

Abstract
Thin-layer cell spectroelectrochemistry, featuring rigorous potential control and rapid redox equilibration within the cell, was used to measure the redox potential Em(Phe a/Phe a−) of pheophytin (Phe) a, the primary electron acceptor in an oxygen-evolving photosystem (PS) II core complex from a thermophilic cyanobacterium Thermosynechococcus elongatus. Interferences from dissolved O2 and water reductions were minimized by airtight sealing of the sample cell added with dithionite and mercury plating on the gold minigrid working electrode surface, respectively. The result obtained at a physiological pH of 6.5 was Em(Phe a/Phe a−) = −505 ± 6 mV vs. SHE, which is by ≈100 mV more positive than the values measured ≈30 years ago at nonphysiological pH and widely accepted thereafter in the field of photosynthesis research. Using the P680* − Phe a free energy difference, as estimated from kinetic analyses by previous authors, the present result would locate the Em(P680/P680+) value, which is one of the key parameters but still resists direct measurements, at approximately +1,210 mV. In view of these pieces of information, a renewed diagram is proposed for the energetics in PS II.
Keywords: charge separation, photosynthesis, spectroelectrochemistry, water oxidation
In the photosynthetic primary process of higher plants, algae, and cyanobacteria, photosystem (PS) I and PS II cooperate in series to convert photon energy into chemical energy through light-induced charge separation and subsequent electron transfers. PS II appears to catalyze water oxidation at a pentanuclear Mn4Ca cluster that accumulates oxidizing equivalents (see recent reviews in refs. 1–5). It is generally supposed that the water oxidation is triggered by charge separation between the primary electron donor, P680, and the primary electron acceptor, pheophytin (Phe) a. The initial radical pair P680+ Phe a− formed by the charge separation, drives forward electron transfer from Phe a− to the first plastoquinone QA, and hole transfer from P680+ to the Mn4Ca cluster through a redox-active tyrosine residue denoted YZ, thus preventing charge recombination. Recent X-ray crystallography clarified the arrangement of protein subunits and cofactors in PS II with 2.9–3.7-Å resolution (6–9), visualizing the electron transfer pathway.
To date, the nature of P680 remains controversial. Available experimental data and theoretical analyses suggest that P680 is assignable to the pigment cluster of the four chlorophyll a molecules (denoted PD1, PD2, ChlD1, and ChlD2) in the PS II reaction center (1, 5). Further uncertainty surrounds the value of the redox potential Em(P680/P680+). Although the Em(P680/P680+) value is an essential parameter to draw a whole picture of the PS II energetics, its direct measurement has never been attained (10) because water, present as dominant (solvent) molecules in most sample solutions, is oxidized first heavily and tends to mask the subsequent oxidation of the target entity P680 at a higher potential. The Em(P680/P680+) value has hence been estimated from the potentials of PS II electron acceptors (10–12) or assessed by computation (13–17).
In the former case, the Phe a redox potential Em(Phe a/Phe a−), together with the P680* − Phe a free energy difference, is a good clue to estimate Em(P680/P680+). An attempt in this direction was made in 1979 by Klimov et al. (11), who had detected Phe a as the primary electron acceptor in PS II membranes from pea (18). They obtained −610 ± 30 mV as Em(Phe a/Phe a−) and, using the P680* − Phe a energy difference of ≈80 meV from delayed fluorescence measurements (19), estimated the Em(P680/P680+) value to be + 1,120 ± 50 mV (11). In 1981 Rutherford et al. (20) indirectly assessed the Em(Phe a/Phe a−) value from an EPR study of the P680 triplet states, to obtain −604 mV, being close to that by Klimov et al. For almost three decades thereafter, the values of approximately −600 mV for the Em(Phe a/Phe a−) and approximately +1.1 V for Em(P680/P680+) have been widely accepted in the discussions on PS II energetics, with no further attempts to measure Em(Phe a/Phe a−).
Recently Rappaport et al. (12) disputed the validity of the Em(Phe a/Phe a−) value, on the ground that the Phe a − QA potential difference did not fit their kinetic analytical data. A value of −80 mV or −30 mV in the presence of DCMU (an inhibitor blocking electron transfer from QA to the second plastoquinone QB) has been assumed to be reasonable for Em(QA/QA−) since the thorough studies of Krieger et al. (21–23), giving the Phe a − QA potential difference of ≈600 meV, although scattered Em(QA/QA−) values in three ranges, approximately −250, 0, and 125 mV, were reported (reviewed in ref. 21). However, the fluorescence decay data by Rappaport et al. on the charge recombination of QA− at the S2 state in the presence of DCMU in Synechocystis PCC 6803 mutants PS II indicated that the Phe a − QA potential difference should be smaller by ≈250 mV. They then estimated the Em(P680/P680+) value to be +1.26 V (12) on the basis of Em(QA/QA−) and kinetic analytical data in the presence of DCMU. Later, Grabolle and Dau (10) analyzed the delayed and prompt Chl fluorescence of PS II membranes in the absence of inhibitor and assessed the Em(P680/P680+) value to be +1.25 V, by citing Em(QA/QA−) to be −80 mV. Since then, the value of approximately +1.25 V is generally accepted for Em(P680/P680+) (1–5) and is used as a reference in computational chemistry (13–16).
