

Abstract
Polymer conjugates of camptothecin (CPT) have been pursued as a solution to the difficulties present in treating cancers with CPT and its derivatives. Covalent attachment of CPT to a polymer can improve solubility, increase blood circulation time, enhance tumor uptake, and significantly improve efficacy of the drug. In this report, we describe a novel polymer conjugate of CPT using a core-functionalized, symmetrically PEGylated poly(L-lysine) (PLL) dendrimer. The PEGylated dendrimer consisted of a lysine dendrimer functionalized with aspartic acid, which was used as an attachment site for poly(ethylene glycol) (PEG) and CPT. The final conjugate had a molecular weight of 40 kDa and was loaded with 4-6 wt% CPT. Polymer-bound CPT was shown to have a long blood circulation half-life of 30.9 ± 8.8 h and a tumor uptake of 4.2 ± 2.3% of the injected dose/g tissue, compared to free CPT in which less than 1% was retained in the blood after 30 min and had a tumor accumulation of 0.29 ± 0.04% of the injected dose/g tissue. The PEGylated PLL-CPT showed superior efficacy in murine (C26) and human colon carcinoma (HT-29) tumor models when compared with no treatment or treatment with irinotecan. In the C26 tumor model, treatment resulted in significantly prolonged survival (P < 0.05) for all mice treated with a single injection of PEGylated PLL-CPT. In the HT-29 tumor model, all mice treated with multiple injections of a low dose survived to the end of the study, with three mice of eight surviving tumor-free.
Keywords: drug delivery, camptothecin, colon carcinoma, dendrimer, PEG, poly(L-lysine)
INTRODUCTION
20(S)-camptothecin (CPT), a highly toxic alkaloid isolated from the plant Camptotheca acuminata, has been found to have broad antitumor activity in a variety of cancers.1 CPT acts by inhibiting topoisomerase-I, an enzyme that stabilizes and rejoins DNA breaks during replication. However, camptothecin has never successfully been transferred to clinical use due to poor solubility, high systemic toxicity, and decreased activity in vivo. In particular, at physiological pH, the lactone ring of CPT hydrolyzes to form a carboxylate that readily binds with serum albumin, which results in the removal of active CPT from the bloodstream.2 Several small-molecule derivatives of CPT have been designed to improve the solubility and bioavailability of the drug, of which topotecan and irinotecan (CPT-11) are used clinically to treat certain human cancers.3 Nevertheless, topotecan and irinotecan still have sub-optimal toxicity profiles and pharmacokinetics, and alternative methods of treatment are needed.
A method of addressing the problems associated with CPT is attachment of CPT to a polymer carrier. Polymer drug carriers have been studied extensively as a solution to many of the problems of small-molecule chemotherapeutic agents. Covalent attachment of a drug to a polymer or physical entrapment of a drug within a polymer system has been shown to increase drug solubility, improve pharmacokinetics, and decrease systemic toxicity.4,5 Additionally, Maeda and coworkers have shown that macromolecules passively target solid tumors through the enhanced permeation and retention (EPR) effect, resulting in higher drug concentrations in the tumor.6,7 Furthermore, researchers have shown that covalent modification of CPT by esterification of the alcohol at the 20-position carbon stabilized the lactone ring at physiological pH.8 Therefore, the esterification of CPT with a polymer at the 20-position alcohol could also prevent loss of activity associated with lactone ring opening.
Many researchers have begun work towards a clinically-viable polymer-CPT conjugate, primarily focusing on linear polymer carriers. Several conjugates have entered or progressed to Phase I/II trials, including the cyclodextrin-PEG copolymer CPT conjugate IT-101 (Calando Pharmaceuticals, Pasadena, CA)9,10,11,12, MAG-CPT, a HPMA-CPT conjugate (Pharmacia and Upjohn, Milan, Italy)13,14, CT-2106, a poly(L-glutamic acid)-CPT conjugate (Cell Therapeutics, Seattle, WA)15,16,17, and Prothecan, a PEG-CPT conjugate (Enzon, Piscataway, NY).18,19,20,21 IT-101 has just entered Phase I clinical trials. Phase I studies of MAG-CPT showed prolonged release of CPT over 46 days22, however, subsequent Phase I/II trials resulted in excessive systemic toxicity to patients bearing tumors23 and revealed that MAG-CPT showed no preferential accumulation of polymer-bound CPT within the tumors.24 CT-2106 showed an improved toxicity profile compared to unconjugated CPT in Phase I trials, and is progressing through Phase II.25 Prothecan showed promise in Phase I trials with noted antitumor activity.26 Despite the number of polymer-CPT conjugates in clinical trials, none have been approved for treatment yet.
PEGylated dendrimers are being pursued for drug delivery applications due to their improved in vivo pharmacokinetic properties, decreased toxicity, and enhanced solubility compared to non-PEGylated dendrimers and are particularly promising for delivery of chemotherapeutic agents.27,28,29,30 In contrast to conventional polymers, dendrimers are synthesized in a controlled, stepwise manner, resulting in a well-defined, branched structure with low polydispersity.31 Furthermore, the architecture, type and number of functional handles can be easily controlled. Researchers have PEGylated poly(amidoamine) (PAMAM)32,33, poly(L-lysine) (PLL)34,35,36,37, and melamine38 dendrimers for different biological applications. Our previous work using PEGylated dendrimers explored asymmetrically and symmetrically PEGylated polyester (PE) dendrimers based on bis(2-hydroxymethyl) propionic acid.39,40,41,42 A conjugate of doxorubicin (DOX) and an eight-arm, 40 kDa asymmetrically PEGylated PE dendrimer was synthesized, and studied in tumored mice. The study revealed the PEGylated dendrimer-DOX conjugate was remarkably effective at delivering DOX to the tumor, with a single, non-toxic dose resulting in complete remission of the tumor in all mice tested.41 The efficacy of this carrier was thought to be due to the core-shell architecture that results from PEGylating the periphery of a globular dendrimer. Drug was covalently attached to the low molecular weight dendrimer core and surrounded by a “shell” of long, solubilizing PEG chains which radiate from the core. Guillaudeu and researchers reported a simple and rapid method for synthesizing symmetrically PEGylated PE dendrimers that retains the core-shell architecture while incorporating multiple functional handles at the dendrimer core.42
As prior work has shown the potential of PEGylated dendrimer drug carriers to significantly improve the efficacy of a chemotherapeutic drug, we have synthesized and evaluated a novel PEGylated PLL-CPT conjugate for cancer treatment. Two generation two lysine dendrons were constructed around a diaminopropane core. The dendrimer periphery was modified with aspartic acid to incorporate two functional handles - a carboxylic acid and an amine. The amine of the aspartic acid was PEGylated with 5kDa PEG chains, and the carboxylate esterified with glycine-20(O)-CPT prodrug. The resulting PEGylated PLL-CPT conjugate had eight arms, a molecular weight of 40 kDa, and contained 6 wt% CPT. Herein is presented the synthesis, in vitro and in vivo analysis, and chemotherapeutic effectiveness of this new polymer-CPT conjugate.
