Patterson AW, Wood WJL, Hornsby M, Lesley S, Spraggon G, Ellman JA. J. Med. Chem. 2006;49:6298–6307. doi: 10.1021/jm060701s. [DOI] [PubMed] [Google Scholar]
- 13.Inagaki H, Tsuruoka H, Hornsby M, Lesley SA, Spraggon G, Ellman JA. J. Med. Chem. 2007;50:2693–2699. doi: 10.1021/jm070111+. [DOI] [PubMed] [Google Scholar]
- 14.Soellner MB, Rawls KA, Grundner C, Alber T, Ellman JA. J. Am. Chem. Soc. 2007;129:9613–9615. doi: 10.1021/ja0727520. [DOI] [PubMed] [Google Scholar]
- 15.Bromme D, McGrath ME. Protein Science. 1996;5:789–791. doi: 10.1002/pro.5560050426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer JT, Rasnick D, Klaus JL, Bromme D. J. Med. Chem. 1995;38:3193–3196. doi: 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- 17.Roush WR, Gwaltney SL, II, Cheng J, Scheidt KA, McKerrow JH, Hansell E. J. Am. Chem. Soc. 1998;120:10994–10995. [Google Scholar]
- 18.For an example of vinyl sulfone inhibitors of cruzain with reversible nature see: Scheidt KA, Roush WR, McKerrow JH, Selzer PM, Hansell E, Rosenthal PJ. Biorg. Med. Chem. 1998;6:2477–2494. doi: 10.1016/s0968-0896(98)80022-9.
- 19.Powers JC, Asgian JL, Ekici OD, James KE. Chem. Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 20.Govardhan CP, Abeles RH. Arch. Biochem. Biophys. 1996;330:110–114. doi: 10.1006/abbi.1996.0231. [DOI] [PubMed] [Google Scholar]
- 21.Smith RA, Copp LJ, Coles PJ, Pauls HW, Robinson VJ, Spencer RW, Heard SB, Krantz A. J. Am. Chem. Soc. 1988;110:4429–4431. [Google Scholar]
- 22.Krantz A, Copp LJ, Coles PJ, Smith RA, Heard SB. Biochemistry. 1991;30:4678–4687. doi: 10.1021/bi00233a007. [DOI] [PubMed] [Google Scholar]
- 23.Krantz A. Methods Enzymol. 1994;244:656–671. doi: 10.1016/0076-6879(94)44049-2. [DOI] [PubMed] [Google Scholar]
- 24.Bieth JG. Methods Enzymol. 1995;248:59–84. doi: 10.1016/0076-6879(95)48007-2. [DOI] [PubMed] [Google Scholar]
- 25.For an analysis of the rate of inactivation dependence on leaving group orientation for caspases see: Brady KD, Giegel DA, Grinnell C, Lunney E, Talanian RV, Wong W, Walker N. Biorg. Med. Chem. 1999;7:621–631. doi: 10.1016/s0968-0896(99)00009-7.
- 26.Brady KD. Biochemistry. 1998;37:8508–8515. doi: 10.1021/bi9803325. [DOI] [PubMed] [Google Scholar]
- 27.Linton SD, et al. J. Med. Chem. 2005;48:6779–6782. doi: 10.1021/jm050307e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.