In view of these finding, a renewed attempt to determine the Em(Phe a/Phe a−) value would be of much significance. Actually, a number of questions have been raised (ref. 12; see also Discussion below) as to the reliability of the Em(Phe a/Phe a−) measurements reported only in the 1978–1981 period (11, 20), carried out with a chemical titration mode at nonphysiological pH values to render the H2O/H2 equilibrium potential negative enough (24, 25): Klimov et al. performed the measurement while changing the pH of a solution from 8 to 11 (11) and Rutherford et al. adjusted the pH at 11 through the titration (20).
Spectroelectrochemistry is a powerful means to probe into the redox behavior of biological molecules (26) because it (i) requires only a small amount of sample, (ii) allows for strict potential control, and (iii) facilitates rapid redox equilibration within a sample cell. These features enabled us to determine the redox potential Em of P700 (primary donor in PS I) and other components to within ±2 mV (27–31) and to unveil the species dependence of the Em value of P700 (28, 29). This has been extended in the present work to measure the Em(Phe a/Phe a−) value in PS II complexes at physiological pH, by overcoming several difficulties inherent in the strongly negative potential range. Use of electrode material exhibiting a large hydrogen overpotential, of which mercury is a typical example, has a possibility to poise solution potential at values more negative than possible by titration with chemical reductants (24, 25). The Em(Phe a/Phe a−) value thus obtained is compared with the previous value and kinetic analytical data, and the energetics for PS II is discussed in the light of the renewed Em(Phe a/Phe a−) value.
Results
In this work, we used PS II core complexes from a thermophilic cyanobacterium Thermosynechococcus elongatus WT* (32), where D1 is encoded by only psbA3. The T. elongatus genome possesses three variant copies of D1 genes (psbA1, psbA2, and psbA3), and a typical difference among them lies at residue 130, which is within an H-bonding distance of the 9-keto carbonyl of Phe a in the D1 branch; the residue 130 is Gln in D1:1, whereas it is Glu in D1:2 and D1:3. Because D1-130 in higher plants as well as D1:3 in T. elongatus is Glu, the PS II complexes from WT* should be appropriate for comparison of Em(Phe a/Phe a−) to be obtained with that in previous reports on higher plants.
Continuous illumination to a solution containing PS II core complexes under reductive conditions, where QA is in the reduced form, induces charge separation causing buildup (photoaccumulation) of Phe a−, enabling one to observe the Phe a/Phe a− redox reaction via absorption spectrum or EPR signal changes (33). Such a reductive condition was usually achieved by adding sodium dithionite into a sample solution under anaerobic conditions (18, 33, 34), leading to the solution's equilibrium potential of −360 to −480 mV at pH 6–8.
In spectroelectrochemistry, we observed light-induced difference absorption spectra of the PS II core complexes at pH 6.5 in an airtight optically transparent thin-layer elctrode (OTTLE) cell equipped with a mercury-electroplated Au mesh as a working electrode. A typical result is depicted in Fig. 1, where the potential of the working electrode was set at −350 mV or −500 mV. The spectral shape and characteristic peaks at 428, 450, 517, and 547 nm are practically the same as those previously reported for PS II complexes (18, 33–36). The two bleaches at 517 and 547 nm arise from the Qx bands of Phe a, the bleach at 428 nm is the Soret band (18, 33), and the 450 nm peak is due to the Phe a− absorption (33, 37). Our result hence represents the light-induced difference spectra due to the redox reaction of photoactive Phe a (38–42), which is in the D1 branch and denoted Phe aD1, in the PS II core complexes. The spectral intensity at −500 mV is approximately half that at −350 mV. This demonstrates that approximately half of the Phe a was reduced electrochemically before illumination and therefore did not participate in the charge separation. When the potential was returned to −350 mV after a measurement at −500 mV, the light-induced difference spectrum was almost identical with the counterpart shown in Fig. 1A; this ensures that the redox reaction of Phe aD1 is fully reversible.
Fig. 1.

Light-minus-dark difference spectra demonstrating the photoaccumulation of Phe a− in the PS II complexes in the OTTLE cell, where the potential of the working electrode was set at −350 mV (solid curve) and −500 mV (dashed curve).
Fig. 2 gives the time courses of absorbance change at 450 nm during and after continuous illumination (photoaccumulation of Phe a−) at a series of negative potentials applied to the working electrode. During illumination, the absorbance increased and approached a steady-state value in 200–300 s, although a gradual decrease from the plateau is observed at lower potentials, typically at −525 mV. After turning off the light, the absorbance generally tended the initial baseline. The gradual decrease during illumination and partial irreversibility after illumination might be due to some irreversible photoreactions of peripheral chlorophylls or carotenes. However, the amplitude of absorbance change at −350 mV was almost identical before and after the potential steps, and this strongly suggests that the redox reaction of Phe aD1 was reversible. The amplitude of absorbance change at −350 mV was also the same as that at −300 mV, indicating that Phe aD1 was electroneutral at −350 mV.