EXPERIMENTAL SECTION
Reagents
Chemicals were purchased from Aldrich and were used without further purification. ONp-activated, Boc-protected lysine was purchased from Bachem (Bubendorf, Switerland). Boc-protected, Bn-ester-protected aspartic acid was purchased from Novabiochem (Darmstadt, Germany). ONp-activated PEG5000 (PEG5k) was purchased from Laysan Bio, Inc. (Arab, AL, US).
For intravenous injection, polymer-CPT conjugates were dissolved in Mg++/Ca++-free PBS, and reported doses were given as drug equivalents of polymer. CPT was suspended in a solution of 10% Tween 80/0.9% NaCl (9:1) and sonicated for 20 min to break up aggregates. CPT-11 was dissolved in a solution of 45 mg/mL D-sorbitol and 0.9 mg/mL lactic acid and heated at 50 °C for 20 min. All solutions were made just prior to injection.
Synthesis of Camptothecin-loaded PEGylated Poly(L-lysine) Dendrimers
The scheme for the synthesis of both glycine-linked PEGylated PLL-CPT (11a) and β-alanine-linked PEGylated PLL-CPT (11b) is shown in Scheme 1. Briefly, Boc-protected L-lysine, activated with p-nitrophenol, was reacted with diaminopropane in DMF. The first generation dendrimer (3) was purified by precipitation into a 0.2 M NaOH solution and collection of the solids via filtration through filter paper. After removal of the Boc-protecting groups in a mixture of TFA and DCM, 4 was made in a similar manner. The aspartic acid periphery was added by reacting the deprotected amines of 4 with Boc-Asp(Bn)-OH (5) in DMF with triethylamine. To PEGylate the Boc-deprotected amines of dendrimer 6, the TFA-dendrimer salt was combined with triethylamine and ONp-activated PEG5k (7) in DMF at 30 °C. Unreacted PEG5k was capped with piperidine. To remove the benzyl ester protecting groups, 8 was stirred with 4% acetic acid in methanol and ∼10% by weight palladium-on-carbon catalyst.
Scheme 1.

Synthesis of glycine-linked and β-alanine-linked PEGylated PLL-CPT conjugates.
Boc-Gly-CPT and Boc-β-Ala-CPT were synthesized following a previously described procedure20. After removal of the Boc-protecting groups with TFA, the CPT prodrugs were conjugated to the free carboxylates of 9 using HBTU in the presence of DIPEA and PCP in DMF. CPT content was quantified by UV absorbance at 365 nm in chloroform (ε = 21,856 M-1cm-1). Additional details of polymer and small molecule characterization can be found in the Supporting Information.
Camptothecin Hydrolysis Studies
To quantify the hydrolysis of CPT from the dendritic polymer carrier in various buffers, 11a or 11b was dissolved in either PBS without Mg++/Ca++ (pH 7.4), acetate buffer (30 mM with 70 mM NaNO3, pH 5.0), or reconstituted human plasma (adjusted to pH 7.4 with HEPES buffer (1 M HEPES, 13.5 g/L NaOH, 9 g/L NaCl)) at a concentration of 10 mg polymer/mL and incubated in the dark at 37 °C. At each timepoint, a 50 μL aliquot was combined with 250 μL 90% MeOH/10% AcOH and 200 μL chloroform and briefly vortexed. The sample was centrifuged at 12,000 rpm for 15 min, and the supernatant injected onto a RP-HPLC for analysis. The RP-HPLC system consisted of a C-8 Zorbax Exlipse XDB-C8 4.6 × 150mm, 5 μm column (Agilent, LOCATION) in line with a Waters 996 Photodiode Array Detector and a Waters 2414 Refractive Index Detector, using gradient elution with distilled water (with 0.05% TFA) and acetonitrile (with 0.05% TFA).
Tumor and Animal Models
C26 murine colon carcinoma cells obtained from the University of California, San Francisco cell culture facility were cultured in RPMI medium 1640 containing 10% FBS. HT-29 human colon carcinoma cells were acquired from Dr. B. Mark Evers at the University of Texas and were cultured in DMEM-1 with Glutamax containing 10% FBS. Female Balb/C mice (16-20 g) and female Athymic nu/nu mice (16-20g) were obtained from Simonsen Laboratories, Inc. (Gilroy, CA). All animal maintenance and experiments performed were in compliance with National Institutes of Health guidelines for animal research under a protocol approved by the Committee on Animal Research at the University of California, San Francisco.
Cytotoxicity Studies in Cells
Cytotoxicity studies were completed as previously described.41 Briefly, cells (C26 or HT-29) were incubated in medium (C26: RPMI 1650 with 10% FBS, 1% penicillin/streptomycin, 1% Glutamax; HT-29: DMEM-1 with Glutamax and 10% FBS) containing varying concentrations of CPT, 11a, or 11b. After a 72-hr incubation, the medium was replaced with medium containing thiazolyl blue tetrazolium bromide (0.83 mg/mL) and incubated an additional 3 hrs. The medium was replaced with dimethyl sulfoxide (200 μL) and 0.1 M glycine buffer (25 μL). Optical densities were measured at 570 nm by a SpectraMAX 190 microplate reader with Softmax Pro software (Molecular Devices, Sunnyvale, CA). IC50 values were obtained from sigmoidal fits of the data using Origin 7 SR4 8.0552 software (OriginLab, Northhampton, MA).
Acute Toxicity Studies in Healthy Mice
To determine the in vivo maximum tolerated dose for PEGylated PLL-CPT, the conjugate was administered intravenously (i.v.) to BALB/c mice in doses of 0 (PBS), 8, 16, 32, 48, and 64 mg CPT equivalents/kg mouse (3 mice per treatment group). The weights and general health of each mouse was monitored. When a mouse showed severe signs of toxicity, which included weight loss greater than 25% of the initial weight, lethargy, a hunched back, and ruffled fur, the mouse was sacrificed. The mice were monitored for 30 days.
Pharmacokinetic and Biodistribution Studies in C26-tumored Balb/C mice
Sixteen to twenty-five gram female Balb/C mice were injected in the right hind flank with 3×105 C26 cells. Nineteen days after tumor inoculation, mice were distributed randomly among four groups. Mice were injected i.v. (three mice per group) with either 11a (10 mg CPT equiv/kg) or with CPT (10 mg/kg) in ∼200 μL solution. The mice injected with polymer were sacrificed at 90 min, 360 min, or 2880 min postinjection. Additional timepoints for the blood concentration were collected by submandibular bleeds. The mice injected with CPT were sacrificed at 1440 min. At the time of sacrifice, the blood (collected by heart puncture), lungs, liver, spleen, intestines, stomach, kidney, muscle, and tumor were collected for analyses. Organs were weighed, and 200-300 mg of the collected organs were homogenized with zirconium beads and one mL acidified isopropyl alcohol (0.075 M HCl, 90% IPA). The samples then incubated at 4° C for 24 hrs. Serum was collected using Microtainer™ serum separator vials and processed in the same manner as the organs. All samples were then placed in a -80° C freezer until measurements could be made. At measurement time, samples were thawed and briefly vortexed. One hundred twenty-five microliters homogenate was combined with 875 μL acidified IPA, homogenized a second time, and centrifuged for 8 min at 14,000 rpm. Two hundred fifty microliters of supernatant was combined with 750 μL acidified IPA for fluorescence measurements. Sample fluorescence was measured on a Fluorolog fluorimeter (HORIBA Jobin Yvon, Edison, NY, USA), with an excitation of CPT at 372 nm and collection of fluorescence at 424 nm. Calibration curves were made from organ samples collected from an untreated mouse. The pharmacokinetic parameters of 11a were calculated assuming the two-compartment model.43
Antitumor Efficacy Studies
C26 murine colon carcinoma model
While under anesthesia, female Balb/C mice were shaved, and C26 cells (3 × 105 cells in 50 μL) were injected subcutaneously on the right hind flank. At eight days post-tumor implantation, mice were randomly distributed into treatment groups of 5-10 mice and treated intravenously (i.v.) (tail vein) as described in the text. All doses were administered in approximately 200 μL of solution. Animals were weighed and tumors measured every other day. The tumor volume was determined by measuring the tumor in three dimensions with calipers and calculated using the formula: tumor volume = length × width × height. Mice were removed from the study when 1) a mouse lost 15% of its body weight from the 1st day of treatment 2) any tumor dimension was >20 mm, or 3) the mouse was found dead. The study was continued until day 60 post-tumor inoculation.
HT-29 tumor model
Female athymic nu/nu mice were injected subcutaneously on the right hind flank with HT-29 cells (1 × 106 cells/mL in 50 μL). Twelve days later, mice were randomized into groups of eight animals per group and treated i.v. (tail vein) as described in the text. The tumor volume was calculated using the following formula: tumor volume = (length × width2)/2. The mice were followed until day 70 post-tumor inoculation.
Statistical analyses
The tumor growth delay (TGD) was calculated from the equation TGD = (T-C)/C × 100, where T is the mean time for the tumor volume of a treatment group to reach a designated volume and C is the mean time of the control group to reach the designated volume (500 mm3 for the C26 tumor model, 400 mm3 for the HT-29 tumor model). Mice that were removed from the study from treatment-related weight loss before the end of the study were not included in the statistical analysis. Mice that did not reach the designated tumor volume but were alive at the end of the study were assigned the last day of the study as the time to tumor volume or day of survival. An independent Student’s t-test was used to analyze the TGD and survival data for all animals. All statistical analyses were performed using MedCalc 8.2.1.0 for Windows (MedCalc Software, Mariakerke, Belgium).
RESULTS
Preparation of PEGylated PLL-CPT conjugates
The synthesis of the PEGylated PLL dendrimer is shown in Scheme 1. Synthesis of the unPEGylated, aspartic acid-terminated lysine dendrimer is step-wise and results in a complete, monodisperse molecule. The final molecular weight by SEC was measured as ∼35 kDa, although the theoretical molecular weight was 40 kDa. As previously reported, SEC underestimates the hydrodynamic volume, and thus molecular weight, of PEGylated dendrimers due to the branched nature of the polymer.39 The final polydispersity of the PEGylated PLL dendrimers was ∼1.1, which was essentially the polydispersity of the 5,000 Da PEG chains.
Based on reports in the literature, glycine was chosen as a linker between the PEGylated PLL dendrimer and CPT because of its favorable release profile and improved efficacy in polymer-CPT systems compared to other CPT linkers.17,20 The second linker, β-alanine, was chosen for use as a non-releasable polymer-CPT control, as β-alanine-linked CPT was shown to have a much slower release from polymer compared to glycine-linked CPT.17 The CPT conjugate 11b was made to test the requirement of drug release on polymer-CPT efficacy. After synthesis of the linker-CPT prodrugs, conjugation of the prodrug was achieved through standard protein coupling chemistry using HBTU. The theoretical maximum weight loading of CPT on the PEGylated PLL dendrimer is 6.5 wt%. Weight-loadings of 6% and 4.5% were achieved for 11a and 11b, respectively.
In vitro hydrolysis of CPT from the PEGylated PLL dendrimer
To mimic the potential in vivo release of CPT, hydrolysis of CPT from the PEGylated PLL dendrimer was monitored in three physiologically-relevant solutions: PBS (pH 7.4), acetate buffer (pH 5.0), and human plasma (buffered to pH 7.4 with HEPES buffer) (Figure 1). We expected that free CPT would be released via hydrolysis of the ester bond linking the drug to the amino acid linker. This was verified by analyzing a sample of free CPT, which corresponded to the small molecule released from each polymer (data not shown).
Figure 1.
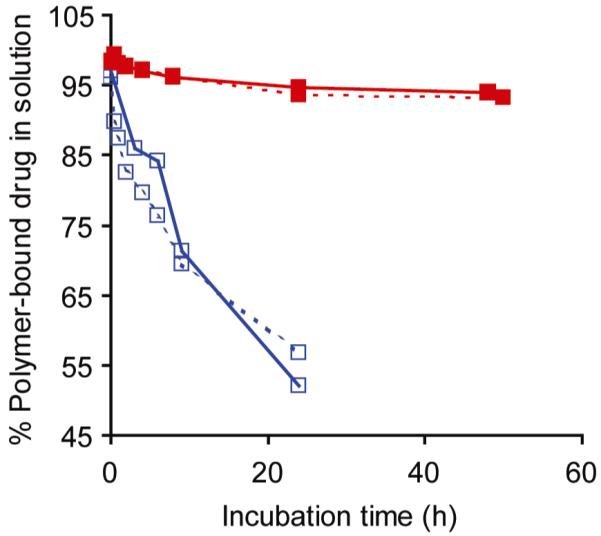
Hydrolysis of 11a and 11b in physiological solutions, given as the % of the polymer-bound CPT in solution as a function over time. 11a (∀) and 11b (!) were incubated in either PBS without Mg++/Ca++ salts, pH 7.4 (—) or reconstituted human plasma, buffered to pH 7.4 with HEPES buffer (--).
Surprisingly, we discovered the hydrolysis of CPT from 11a and 11b was the same in both PBS buffer and human plasma (Figure 1). Many researchers have chosen amino acid linkers for CPT because it is thought that CPT release could be achieved by enzymatic cleavage, and in fact, many observe faster release rates in the presence of Cathepsin B or human plasma.13,18,20,44 However, we observed similar release profiles of CPT in the presence and absence of enzymes. In both buffers, the hydrolysis of CPT from 11a had a half-life of 20 h, and hydrolysis of CPT from 11b had half-life of ∼200 h. The release of CPT in acetate buffer was less than 2% after 24 hrs for both polymers (data not shown). The faster hydrolysis of CPT at pH 7.4 compared to hydrolysis at pH 5.0 was expected, as ester bonds are less stable at higher pH.
In vitro toxicity of PEGylated PLL-CPT conjugates
The cytotoxicity of CPT and each polymer-CPT conjugate in C26 cells and HT-29 cells is given in Table 1. In the C26 line, conjugation of CPT via the glycine linker (11a) reduced the toxicity of CPT to the cells by approximately two-fold. Conjugation of CPT via the β-alanine linker (11b) reduced the toxicity by nearly two orders of magnitude. This decrease in toxicity of 11b reflected the slower hydrolysis of CPT from the polymer. In the HT-29 cell line, the toxicity of 11a was reduced by more than one order of magnitude, compared to free CPT.
Table 1.