Fig. 2.

Time courses of absorbance change at 450 nm ΔA450 during photoaccumulation of Phe a− in the PS II complexes at a series of electrode potentials in the OTTLE cell.
The amplitudes of absorbance change at 450 nm at a series of electrode potentials relative to that at −350 mV were used to construct two types of Nernstian plots as shown in Fig. 3 A and B. A least-squares fit of the data in Fig. 3A gave a slope of 55 mV per decade, being sufficiently close to 59 mV per decade expected for a reversible one-electron redox process. The Em (Phe a/Phe a−) value, corresponding to the intercept on the ordinate, is seen to be −502 mV at pH 6.5. A titration-type display given in Fig. 3B is also in line with a one-electron redox reaction at Em = −502 mV; little hysteresis between the reductive and oxidative directions supports the reversibility of the Phe a redox reaction. By summarizing four independent measurements (Fig. S1), we conclude that the Em(Phe a/Phe a−) value is −505 mV with a standard deviation of 6 mV (Fig. S2). We also performed spectroelectrochemical measurements of Em(Phe a/Phe a−) at pH 7.5 (Fig. S3), and the average value of two independent measurements was −497 mV (Fig. S4).
Fig. 3.

Nernstian plots for the redox reaction of Phe a in PS II complexes based on ΔA450 values. The dashed line in A drawn by a least-squares fit to the plots gives a slope of 55 mV per decade. The curve in B denotes a theoretical one for one-electron redox process with Em = −502 mV.
Discussion
By use of a mercury-electroplated Au mesh electrode on which water reduction (hydrogen evolution) is minimized and the use of dithionite-containing sample solution in an airtight OTTLE cell with minimal disturbance from dissolved O2 reduction, our spectroelectrochemical measurement has yielded a value of −505 ± 6 mV at a physiological pH of 6.5 for the photoactive Phe aD1 in the PS II complexes prepared from T. elongatus WT*. The standard deviation of 6 mV for the determination of Em(Phe a/Phe a−) seems to be rather large (seen in Fig. S2), especially compared with our previous measurements on redox species in PS I and II yielding values within an error range of ±2 mV (27–31); this might be partially due to an interference from irreversible photoreactions of peripheral species during photoaccumulation of Phe a−. The Em(Phe a/Phe a−) determined in the present work is by as much as ≈100 mV positive of those reported in 1978–1981, namely −610 ± 30 mV by Klimov et al. (11) and −604 mV by Rutherford et al. (20) from measurements in nonphysiological aqueous media (pH 8–11).
The origin for the discrepancy may be sought in (i) the pH dependence of Em(Phe a/Phe a−) and/or (ii) possible denaturation of PS II complexes at nonphysiological pH. A pH dependence of the Em(Phe a/Phe a−) on a Hg electrode in dimethylformamide was noted by Kazakova et al. (43): by acidification the Em(Phe a/Phe a−) value tended to shift positively by 30 mV/pH and 60 mV/pH in the pH regions 9.0–10.0 and 6.0–9.0, respectively. Because the redox potential of Chl a does not depend on pH in aqueous media (44), the pH dependence of the Em value may be characteristic of Phe a. In addition to the pH dependence itself, an electrostatic influence from amino acid residues around Phe a in the PS II complex should be taken into account for the Em(Phe a/Phe a−) value. Our results of Em(Phe a/a−) at pH 6.5 and 7.5 revealed, however, that Em(Phe a/Phe a−) is almost independent of pH in this physiological pH range, suggesting that one cannot simply assume a pH dependence of Em(Phe a/Phe a−) in the nonphysiological high pH region. In any event some denaturation of PS II complexes at pH 10.0–11.0 is inevitable; the oxygen-evolving activity has an optimum pH ≈6, and is lost at pHs higher than 9 (36, 45). These factors may have shifted the previously measured values from the actual one. Furthermore, the Em(Phe a/Phe a−) value at a physiological pH should be used in considering the energetics within PS II.
It should be noted that the Em(Phe a/Phe a−) value was measured under the reductive condition by controlling the working electrode's potential in sample solutions added with dithionite in this work. Such a highly reductive condition would release the Mn cluster, impairing oxygen evolution activity and thus may shift Em(Phe a/Phe a−); indeed, it was found that the loss of oxygen evolution activity shifts Em(QA/QA−) positively by 150–190 mV (21, 22). However, we confirmed that the PS II complexes prepared from T. elongatus WT* strain can keep oxygen evolution activity entirely even after incubation in the dark in the presence of dithionite, and it can still keep as much as 70% of the activity under illumination for 15 min in the presence of dithionite, which simulated the first measurement of the Phe a− photoaccumulation. Although the oxygen evolution activity would decline gradually during the spectroelectrochemical measurements, if Em(Phe a/Phe a−) shifts by the lack of the oxygen evolution activity, the Nernstian plots (Fig. 3) would not present a single one-electron redox process at a redox potential. Therefore, the Em(Phe a/Phe a−) value might not be different so much as the case seen in Em(QA/QA−) from the actual one in fully intact PS II complexes. This also implies that adjusting potentials for photoprotection in the case of oxygen evolution activity loss should perform on Em(QA/QA−) only, inducing direct charge recombination from QA− to P680+ (22).