IC50 values of polymer-CPT conjugates and free CPT in cells. Values determined by the MTT assay
| Polymer/Drug | C26 (nM) | HT-29 (nM) |
|---|---|---|
| 11a | 67.6 ± 6.5 | 522 ± 95 |
| 11b | 1030 ± 80 | NA |
| CPT | 34.4 ± 7.0 | 38.3 ± 5.0 |
Pharmacokinetics and biodistribution in tumored mice
The pharmacokinetics and body distribution of 11a and CPT were determined in C26-tumored female Balb/C mice (Figure 2). Less than 1% of the injected dose of free CPT was retained in the blood after 30 min. In contrast, conjugation of CPT to the PEGylated PLL dendrimer significantly prolonged blood circulation, resulting in a blood circulation half-life of 30.9 ± 8.8 h. Furthermore, area-under-the-curve of 11a was calculated to be 460 ± 140% of the injected dose-h/gram tissue (%ID-h/g tissue). For polymer-drug conjugates, a large area-under-the-curve is desirable because increased systemic exposure results in increased tumor exposure to the drug, and can result in high drug accumulation in the tumor.
Figure 2.

(a) Blood circulation profile of 11a (●) and free CPT (○) given as the average %ID/g blood over time. The solid line represents the calculated blood curve of 11a, using the two-compartment model. (b) Biodistribution of 11a and free CPT given as the average %ID/g tissue collected at 90 min, 6 hrs, and 48 hrs for 11a and collected at 24 hrs for free CPT. Negative values for the average %ID/g tissue are a result of measurements at or near the background fluorescence of CPT.
Given the long blood circulation half-life, the biodistribution of 11a in tumored mice was collected at three timepoints post-injection, with the last timepoint at 48 h post-injection. However, only a 24 h post-injection timepoint was collected for free CPT (Figure 2b). As can be seen, free CPT accumulated in the lungs, liver, and spleen, with accumulations of 1.75 ± 0.65 %ID/g tissue, 4.07 ± 0.72 %ID/g tissue, and 7.33 ± 1.77 %ID/g tissue, respectively. Accumulation of free CPT in the tumor tissue was much less, with only an accumulation of 0.29 ± 0.04 %ID/g tissue. Conjugation of CPT to the PEGylated PLL dendrimer decreased accumulation of CPT in the lungs, liver, and spleen, and significantly improved accumulation of CPT in the tumor. After 48 h, the accumulation of CPT in the tumor was 4.2 ± 2.3 %ID/g tissue. In fact, the accumulation of CPT in the tumor from polymer-bound CPT was significantly improved after only six h, when compared with the accumulation of free CPT after 24 h.
The biodistribution of both free CPT and the polymer-CPT conjugate was determined using the fluorescence of CPT at 424 nm. While the pharmacokinetics and biodistribution of polymer-bound CPT could be that of free CPT and 11a, we feel confident that we are observing primarily polymer-bound CPT given the rapid removal of free CPT from the body. Furthermore, we note that negative values for tissue accumulation were a result of measurements that were at or below the background level of fluorescence signal at 424 nm. Thus, we concluded very little CPT was present in these measurements.
Efficacy of PEGylated PLL-CPT in C26-tumored mice
CPT derivatives are used clinically to treat colorectal cancers. We chose to test our PEGylated PLL-CPT conjugates in the murine colon carcinoma model C26. After determining the maximum tolerated dose in healthy Balb/C mice to be between 32 mg CPT/kg and 48 mg CPT/kg (data not shown), two chemotherapy experiments were performed in C26-tumored Balb/C mice, and are detailed in Figure 3 and Table 2. As CPT is not typically tested in the C26 tumor model, an initial study was completed to compare 11a against free drug controls. In the drug controls study, a single, high dose of 11a was tested against free CPT and irinotecan at either a single or multiple injections (Table 2a, Figure 3a). The dose of 24 mg CPT/kg was tested, as it was 75% of the highest tolerated dose in healthy mice. A single dose of 10 mg CPT/kg was chosen based on the toxicity of CPT in the Balb/C mouse strain. Irinotecan was given at a dose of 50 mg CPT-11/kg for a total of four doses, and was included as a second free drug control. While it was found that neither CPT nor irinotecan had any significant effect on the survival of the C26-tumored mice, irinotecan, given twice weekly for four total doses, significantly delayed tumor growth (18% TGD) compared to no treatment (P ≤ 0.01). Despite no significant survival response from either free drug, mice treated with 11a showed significantly increased survival (P ≤ 0.0001) and improved TGD (72%) compared to both PBS and irinotecan.
Figure 3.

(a) Antitumor efficacy in the C26 tumor model, free drug control experiment. (b) Antitumor efficacy in the C26 tumor model, dose escalation and multiple doses schedule. (c) Antitumor activity in the HT-29 tumor model.
Table 2.
Antitumor activity of 11a in the C26 and HT-29 colon carcinoma models
| Experiment / Treatment | n | Dose (mg/kg) | Number of injections | Treatment days | Mean TGD (%) | Median Survival Time (d) | TRD | TFS | LTS |
|---|---|---|---|---|---|---|---|---|---|
| (a) C26 - Free drug controls | |||||||||
| PBS | 9 | - | 1 | 8 | - | 26 | 0 | 0 | 0 |
| CPT | 10 | 10 | 1 | 8 | < 5 | 25 | 0 | 0 | 0 |
| CPT-11 | 10 | 50 | 4 | 8, 11, 13, 15 | 18† | 30 | 0 | 0 | 0 |
| 11a | 10 | 24 | 1 | 8 | 72*‡ | 41*‡ | 0 | 0 | 0 |
| (b) C26 - Dose escalation and multiple doses | |||||||||
| PBS | 8 | - | 1 | 8 | - | 18 | 0 | 0 | 0 |
| 8 | 8 | 406 | 1 | 8 | 1.5 | 18 | 0 | 0 | 0 |
| 11b | 5 | 24 | 1 | 8 | 10 | 20 | 0 | 0 | 0 |
| 11a | 8 | 24 | 1 | 8 | 89* | 34* | 0 | 0 | 0 |
| 11a | 8 | 18 | 1 | 8 | 59* | 30* | 0 | 0 | 0 |
| 11a | 8 | 12 | 1 | 8 | 47* | 26* | 0 | 0 | 0 |
| 11a | 8 | 6 | 1 | 8 | 39* | 24† | 0 | 0 | 0 |
| 11a | 8 | 18 | 3 | 8, 15, 22 | NA | 23 | 8 | 0 | 0 |
| 11a | 8 | 12 | 3 | 8, 15, 22 | 155* | 49* | 0 | 1 | 1 |
| (c) HT-29 - Dose escalation, multiple doses, and free drug controls | |||||||||
| PBS | 8 | - | 1 | 8 | - | 45 | 0 | 0 | 0 |
| CPT-11 | 8 | 50 | 4 | 12, 14, 19, 21 | 60† | 63† | 0 | 0 | 4 |
| 11a | 8 | 24 | 1 | 12 | NA | 16 | 8 | 0 | 0 |
| 11a | 8 | 18 | 1 | 12 | 97† | 18 | 5 | 0 | 2 |
| 11a | 8 | 12 | 3 | 12, 19, 26 | 122*£ | 70*£ | 0 | 3 | 8 |
Notes: See Materials and Methods for experimental details and statistical methods. Dose given in d drug equivalents, except for 8, which is given as dose of polymer.
TGD, tumor growth delay, calculated from time of growth to 500 mm3 (C26) or 400 mm3(HT-29); treatment-related death; TFS, tumor-free survivors; LTS, long term survivors
Compared to PBS, P ≤ 0.0001
Compared to PBS, P ≤ 0.05
Compared to CPT-11, P ≤ 0.0001
Compared to CPT-11, P ≤ 0.05
Given the promising results of the first C26 chemotherapy study, a second chemotherapy study in C26-tumored mice was completed, looking at single-injection dose escalation (6, 12, 18, and 24 mg CPT/kg) and multiple-injection schedules (12 mg CPT/kg and 18 mg CPT/kg, given once weekly for a total of 3 injections) of 11a. A treatment of 24 mg CPT/kg of 11b was given as a control in which CPT was “non-releasable.” PEGylated dendrimer 8 was also treated at a dose of 406 mg polymer/kg, equivalent polymer to a dose of 24 mg CPT/kg, as a polymer-only control. Treatment with 11a proved to be remarkably effective at prolonging tumor growth delay and survival of the mice. All groups treated with a single injection of 11a, even down to a dose of 6 mg CPT/kg, showed significant tumor growth delay and increased survival (P ≤ 0.0001). As for treatment with multiple injections of 11a, all mice treated with dose of 18 mg CPT/kg suffered severe weight loss after the second and third injections, and were removed from the study as treatment-related deaths. However, mice treated with three weekly injections of 12 mg CPT/kg had the greatest TGD at 155% and longest median survival time. The median survival time was 49 days, with one mouse being tumor-free at day 60. Additionally, no significant weight loss was observed. As expected, treatment with either 11b or 8 resulted in TGD and survival similar to no treatment (PBS).
Efficacy of 11a in HT-29-tumored mice
Several researchers have tested the efficacy of polymer-CPT conjugates in the HT-29 human colon carcinoma model.12,13,14,19,21 Likewise, we tested the efficacy of 11a in HT-29-tumored nu/nu athymic female mice (Figure 3 and Table 2). Mice were treated with either a single injection of 24 mg CPT/kg or 18 mg CPT/kg or three weekly injections of 12 mg CPT/kg. Irinotecan, given at 50 mg CPT-11/kg twice weekly for four injections, was used as a positive control.45 The irinotecan schedule was found to significantly delay tumor growth (60% TGD) and prolong survival compared to no treatment (P ≤ 0.05), and four mice survived to the end of the study (day 70). Treatment with 11a with the once weekly injection of 12 mg CPT/kg, however, had a significantly prolonged TGD of 122% compared to both PBS (P ≤ 0.0001) and irinotecan (P ≤ 0.05), and all mice survived to the end of the study. Three of eight mice were tumor-free. Mice treated with either 24 mg CPT/kg or 18 mg CPT/kg of 11a showed excessive weight loss within 10 days of treatment and were removed from the study as treatment-related deaths. Of the three mice that survived the 18 mg CPT/kg of 11a treatment, tumor growth was significantly delayed compared to PBS (P ≤ 0.01).
DISCUSSION
Polymer drug carriers are studied for their potential to reduce drug toxicity, improve bioavailability, increase drug solubility and enhance targeting.4 Anticancer drugs covalently attached to polymer via a releasable linkage more effectively target tumor tissue than drug alone through the EPR effect.7 With a careful design of the drug linkage, the drug can remain attached to the polymer while circulating in the blood compartment, and be released after the polymer drug carrier is taken up into the tumor tissue.
Dendrimers have been explored for drug delivery applications due to their well-defined size, shape, and controlled exterior functionality.27,28,29,46,47,48,49 Our group has extensively studied dendritic polymer architectures for the delivery of anticancer drugs to tumors.40-42,50-53 In particular, we had remarkable success treating the C26 colon carcinoma model with a 40 kDa asymmetrically PEGylated polyester dendrimer-DOX conjugate, termed the “bowtie”.41 However, due to its asymmetric nature, the synthesis of this drug carrier was labor intensive, and a simpler symmetric, core-functional PEGylated dendrimer was designed in which the favorable pharmacokinetics and multifunctionality of the “bowtie”-type dendrimers were retained.42 In the present study, we applied the symmetric, core-functional design to PEGylated PLL dendrimers and covalently attached CPT. The PEGylated PLL dendrimer-CPT conjugates were analyzed for their in vitro and in vivo properties, and their antitumor efficacy in the C26 murine and HT-29 human colon carcinoma models studied.
The synthesis of PEGylated PLL dendrimers following the core-functional design is a simple step-wise procedure, resulting in a well-characterized drug carrier with low polydispersity. One of the advantages of this method is the ease in which multiple functionalities are incorporated into the periphery of the dendrimer using simple amino acids. Aspartic acid was chosen to add multifunctionality to the lysine dendrimer. Furthermore, the step-wise, column-free procedure for making peptide dendrimers allows for rapid scale-up of the synthesis, and large quantities of the PEGylated PLL drug carrier can be made easily.
To covalently attach camptothecin to a polymer backbone, multiple linkages based on amino acids and small peptides have been studied.17,18,20,44 Of those studied, polymer-camptothecin conjugates using a linker consisting of a single glycine residue are frequently found the most efficacious due to the favorable stability and release profile of glycine-CPT conjugates.17,20 Based on these prior results, we chose to examine the efficacy of our PEGylated PLL dendrimers in delivering CPT using a glycine linkage between CPT and the aspartic acid carboxylate. The amino acid linker β-alanine was chosen to verify the requirement of a releasable drug linkage in the efficacy of our drug carrier. A previous study indicated that CPT attached to a polymer backbone via β-alanine would have a much slower release rate and lower efficacy.17 This was verified by hydrolysis studies, which showed the release of CPT from β-alanine linkage was an order of magnitude slower than release of CPT from the glycine linkage. Surprisingly, hydrolysis studies of 11a and 11b in PBS and plasma revealed no difference in CPT release in the presence of plasma proteins and enzymes. This suggested to us that the release of CPT from the PEGylated PLL dendrimer core occurred primarily by hydrolysis. It is possible that the core-shell architecture of the PEGylated dendrimers prevents enzymes and plasma proteins from accessing the drug linkage near the core of the PEGylated dendrimer. Similarly, prior work from our group observed nearly identical degradation rates in plasma and PBS buffer observed for PEGylated polyester dendrimers.42
To the best of our knowledge, only a few reports exist testing CPT or its derivatives in a subcutaneously implanted C26 tumor model.54,55 Therefore, we conducted an initial efficacy study in subcutaneously-C26-tumored Balb/C mice using a single high dose of CPT, a single high dose of 11a, and 4 injections of CPT-11, given every two days. Overall, we found neither CPT nor CPT-11 to be effective in prolonging the survival of mice when using these schedules. However, a single, high dose of our 11a significantly prolonged mouse survival time and delayed tumor growth. This improved efficacy of polymer-bound CPT may be a result of not only the increased blood circulation time, but increased tumor accumulation of CPT. Furthermore, a second study in C26-tumored Balb/C mice with 11a showed mice treated with a single injection as low as 6 mg CPT/kg had significantly prolonged survival and TGD when compared to the control mice. As expected, a dose-dependent response was observed, with mice treated with higher doses having higher median survival times and higher TGD. However, we did observe that mice treated at higher doses (18 mg CPT/kg and 24 mg CPT/kg) showed some weight loss in the week following treatment. This weight loss proved to be too extensive when a multiple-injection schedule was used for the dose of 18 mg CPT/kg. All mice were removed from the study due to excessive weight loss from 11a treatment. On the other hand, a multiple-injection schedule at a lower dose of 11a, 12 mg CPT/kg, proved to be the most effective out of all treatment groups at prolonging survival time and delaying tumor growth without any significant weight loss from treatment.