Based on the Em(Phe a/Phe a−) value determined in this work and kinetic analytical data in the literature, the energetic relationships among the electron transfer components in the PS II complexes can be portrayed as follows. From the nature of charge separation between P680* to Phe a, one can indirectly estimate the Em(P680/P680+) value, which still resists direct measurement as mentioned above. Although Klimov et al. (ref. 11; see also Introduction) estimated Em(P680/P680+) simply from the Em(Phe a/Phe a−) value and the free energy difference, according to the Weller's equation (46), which describes an energetic relationship between an electron donor and an acceptor, the free energy correlation surrounding the P680* → Phe a electron transfer should obey the following formula:
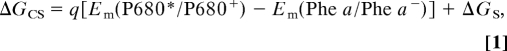 |
where q is the elementary charge, ΔGCS is the free energy difference for the charge separation, and ΔGS is the stabilization energy induced by the separated ion pair formation. In the present context, ΔGCS denotes the energy difference between [P680* Phe a] and [P680+ Phe a−], and have been scrutinized often by time-resolved fluorescence spectroscopy (47–49). Values of approximately −150 meV for ΔGCS in PS II complexes were collected from earlier fluorescence decay kinetic analytical data (47). A similar value of −165 meV was reported from time-resolved photovoltage measurements on unstacked PS II membrane fragments and data analysis in terms of the exciton ion-pair equilibrium model (50). ΔGS is generally referred to as a static Coulombic interaction calculated as −q2/(4πεr), where ε is the permittivity, and r is the ionic center-to-center distance: The interaction energy between P680 and Phe a ranges from −45 to −225 mV for εr (relative permittivity) of 20 to 4, respectively, by assuming that the cation is located predominantly on the D1 side of the special pair (51). Considering an entropic equilibration at an excited state between P680 and antenna Chls (AnChl) and charge separation driven by the equilibrated [AnChl P680]*; however, ΔGS should be within the difference between the equilibrated state and the charge-separated state [P680+ Phe a−]; otherwise the electron transfer step from [AnChl P680]* to Phe a accompanying the free energy change between the energy level of [AnChl P680]* and qEm(Phe a/Phe a−) becomes an uphill reaction. Consequently, ΔGS may be within 0 to −40 meV because the energy difference of [AnChl P680]* and [P680+Phe a−] was estimated to be −40 meV (50) or −50 meV (47).
The free energy difference between [P680+ Phe a− QA] and [P680+ Phe a QA−] is calculated to be −380 to −420 meV (depending on ΔGS) by subtracting a currently accepted value of −80 mV for Em(QA/QA−) (21) from the present result, in considering an electron transfer from Phe a− to QA. This energy difference seems to agree with the reported values based on the charge recombination data, −310 meV by Rappaport et al. (ref. 12: −360 meV added with the DCMU's effect of +50 mV) and −340 meV by Grabolle and Dau (10), more reasonably than by using a value of approximately −600 meV calculated with the Em(Phe a/Phe a−) value reported by Klimov et al. However, by considering an electrostatic interaction between Phe a and anionic QA−, which may be present during the measurements of Em(Phe a/Phe a−), Rappaport et al. claimed that the true value of Em(Phe a/Phe a−) must be more positive than the value of Klimov et al., and the experimental ambiguity must be one of the reasons for the discrepancy in Em(Phe a/Phe a−) (12). This electrostatic effect exerted by QA− was also confirmed by computational chemistry, and was calculated to be ≈90 meV (17). Although these claims might also hold for our result, Gibasiewicz et al. reported on the electrostatic effects of the redox states of QA on the energy difference ΔGCS by time-resolved photovoltage measurements (50): Indeed the anionic QA− (singly reduced QA) generate a large increase (90 meV) in ΔGCS, whereas the effect of QAH2 (doubly reduced QA) on ΔGCS is similar (at most 4 meV) to that of QA at the neutral state. Because experimental conditions for the double reduction of QA (50, 52–55) are similar to that in our spectroelectrochemical measurement for Phe a− photoaccumulation (see Materials and Methods), our result may have not been affected much by the electrostatic problems.
Fig. 4 summarizes the energetics within PS II based on the Em(Phe a/Phe a−) value obtained in this work and the relationships among the electron transfer components from kinetic analyses. The Em(P680/P680+) value was estimated to be less positive than +1,210 mV. The Em(Phe a/Phe a−) value for the PS II complexes from T. elongatus WT*, where only the psbA3 gene expressed, was −505 mV, whereas a site-directed mutagenesis study with Synechocystis PCC 6803 (56) showed that substitution of Glu at position 130 of D1 with Gln decreased the free energy difference ΔGCS, corresponding to a shift in the Em(Phe a/Phe a−) value by +33 mV. Such a mutational effect on the energetics was also confirmed by thermoluminescence measurements (57). However, a thermoluminescence study performed on PS II complexes from T. elongatus WT* and WT' (psbA1 dominantly expressed) did not show a large difference, suggesting that the substitution of residue 130 has negligible effect in T. elongatus when compared with the situation in Synechocystis PCC 6803 (32). In any event, kinetic analyses such as thermoluminescence and delayed fluorescence provide only an estimate of the energy difference between Phe a and P680 or Phe a and QA, whereas a direct measurement of the Em(Phe a/Phe a−) value by spectroelectrochemistry would reveal not only mutational effects but also species dependence more clearly. Because the image in Fig. 4 is a compilation of results from cyanobacteria and higher plants, more systematic analyses on a species and probing into differences among species are still required to draw a detailed and final conclusion on the energetics within PS II.