A multiple-injection treatment schedule at a dose of 12 mg CPT/kg of 11a was also found to be the most effective in delaying tumor growth and prolonging survival in the HT-29 tumor model. At the end of the study, all mice had survived with an average tumor volume <300 mm3 in mice bearing tumors, and three mice were tumor-free. Additionally, we observed that a multiple-injection treatment of irinotecan had significant tumor delay and prolonged survival in the HT-29 tumor model, as predicted.45 However, a multiple-injection treatment of 11a at the dose of 12 mg CPT/kg significantly improved tumor delay and survival over CPT-11. This result is comparable to results obtained by Schluep and coworkers with their cyclodextrin-CPT conjugate IT-101 using the approximately the same treatment schedule and dose.12 However, our polymer is of a lower molecular weight than IT-101, which could result in less long-term accumulation. Unfortunately, many of the mice treated with a single high dose of either 18 mg CPT/kg or 24 mg CPT/kg of 11a observed excessive weight loss and were removed from the study as treatment-related deaths. The HT-29 model is a human tumor model and must be tested in athymic nude mice, thus we anticipated that the nude mice would be more sensitive to 11a than the Balb/C mice, and we observed greater toxic effects at equivalent doses.
Taken together, these results indicate that the PEGylated PLL dendrimer carrier effectively improved the bioavailability, solubility, and efficacy of the anticancer drug CPT. Furthermore, the glycine-CPT conjugate, 11a, was effective against the colon carcinoma models C26 and HT-29 by prolonging survival time and significantly slowing tumor growth. As seen in previous studies, the PEGylated dendrimer polymer drug carrier has proven to effectively prolong blood circulation time, increase tumor accumulation, and protect its drug cargo from enzymatic access. To the best of our knowledge, this is the first paper reporting the synthesis and in vivo evaluation of a PEGylated poly(L-lysine) dendrimer-drug conjugate. We are continuing to test the PEGylated PLL dendrimer-CPT in additional efficacy studies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the National Institute of Health for financial support of dendritic polymer drug carrier research (R01 EB002047). We also thank Dr. B. Mark Evers at the University of Texas for providing us with HT-29 tumor model.
Abbreviations
- Boc
N-tert-butoxycarbonyl
- Bn
benzyl
- CPT
camptothecin
- CPT-11
irinotecan
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- HBTU
O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate
- IPA
isopropyl alcohol
- LTS
long term survivors
- ONp
p-nitrophenyl ester
- PCP
pentachlorophenol
- PEG
polyethylene glycol
- PLL
poly(L-lysine)
- RP-HPLC
reverse-phase high performance liquid chromatography
- TFA
trifluoroacetic acid
- TFS
tumor-free survivors
- TGD
tumor growth delay
- TRD
treatment-related death
REFERENCES
- (1).Wall M, Wani M, Cook C, Palmer K, McPhail A, Sims G. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloid Leukemia and Tumor Inhibitor From Camptotheca Acuminata. J. Am. Chem. Soc. 1966;88:3888–3890. [Google Scholar]
- (2).Mi Z, Burke T. Differential Interactions of Camptothecin Lactone and Carboxylate Forms With Human Blood Components. Biochemistry (Mosc) 1994;33:10325–10336. doi: 10.1021/bi00200a013. [DOI] [PubMed] [Google Scholar]
- (3).Kehrer D, Soepenberg O, Loos W, Verweij J, Sparreboom A. Modulation of Camptothecin Analogs in the Treatment of Cancer: a Review. Anticancer Drugs. 2001;12:89–105. doi: 10.1097/00001813-200102000-00002. [DOI] [PubMed] [Google Scholar]
- (4).Haag R, Kratz F. Polymer Therapeutics: Concepts and Applications. Angew. Chem. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- (5).Pasut G, Veronese FM. Polymer-Drug Conjugation, Recent Achievements and General Strategies. Progress in Polymer Science. 2007;32:933–961. [Google Scholar]
- (6).Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer-Chemotherapy - Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- (7).Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: a Review. J. Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- (8).Zhao H, Lee C, Sai PK, Choe YH, Boro M, Pendri A, Guan SY, Greenwald RB. 20-O-Acylcamptothecin Derivatives: Evidence for Lactone Stabilization. J. Org. Chem. 2000;65:4601–4606. doi: 10.1021/jo000221n. [DOI] [PubMed] [Google Scholar]
- (9).Cheng JJ, Khin KT, Jensen GS, Liu AJ, Davis ME. Synthesis of Linear, Beta-Cyclodextrin-Based Polymers and Their Camptothecin Conjugates. Bioconjug. Chem. 2003;14:1007–1017. doi: 10.1021/bc0340924. [DOI] [PubMed] [Google Scholar]
- (10).Cheng J, Khin KT, Davis ME. Antitumor Activity of B-Cyclodextrin Polymer-Camptothecin Conjugates. Mol. Pharmaceutics. 2004;1:183–193. doi: 10.1021/mp049966y. [DOI] [PubMed] [Google Scholar]
- (11).Schluep T, Cheng J, Khin K, Davis ME. Pharmacokinetics and Biodistribution of the Camptothecin-Polymer Conjugate IT-101 in Rats and Tumor-Bearing Mice. Cancer Chemother. Pharmacol. 2006;57:654–662. doi: 10.1007/s00280-005-0091-7. [DOI] [PubMed] [Google Scholar]
- (12).Schluep T, Hwang J, Cheng JJ, Heidel JD, Bartlett DW, Hollister B, Davis ME. Preclinical Efficacy of the Camptothecin-Polymer Conjugate IT-101 in Multiple Cancer Models. Clin. Cancer Res. 2006;12:1606–1614. doi: 10.1158/1078-0432.CCR-05-1566. [DOI] [PubMed] [Google Scholar]
- (13).Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d’Argy R, Ghiglieri A, Castelli MG, Farao M, Pesenti E, Gigli M, Angelucci F, Suarato A. Polymer-Bound Camptothecin: Initial Biodistribution and Antitumour Activity Studies. J. Controlled Release. 2000;65:105–119. doi: 10.1016/s0168-3659(99)00243-6. [DOI] [PubMed] [Google Scholar]