Fig. 4.

Energetics for electron transfer components in PS II based on the redox potentials. The gray boxes indicate energy level uncertainties depending on ΔGS. The values of ΔGCS and ΔGS derive from the literature (47, 48) and the value of Em(QA/QA−) was cited from ref. 21.
Materials and Methods
Purification of PS II Core Complexes.
Oxygen-evolving PS II core complexes were purified from T. elongatus WT* (32) in which the psbA1 and psbA2 genes encoding D1 have been deleted from genome of 43H strain (58) using Ni2+-affinity column chromatography as described elsewhere. The eluted out PS II core complexes were concentrated by use of an Amicon Ultra-15 concentrator device (Millipore). The PS II core complexes were stored until use in liquid nitrogen in a medium containing 40 mM Mes-NaOH (pH 6.5), 15 mM CaCl2, 15 mM MgCl2, 10% glycerol, 0.03% dodecyl-β-D-maltoside (DM) and 1 M glycine-betaine. Water oxidation function of the PS II core complexes were active as ≈5,000 μmol of O2 mg Chl−1 h−1.
Spectroelectrochemistry of Phe a/Phe a−.
An airtight OTTLE cell, being an adapted version of the basic design reported by Hawkridge and Ke (24), was used for the spectroelectrochemical measurements. The optical path length was ≈300 μm, and the effective cell volume was 500 μL. A gold minigrid (100 mesh per inch) electrode, rinsed by ultrasonification in dilute HNO3 and then in ultrapure water, was electroplated cathodically with mercury by applying a potential of approximately −1.0 V in a solution of saturated Hg(NO3)2 by using a potentiostat until the entire electrode surface turned to shiny gray (59). The Hg–Au mesh served as a working electrode, a Pt black wire as a counter electrode, and an Ag–AgCl (in saturated KCl) as a reference electrode in the thin-layer cell. In the text body, the electrode potential is referred to a standard hydrogen electrode (SHE) (+199 mV vs. Ag–AgCl).
In spectroelectrochemical measurements, the PS II core complexes were suspended at a Chl a concentration of 0.6 mM, corresponding to ≈15 μM Phe aD1, in a buffer containing 50 mM Mes-NaOH (pH 6.5), 0.2 M KCl, 0.2% dodecyl-β-D-maltoside (DM), 1 M glycine-betaine, and the following redox mediators: 500 μM anthraquinone (Em = −225 mV), 500 μM methyl viologen (Em = −443 mV), and 1,1′-propylene-2,2′-bypyridylium (Triquat, Em = −556 mV). For spectroelectrochemical measurements at pH 7.5, the buffer was exchanged with Hepes-NaOH (pH 7.5) instead of Mes-NaOH at a final concentration of 50 mM using Microcon-100 (Amicon). The PS II sample solution was, after addition of 5 mg·mL−1 sodium dithionite, transferred to the spectroelectrochemical cell filled with Ar. The buffering capacity of 50 mM Mes-NaOH (pH 6.5) or Hepes-NaOH (pH 7.5) is sufficient to keep an intended pH even after addition of sodium dithionite, which was also confirmed by an open-circuit potential of approximately −380 mV and −440 mV in the case of using Mes and Hepes, respectively, in the OTTLE cell.
Light-induced difference absorption spectra of Phe a/Phe a− were measured by using a dual-wavelength spectrophotometer Model V670 (JASCO) modified for lateral illumination (60). The electrode potential was controlled with a potentiostat Model 2020 (Toho Technical Research). Photoreduction of Phe a was induced by red actinic light at an intensity of 40 μE·m−2·s−1 from a 500-W Xe lamp (UXL 500 D-O; Ushio) with a Toshiba R-65 filter, and the photomultiplier inlet port was protected from the actinic light by two plates of Corning 4-96 filter. Absorbance changes due to Phe a− photoaccumulation at a series of electrode potentials were measured at not shorter than 40 min after each potential stepping. The potential step was started first to a negative (reductive) direction, and then to a positive (oxidative) direction.
Supplementary Material
Acknowledgments.
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (No. 19614003 to T.W.) from the Japan Society for the Promotion of Science, a Grant-in-Aid for Young Scientists (B) (18770116 to M.S.) and a global Centers of Excellence Program for “Chemistry Innovation through Cooperation of Science and Engineering” (to T.W.) from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.B. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0905388106/DCSupplemental.
References
- 1.Renger G. In: Primary Processes of Photosynthesis: Principles and Apparatus, Part II. Renger G, editor. Cambridge, UK: Royal Society Chemistry; 2008. pp. 237–290. [Google Scholar]
- 2.Rappaport F, Diner BA. Primary photochemistry and energetics leading to the oxidation of the (Mn)4Ca cluster and to the evolution of molecular oxygen in photosystem II. Coord Chem Rev. 2008;252:259–272. [Google Scholar]
- 3.Dau H, Haumann M. The manganese complex of photosystem II in its reaction cycle—Basic framework and possible realization at the atomic level. Coord Chem Rev. 2008;252:273–295. [Google Scholar]
- 4.McEvoy JP, Brudvig GW. Water-splitting chemistry of photosystem II. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 5.Renger G, Holzwarth AR. In: Photosystem II: The Water:Platoquinone Oxido-Reductase in Photosynthesis. Wydrqynski T, Satoh K, editors. The Netherlands: Springer, Dordrecht; 2005. pp. 139–175. [Google Scholar]
- 6.Kamiya N, Shen JR. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7-Å resolution. Proc Natl Acad Sci USA. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira KN, Iverson TM, Maghlaoui K, Berber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 8.Loll B, Kern J, Saenger W, Zouni A, Biesadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 9.Gukov A, Kern J, Gabdulkhakov A, Broser M, Zouni A, Saenger W. Cyanobacterial photosystem II at 2.9-Å resolution and the role of quinones, lipids, channels and chloride. Nat Struct Mol Biol. 2009;16:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 10.Grabolle M, Dau H. Energetics of primary and secondary electron transfer in photosystem II membrane particles of spinach revisited on basis of recombination-fluorescence measurements. Biochim Biophys Acta. 2005;1708:209–218. doi: 10.1016/j.bbabio.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Klimov VV, Allakhverdiev SI, Demeter S, Krasnovskii AA. Photoreduction of pheophytin in the photosystem 2 of chloroplasts with respect to the redox potential of the medium. Dokl Akad Nauk SSSR. 1979;249:227–230. [Google Scholar]
- 12.Rappaport F, Guervova-Kuras M, Nixon PJ, Diner BA, Lavergne J. Kinetics and pathways of charge recombination in photosystem II. Biochemistry. 2002;41:8518–8527. doi: 10.1021/bi025725p. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa K, Noguchi T. Density functional theory calculations on the dielectric constant dependence of the oxidation potential of chlorophyll: Implication for the high potential of P680 in photosystem II. Biochemistry. 2005;44:8865–8872. doi: 10.1021/bi050273c. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi R, Hasegawa K, Noguchi T. Effect of charge distribution over a chlorophyll dimer on the redox potential of P680 in photosystem II as studied by Density Functional Theory calculations. Biochemistry. 2008;47:6289–6291. doi: 10.1021/bi8007998. [DOI] [PubMed] [Google Scholar]
- 15.Ishikita H, Loll B, Biesiadka J, Saenger W, Knapp E-W. Redox potentials of chlorophylls in the photosystem II reaction center. Biochemistry. 2005;44:4118–4124. doi: 10.1021/bi047922p. [DOI] [PubMed] [Google Scholar]
- 16.Ishikita H, Saenger W, Biesiadka J, Loll B, Knapp E-W. How photosynthetic reaction center control oxidation power in chlorophyll pairs P680, P700, and P870. Proc Natl Acad Sci USA. 2006;103:9855–9860. doi: 10.1073/pnas.0601446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishikita H, Biesiadka J, Loll B, Saenge W, Knapp E-W. Cationic state of accessory chlorophyll and electron transfer through pheophytin to plastoquinone in photosystem II. Angew Chem Int Ed. 2006;45:1964–1965. doi: 10.1002/anie.200503804. [DOI] [PubMed] [Google Scholar]
- 18.Klimov VV, Klevanik AV, Shuvalov VA, Krasnovsky AA. Reduction of pheophytin in the primary light reaction of photosystem II. FEBS Lett. 1977;82:183–186. doi: 10.1016/0014-5793(77)80580-2. [DOI] [PubMed] [Google Scholar]
- 19.Klimov VV, Allakhverdiev SI, Pashchenko Measurement of the activation energy and lifetime of fluorescence of photosystem 2 chlorophyll. Dokl Akad Nauk SSSR. 1978;242:1204–1207. [Google Scholar]
- 20.Rutherford AW, Mullet JE, Crofts AR. Measurement of the midpoint potential of the pheophytin acceptor of photosystem II. FEBS Lett. 1981;123:235–237. [Google Scholar]
- 21.Krieger A, Rutherford AW, Johnson GN. On the determination of redox midpoint potential of the primary quinone electron acceptor, QA, in photosystem II. Biochim Biophys Acta. 1995;1229:193–201. [Google Scholar]
- 22.Johnson GN, Rutherford AW, Krieger A. A change in the midpoint potential of the quinone QA in photosystem II associated with photoactivation of oxygen evolution. Biochim Biophys Acta. 1995;1229:202–207. [Google Scholar]