- (14).Zamai M, vandeVen M, Farao M, Gratton E, Ghiglieri A, Castelli MG, Fontana E, d’Argy R, Fiorino A, Pesenti E, Suarato A, Caiolfa VR. Camptothecin Poly[N-(2-Hydroxypropyl) Methacrylamide] Copolymers in Antitopoisomerase-1 Tumor Therapy: Intratumor Release and Antitumor Efficacy. Mol. Cancer Ther. 2003;2:29–40. [PubMed] [Google Scholar]
- (15).Singer J, Bhatt R, Tulinsky J, Buhler K, Heasley E, Klein P, de Vries EGE. Water-Soluble Poly-(L-Glutamic Acid)-Gly-Camptothecin Conjugates Enhance Camptothecin Stability and Efficacy in Vivo. J. Controlled Release. 2001;74:243–247. doi: 10.1016/s0168-3659(01)00323-6. [DOI] [PubMed] [Google Scholar]
- (16).Zou J, Wu Q, Tansey W, Chou D, Hung M, Charnsangavej C, Wallace S, Li C. Effectiveness of Water Soluble Poly(L-Glutamic Acid)-Camptothecin Conjugate Against Resistant Human Lung Cancer Xenografted in Nude Mice. Int. J. Oncol. 2001;18:331–336. doi: 10.3892/ijo.18.2.331. [DOI] [PubMed] [Google Scholar]
- (17).Bhatt R, de Vries P, Tulinsky J, Bellamy G, Baker B, Singer JW, Klein P. Synthesis and in Vivo Antitumor Activity of Poly(L-Glutamic Acid) Conjugates of 20(S)-Camptothecin. J. Med. Chem. 2003;46:190–193. doi: 10.1021/jm020022r. [DOI] [PubMed] [Google Scholar]
- (18).Conover CD, Greenwald RB, Pendri A, Shum KL. Camptothecin Delivery Systems: the Utility of Amino Acid Spacers for the Conjugation of Camptothecin With Polyethylene Glycol to Create Prodrugs. Anticancer. Drug Des. 1999;14:499–506. [PubMed] [Google Scholar]
- (19).Conover CD, Greenwald RB, Pendri A, Gilbert CW, Shum KL. Camptothecin Delivery Systems: Enhanced Efficacy and Tumor Accumulation of Camptothecin Following Its Conjugation to Polyethylene Glycol Via a Glycine Linker. Cancer Chemother. Pharmacol. 1998;42:407–414. doi: 10.1007/s002800050837. [DOI] [PubMed] [Google Scholar]
- (20).Greenwald RB, Pendri A, Conover CD, Lee C, Choe YH, Gilbert C, Martinez A, Xia J, Wu D, Hsue M. Camptothecin-20-PEG Ester Transport Forms: the Effect of Spacer Groups on Antitumor Activity. Bioorg. Med. Chem. 1998;6:551–562. doi: 10.1016/s0968-0896(98)00005-4. [DOI] [PubMed] [Google Scholar]
- (21).Yu DS, Peng P, Dharap SS, Wang Y, Mehlig M, Chandna P, Zhao H, Filpula D, Yang K, Borowski V, Borchard G, Zhang ZH, Minko T. Antitumor Activity of Poly(Ethylene Glycol)-Camptothecin Conjugate: The Inhibition of Tumor Growth in Vivo. J. Controlled Release. 2005;110:90–102. doi: 10.1016/j.jconrel.2005.09.050. [DOI] [PubMed] [Google Scholar]
- (22).Bissett D, Cassidy J, de Bono JS, Muirhead F, Main M, Robson L, Fraier D, Magne ML, Pellizzoni C, Porro MG, Spinelli R, Speed W, Twelves C. Phase I and Pharmacokinetic (PK) Study of MAG-CPT (PNU 166148): a Polymeric Derivative of Camptothecin (CPT) Br. J. Cancer. 2004;91:50–55. doi: 10.1038/sj.bjc.6601922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wachters FM, Groen HJM, Maring JG, Gietema JA, Porro M, Dumez H, de Vries EGE, van Oosterom AT. A Phase I Study With MAG-Camptothecin Intravenously Administered Weekly for 3 Weeks in a 4-Week Cycle in Adult Patients With Solid Tumours. Br. J. Cancer. 2004;90:2261–2267. doi: 10.1038/sj.bjc.6601811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sarapa N, Britto M, Speed W, Jannuzzo M, Breda M, James C, Porro M, Rocchetti M, Wanders A, Mahteme H, Nygren P. Assessment of Normal and Tumor Tissue Uptake of MAG-CPT, a Polymer-Bound Prodrug of Camptothecin, in Patients Undergoing Elective Surgery for Colorectal Carcinoma. Cancer Chemother. Pharmacol. 2003;52:424–430. doi: 10.1007/s00280-003-0685-x. [DOI] [PubMed] [Google Scholar]
- (25).Homsi J, Simon GR, Garrett CR, Springett G, De Conti R, Chiappori A, Munster PN, Burton MK, Stromatt S, Allievi C, Angiulli P, Eisenfeld A, Sullivan DM, Daud AI. Phase I Trial of Poly-L-Glutamate Camptothecin (CT-2106) Administered Weekly in Patients With Advanced Solid Malignancies. Clin. Cancer Res. 2007;13:5855–5861. doi: 10.1158/1078-0432.CCR-06-2821. [DOI] [PubMed] [Google Scholar]
- (26).Rowinsky EK, Rizzo J, Ochoa L, Takimoto CH, Forouzesh B, Schwartz G, Hammond LA, Patnaik A, Kwiatek J, Goetz A, Denis L, McGuire J, Tolcher AW. A Phase I and Pharmacokinetic Study of PEGylated Camptothecin As a 1-Hour Infusion Every 3 Weeks in Patients With Advanced Solid Malignancies. J. Clin. Oncol. 2003;21:148–157. doi: 10.1200/JCO.2003.03.143. [DOI] [PubMed] [Google Scholar]
- (27).Gillies ER, Frechet JMJ. Dendrimers and Dendritic Polymers in Drug Delivery. Drug Discov. Today. 2005;10:35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- (28).Lee CC, Mackay JA, Frechet JMJ, Szoka FC. Designing Dendrimers for Biological Applications. Nat. Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- (29).Gajbhiye V, Kumar PV, Tekade RK, Jain NK. Pharmaceutical and Biomedical Potential of PEGylated Dendrimers. Curr. Pharm. Des. 2007;13:415–429. [Google Scholar]
- (30).Majoros IJ, Williams CR, Baker JR. Current Dendrimer Applications in Cancer Diagnosis and Therapy. Current Topics in Medicinal Chemistry. 2008;8:1165–1179. doi: 10.2174/156802608785849049. [DOI] [PubMed] [Google Scholar]
- (31).Fréchet JMJ, Tomalia DA. Dendrimers and Other Dendritic Polymers. Wiley; New York: 2001. [Google Scholar]
- (32).Luo D, Haverstick K, Belcheva N, Han E, Saltzman WM. Poly(Ethylene Glycol)-Conjugated PAMAM Dendrimer for Biocompatible, High-Efficiency DNA Delivery. Macromolecules. 2002;35:3456–3462. [Google Scholar]
- (33).Kono K, Kojima C, Hayashi N, Nishisaka E, Kiura K, Wataral S, Harada A. Preparation and Cytotoxic Activity of Poly(Ethylene Glycol)-Modified Poly(Amidoamine) Dendrimers Bearing Adriamycin. Biomaterials. 2008;29:1664–1675. doi: 10.1016/j.biomaterials.2007.12.017. [DOI] [PubMed] [Google Scholar]