- 23.Krieger-Liszkay A, Rutherford AW. Influence of herbicide binding on the redox potential of the quinone acceptor in photosystem II: Relevance to photodamage and phytotoxicity. Biochemistry. 1998;37:17339–17344. doi: 10.1021/bi9822628. [DOI] [PubMed] [Google Scholar]
- 24.Hawkridge FM, Ke B. An electrochemical thin-layer cell for spectroscopic studies of photosynthetic electron-transport components. Anal Biochem. 1977;78:76–85. doi: 10.1016/0003-2697(77)90010-0. [DOI] [PubMed] [Google Scholar]
- 25.Rickard LM, Handrum HL, Hawkridge FM. A mediated electrochemical redox study of soluble spinach ferredoxin using optically coupled methods. Bioelectrochem Bioenerg. 1978;5:686–696. [Google Scholar]
- 26.Burgess JD, Hawkridge FM. In: Electroanalytical Methods for Biological Materials. Brajter-Toth A, Chambers J Q, editors. New York: Marcel Dekker; 2002. pp. 109–142. [Google Scholar]
- 27.Nakamura A, Suzawa T, Watanabe T. Spectroelectrochemical determination of the redox potential of P700 in spinach with an optically transparent thin-layer electrode. Chem Lett. 2004;33:688–689. [Google Scholar]
- 28.Nakamura A, Suzawa T, Kato Y, Watanabe T. Significant species-dependence of P700 redox potential as verified by spectroelectrochemistry: Comparison of spinach and Thermosynechococcus elongatus. FEBS Lett. 2005;579:2273–2276. doi: 10.1016/j.febslet.2005.02.076. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Nakamura A, Yoshinori K, Kato Y, Watanabe T. Spectroelectrochemistry of P700 in native photosystem I particles and diethyl ether-treated thylakoid membranes from spinach and Thermosynechococcus elongatus. FEBS Lett. 2008;582:1123–1128. doi: 10.1016/j.febslet.2008.02.075. [DOI] [PubMed] [Google Scholar]
- 30.Shibamoto T, Kato Y, Watanabe T. Spectroelectrochemistry of cytochrome b559 in the D1–D2-Cyt b559 complex from spinach. FEBS Lett. 2008;582:1490–1494. doi: 10.1016/j.febslet.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 31.Tomo T, et al. Characterization of highly purified photosystem I complexes from the chlorophyll d-dominated cyanobacterium Acaryochloris marina MBIC 11017. J Biol Chem. 2008;283:18198–18209. doi: 10.1074/jbc.M801805200. [DOI] [PubMed] [Google Scholar]
- 32.Sugiura M, Boussac A, Noguchi T, Rappaport F. Influence of histidine-198 of the D1 subunit on the properties of the primary electron donor, P680, of photosystem II in Thermosynechococcus elongatus. Biochim Biophys Acta. 2008;1777:331–342. doi: 10.1016/j.bbabio.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Ke B. In: Photosynthesis Photobiochemistry and Photobiophysics. Ke B, editor. The Netherlands: Kluwer Academic Publishers, Dordrecht; 2001. pp. 305–322. [Google Scholar]
- 34.Ohnishi N, Kashino Y, Satoh K, Ozawa S, Takahashi Y. Chloroplast-encoded polypeptide PsbT is involved in the repair of primary electron acceptor QA of photosystem II during photoinhibition in Chlamydomonas reinhardtii. J Biol Chem. 2007;282:7107–7115. doi: 10.1074/jbc.M606763200. [DOI] [PubMed] [Google Scholar]
- 35.Klimov VV, Dolan E, Ke B. EPR properties of an intermediary electron acceptor (pheophytin) in photosystem-II reaction centers at cryogenic temperatures. FEBS Lett. 1980;112:97–100. [Google Scholar]
- 36.Ke B, Inoue H, Babcock GT, Fang Z-X, Dolan E. Optical and EPR characterization of oxygen-evolving photosystem II subchloroplast fragments isolated from the thermophilic blue-green alga Phormidium laminosum. Biochim Biophys Acta. 1982;682:297–306. [Google Scholar]
- 37.Fujita I, Davis MS, Fajer J. Anion radicals of pheophytin and chlorophyll a: their role in the primary charge separations of plant photosynthesis. J Am Chem Soc. 1978;100:6280–6282. [Google Scholar]
- 38.Lubitz W, et al. ENDOR studies of the intermediate electron acceptor radical anion I− in photosystem II reaction centers. Biochim Biophys Acta. 1989;977:227–232. doi: 10.1016/s0005-2728(89)80076-3. [DOI] [PubMed] [Google Scholar]
- 39.Moenne-Loccoz P, Robert B, Lutz M. A resonance Raman characterization of the primary electron acceptor in photosystem II. Biochemistry. 1989;28:3641–3645. [Google Scholar]
- 40.Nebedryk E, et al. Characterization of bonding interactions of the intermediary electron acceptor in the reaction center of photosystem II by FTIR spectroscopy. Biochim Biophys Acta. 1990;1016:49–54. [Google Scholar]