- (34).Okuda T, Kawakami S, Maeie T, Niidome T, Yamashita F, Hashida M. Biodistribution Characteristics of Amino Acid Dendrimers and Their PEGylated Derivatives After Intravenous Administration. J. Controlled Release. 2006;114:69–77. doi: 10.1016/j.jconrel.2006.05.009. [DOI] [PubMed] [Google Scholar]
- (35).Okuda T, Kawakami S, Akimoto N, Niidome T, Yamashita F, Hashida M. PEGylated Lysine Dendrimers for Tumor-Selective Targeting After Intravenous Injection in Tumor-Bearing Mice. J. Controlled Release. 2006;116:330–336. doi: 10.1016/j.jconrel.2006.09.012. [DOI] [PubMed] [Google Scholar]
- (36).Bhadra D, Bhadra S, Jain NK. PEGylated Peptide Dendrimeric Carriers for the Delivery of Antimalarial Drug Chloroquine Phosphate. Pharm. Res. 2006;23:623–633. doi: 10.1007/s11095-005-9396-9. [DOI] [PubMed] [Google Scholar]
- (37).Kaminskas LM, Boyd BJ, Karellas P, Krippner GY, Lessene R, Kelly B, Porter CJH. The Impact of Molecular Weight and PEG Chain Length on the Systemic Pharmacokinetics of PEGylated Poly L-Lysine Dendrimers. Mol. Pharmaceutics. 2008;5:449–463. doi: 10.1021/mp7001208. [DOI] [PubMed] [Google Scholar]
- (38).Umali AP, Crampton HL, Simanek EE. Triazine Dendrimers With Orthogonally Protected Amines on the Periphery. Masking Amines With Dde and BOC Groups Provides an Alternative to Carrying Protected Alcohols and Disulfides Through an Iterative Synthesis. J. Org. Chem. 2007;72:9866–9874. doi: 10.1021/jo701320h. [DOI] [PubMed] [Google Scholar]
- (39).Gillies ER, Fréchet JMJ. Designing Macromolecules for Therapeutic Applications: Polyester Dendrimer-Poly(Ethylene Oxide) “Bow-Tie” Hybrids With Tunable Molecular Weight and Architecture. J. Am. Chem. Soc. 2002;124:14137–14146. doi: 10.1021/ja028100n. [DOI] [PubMed] [Google Scholar]
- (40).Gillies ER, Dy EE, Fréchet JMJ, Szoka FC. Biological Evaluation of Polyester Dendrimer:Poly(Ethylene Oxide) “Bow-Tie” Hybrids With Tunable Molecular Weight and Architecture. Mol. Pharmaceutics. 2005;2:129–138. doi: 10.1021/mp049886u. [DOI] [PubMed] [Google Scholar]
- (41).Lee CC, Gillies E, Fox ME, Dy E, Fréchet JMJ, Szoka FC. A Single Dose of Bowtie-Conjugated Doxorubicin Cures Mice Tumored With C26 Colon Carcinoma. Proc. Natl. Acad. Sci. U. S. A. 2006;103:16649–16654. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Guillaudeu SJ, Fox ME, Haidar YM, Dy EE, Szoka FC, Fréchet JMJ. PEGylated Dendrimers With Core Functionality for Biological Applications. Bioconjug. Chem. 2008;19:461–469. doi: 10.1021/bc700264g. [DOI] [PubMed] [Google Scholar]
- (43).Welling PG. Pharmacokinetics: Processes, Mathematics, and Applications. American Chemical Society; Washington, D.C.: 1997. The Two-Compartment Open Model With Intravenous or Oral Administration. Chapter 17. [Google Scholar]
- (44).Jensen E, Bundgaard H. Peptide Esters As Water-Soluble Prodrugs for Hydroxyl Containing Agents: Chemical Stability and Enzymatic Hydrolysis of Benzyl Esters of Glycine, Diglycine and Triglycine. Int. J. Pharm. 1991;71:117–125. [Google Scholar]
- (45).Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a Highly Active Nanoliposomal Irinotecan Using a Novel Intraliposomal Stabilization Strategy. Cancer Res. 2006;66:3271–3277. doi: 10.1158/0008-5472.CAN-05-4007. [DOI] [PubMed] [Google Scholar]
- (46).Kukowska-Latallo JF, Candido KA, Cao ZY, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR. Nanoparticle Targeting of Anticancer Drug Improves Therapeutic Response in Animal Model of Human Epithelial Cancer. Cancer Res. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- (47).Dufes C, Keith WN, Bilsland A, Proutski I, Uchegbu JF, Schatzlein AG. Synthetic Anticancer Gene Medicine Exploits Intrinsic Antitumor Activity of Cationic Vector to Cure Established Tumors. Cancer Res. 2005;65:8079–8084. doi: 10.1158/0008-5472.CAN-04-4402. [DOI] [PubMed] [Google Scholar]
- (48).Singh P, Gupta U, Asthana A, Jain NK. folate and Foalte-PEG-PAMAM Dendrimers: Synthesis, Characterization, and Targeted Anticancer Drug Delivery Potential in Tumor Bearing Mice. Bioconjug. Chem. 2008;19:2239–2252. doi: 10.1021/bc800125u. [DOI] [PubMed] [Google Scholar]
- (49).Svenson S, Tomalia DA. Commentary-Dendrimers in Biomedical Applications - Reflections on the Field. Advanced Drug Delivery Reviews. 2005;57:2106–2129. doi: 10.1016/j.addr.2005.09.018. [DOI] [PubMed] [Google Scholar]
- (50).De Jesus OLP, Ihre HR, Gagne L, Fréchet JMJ, Szoka FC. Polyester Dendritic Systems for Drug Delivery Applications: In Vitro and in Vivo Evaluation. Bioconjug. Chem. 2002;13:453–461. doi: 10.1021/bc010103m. [DOI] [PubMed] [Google Scholar]
- (51).Lee CC, Yoshida M, Fréchet JMJ, Dy EE, Szoka FC. In Vitro and in Vivo Evaluation of Hydrophilic Dendronized Linear Polymers. Bioconjug. Chem. 2005;16:535–541. doi: 10.1021/bc0497665. [DOI] [PubMed] [Google Scholar]
- (52).Fox ME, Szoka FC, Fréchet JMJ. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture. Acc. Chem Res. 2009 doi: 10.1021/ar900035f. ACS-ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Nasongkla N, Chen B, Macaraeg N, Fox ME, Fréchet JMJ, Szoka FC. Dependence of Pharmacokinetics and Biodistribution on Polymer Architecture: Effect of Cyclic versus Linear Polymers. J. Am. Chem. Soc. 2009;131:3842–3843. doi: 10.1021/ja900062u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kawano K, Watanabe M, Yamamoto T, Yokoyama M, Opanasopit P, Okano T, Maitani Y. Enhanced Antitumor Effect of Camptothecin Loaded in Long-Circulating Polymeric Micelles. J. Controlled Release. 2006;112:329–332. doi: 10.1016/j.jconrel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- (55).Watanabe M, Kawano K, Toma K, Hattori Y, Maitani Y. In Vivo Antitumor Activity of Camptothecin Incorporated in Liposomes Formulated With an Artificial Lipid and Human Serum Albumin. J. Controlled Release. 2008;127:231–238. doi: 10.1016/j.jconrel.2008.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.