- 41.Groot ML, et al. Initial electron donor and acceptor in isolated photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc Natl Acad Sci USA. 2005;102:13087–13092. doi: 10.1073/pnas.0503483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holzwarth AR, et al. Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: Pheophytin is the primary electron acceptor. Proc Natl Acad Sci USA. 2006;103:6895–6900. doi: 10.1073/pnas.0505371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kazakova AA, Kisselev BA, Kozlov YN. Electrochemical reduction of pheophytin and its participation in the functioning of photosystem II. Bioelectrochem Bioenerg. 1989;21:367–372. [Google Scholar]
- 44.Kozlov YN, Kiselev BA, Evstigneev VB. Electrochemical study of chlorophyll. II. Reduction of chlorophyll and further transformation of anion-radicals. Biofizika. 1973;18:59–63. [PubMed] [Google Scholar]
- 45.Schiller H, Dau H. Preparation protocols for high-activity photosystem II membrane particles of green algae and higher plants, pH dependence of oxygen evolution and comparison of the S2-state multiline signal by X-band EPR spectroscopy. J Photochem Photobiol B Biol. 2000;55:138–144. doi: 10.1016/s1011-1344(00)00036-1. [DOI] [PubMed] [Google Scholar]
- 46.Weller A. Photoinduced electron transfer in solution: Exciplex and radical ion pair formation free enthalpies and their solvent dependence. Z Phys Chem. 1982;133:93–98. [Google Scholar]
- 47.Vasil'ev S, Bergmann A, Redlin H, Eichler H-J, Renger G. On the role of exchangeable hydrogen bonds for the kinetics of P680+ QA− formation and P680+ Phe− recombination in photosystem II. Biochim Biophys Acta. 1996;1276:35–44. [PubMed] [Google Scholar]
- 48.Dau H, Sauer K. Exciton equilibration and photosystem II exciton dynamics: A fluorescence study on photosystem II membrane particles of spinach. Biochim Biophys Acta. 1996;1273:175–190. [Google Scholar]
- 49.Vassiliev S, Lee C, Brudvig GW, Bruce D. Structure-based modeling of excited-state transfer and trapping in histidine-tagged photosystem II core complexes from Synechocystis. Biochemistry. 2002;41:12236–12243. doi: 10.1021/bi0262597. [DOI] [PubMed] [Google Scholar]
- 50.Gibasiewicz K, Dobek A, Breton J, Leible W. Modulation of primary radical pair kinetics and energetics in photosystem II by the redox state of the quinone electron acceptor QA. Biophys J. 2001;80:1617–1630. doi: 10.1016/S0006-3495(01)76134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diner BA, et al. Site-directed mutations at D1-His198 and D2-His197 of photosystem II in Synechocystis PCC 6803: Sites of primary charge separation and cation and triplet stabilization. Biochemistry. 2001;40:9265–9281. doi: 10.1021/bi010121r. [DOI] [PubMed] [Google Scholar]
- 52.Van Mieghem FJE, Searle GFW, Rutherford AW, Schaafsma TJ. The influence of the double reduction of QA on the fluorescence decay kinetics of photosystem II. Biochim Biophys Acta. 1992;1100:198–206. [Google Scholar]
- 53.Vass I, et al. Reversible and irreversible intermediates during photoinhibition of photosystem II: Stable reduced QA species promote chlorophyll triplet formation. Proc Natl Acad Sci USA. 1992;89:1408–1412. doi: 10.1073/pnas.89.4.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu B, Napiwotzki A, Eckert HJ, Eichler HJ, Renger G. Studies on the recombination kinetics of the radical pair P680+Phoe− in isolated PS II core complexes from spinach. Biochim Biophys Acta. 1993;1142:129–138. [Google Scholar]
- 55.Vass I, Gatzen G, Holzwarth AR. Picosecond time-resolved fluorescence studies on photoinhibition and double reduction of QA in photosystem II. Biochim Biophys Acta. 1993;1183:388–396. [Google Scholar]
- 56.Merry SAP, et al. Modulation of quantum yield of primary radical pair formation in photosystem II by site-directed mutagenesis affecting radical cations and anions. Biochemistry. 1998;37:17439–17447. doi: 10.1021/bi980502d. [DOI] [PubMed] [Google Scholar]
- 57.Cser K, Vass I. Radiative and non-radiative charge recombination pathway in photosystem II studied by thermoluminescence and chlorophyll fluorescence in the cyanobacterium Synechocystis 6803. Biochim Biophys Acta. 2007;1767:233–243. doi: 10.1016/j.bbabio.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 58.Sugiura M, Inoue Y. Highly purified thermo-stable oxygen-evolving photosystem II core complex from the thermophilic cyanobacterium Synechococcus elongatus having His-tagged CP43. Plant Cell Physiol. 1999;40:1219–1231. doi: 10.1093/oxfordjournals.pcp.a029510. [DOI] [PubMed] [Google Scholar]
- 59.Meyer ML, DeAngelis TP, Heineman WR. Mercury-gold minigrid optically transparent thin-layer electrode. Anal Chem. 1977;49:602–606. doi: 10.1021/ac50012a025. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura A, Watanabe T. HPLC determination of photosynthetic pigments during greening of etiolated barley leaves. FEBS Lett. 1998;426:201–204. doi: 10.1016/s0014-5793(98)00344-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.