

Abstract
Most patients with acute lung injury (ALI) have reduced alveolar fluid clearance that has been associated with higher mortality. Several mechanisms may contribute to the decrease in alveolar fluid clearance. In this study, we tested the hypothesis that pulmonary edema fluid from patients with ALI might reduce the expression of ion transport genes responsible for vectorial fluid transport in primary cultures of human alveolar epithelial type II cells. Following exposure to ALI pulmonary edema fluid, the gene copy number for the major sodium and chloride transport genes decreased. By Western blot analyses, protein levels of αENaC, α1Na,K-ATPase, and cystic fibrosis transmembrane conductance regulator decreased as well. In contrast, the gene copy number for several inflammatory cytokines increased markedly. Functional studies demonstrated that net vectorial fluid transport was reduced for human alveolar type II cells exposed to ALI pulmonary edema fluid compared with plasma (0.02±0.05 versus 1.31±0.56 μl/cm2/h, p<0.02). An inhibitor of p38 MAPK phosphorylation (SB202190) partially reversed the effects of the edema fluid on net fluid transport as well as gene and protein expression of the main ion transporters. In summary, alveolar edema fluid from patients with ALI induced a significant reduction in sodium and chloride transport genes and proteins in human alveolar epithelial type II cells, effects that were associated with a decrease in net vectorial fluid transport across human alveolar type II cell monolayers.
Impaired alveolar fluid clearance (AFC;2 i.e. the resolution of alveolar edema) is a common characteristic among patients with acute lung injury (ALI) and acute respiratory distress syndrome. The level of AFC impairment has significant prognostic value in determining morbidity and mortality (1, 2). Multiple clinically relevant experimental studies have tried to uncover the underlying mechanisms that reduce AFC in ALI, and several pathways have been implicated (3, 4).
In the alveolar environment, basal AFC is determined predominately by amiloride-sensitive (epithelial sodium channel (ENaC)) and -insensitive sodium channels and the activity of the Na,K-ATPase (3, 5-8). Several stimuli can up-regulate AFC including β-adrenergic agonists via cAMP-dependent mechanisms (3, 4). In the mouse and human lung, cAMP-dependent alveolar epithelial fluid transport is dependent on CFTR activity, especially in mediating β-adrenergic receptor-driven alveolar epithelial fluid transport (9-11).
We and others have reported that, in the early phase of ALI, pulmonary edema fluid contains high levels of several proinflammatory cytokines, including IL-1β, IL-8, TNFα, and TGFβ1 (12-14). Several of these proinflammatory cytokines have been studied in experimental fluid transport experiments. For example, during short in vitro exposures, TNFα increases AFC, which is mediated predominantly by both TNFα receptor-dependent and -independent effects (15, 16). In contrast, for exposures up to 24 h, TNFα decreases the expression of ENaC (α, β, and γ subunits) mRNA and protein levels as well as the amiloride-sensitive current and ouabain-sensitive Rb+ uptake in rat alveolar epithelial cells (17). Similarly we found that IL-1β decreases dexamethasone-induced αENaC mRNA and protein levels and the amiloride-sensitive fraction of the transepithelial current and sodium transport across rat type II cell monolayers (18). This inhibitory effect of IL-1β on αENaC expression is mediated through p38 MAPK phosphorylation. More recently, we reported that TGFβ1 decreases the amiloride-sensitive fraction of Na+ uptake and fluid transport across monolayers of rat and human type II cells as well as αENaC mRNA and protein expression (19). The inhibitory effect is mediated by ERK1/2 MAPKs phosphorylation. In chronic inflammation associated with nasal polyposis, TGFβ1 down-regulates CFTR mRNA and protein expression as well as the cAMP-dependent current in human nasal epithelial cells (20).
Despite these results with individual cytokines, the effect of pulmonary edema fluid itself from patients with ALI on human alveolar epithelial type II cell transport has not yet been studied. Therefore, we used our recently developed in vitro model of polarized human alveolar epithelial type II cells with a high transepithelial resistance, which makes it possible to measure apical to basal net fluid transport (9). The objective was to determine the effect of undiluted pulmonary edema fluid from patients with ALI on AFC across polarized human alveolar epithelial type II cell monolayers. ALI pulmonary edema fluid caused a marked decrease in net transepithelial fluid transport compared with simultaneously collected plasma. To understand the mechanisms by which ALI edema fluid decreased AFC, we first studied the effect of the ALI pulmonary edema fluid on the gene and protein expression of the major inflammatory cytokines and the critical sodium and chloride transport proteins involved in AFC. ALI pulmonary edema had a differential effect, inducing a marked increase in cytokine/chemokine gene expression and, at the same time, a decrease in all of the major sodium and chloride transport gene and protein levels. To further understand which soluble factors were critical, we pretreated human alveolar epithelial type II cells with inhibitors of the major cytokines or two signaling pathways, p38 and ERK1/2 MAPKs, mechanisms that can down-regulate vectorial fluid transport. Our results demonstrate that ALI pulmonary edema fluid contains soluble factors that are capable of adversely affecting the resolution of pulmonary edema in patients who developed ALI from sepsis.
EXPERIMENTAL PROCEDURES
Isolation of Human Alveolar Type II Pneumocytes
Type II epithelial cells were isolated from human donor lungs (preserved at 4 °C for 4-8 h) that were declined for transplantation by the California Transplant Donor Network using established methods (9). For isolation, a lobe was selected that was free of obvious consolidation or hemorrhage by gross inspection. The pulmonary artery was perfused 10 times with PBS, and the distal airspaces were then lavaged 10 times with Ca2+, Mg2+-free PBS solution containing 0.5 mm EGTA and 0.5 mm EDTA. Elastase, 13 units/ml in Ca2+, Mg2+-free Hanks’ balanced salt solution, was instilled into the distal airspaces through segmental bronchial intubation, and the lobe was incubated in a water bath at 37 °C for 45 min. Following digestion, the lobe was minced finely in the presence of fetal bovine serum and DNase at 500 μg/ml. The cell-free fraction was filtered sequentially through one layer of sterile gauze, two layers of gauze, and 150- and 30-μm nylon meshes. The filtrate was then layered onto a Percoll density gradient (1.04-1.09 g/ml) and centrifuged at 1500 rpm for 20 min. The top layer containing both type II pneumocytes and macrophages was collected and centrifuged at 800 rpm for 10 min. The cell pellet was resuspended in Ca2+, Mg2+-free PBS solution containing 5% fetal bovine serum and incubated with magnetic beads coated with anti-CD14 antibodies at 4 °C for 40 min. Macrophages were then selectively depleted with a Dynal magnet (Dynal Biotech, Oslo, Norway) and further incubated on Petri dishes coated with human IgG antibodies against macrophages. The remaining alveolar epithelial type II cells were then plated on collagen I-coated 24-well plates (Biocoat, BD Biosciences) at 5% CO2 at 37 °C at a concentration of 1.0 × 106 cells/well. The cells were exposed to media, Dulbecco’s modified Eagle’s medium-H21 and Ham’s F-12 (1:1), with antibiotics (penicillin, streptomycin, gentamicin, and amphotericin) and 10% fetal bovine serum for 48 h and without fetal bovine serum for 24 h. 72 h after the isolation, the type II cells were washed once with PBS and then exposed to 1 ml of ALI pulmonary edema fluid with and without inhibitors or the corresponding plasma for 24 h. In a separate set of experiments, human alveolar epithelial type II cells were exposed to cytomix, a mixture of the major biologically active cytokines found in ALI pulmonary edema fluid (IL-1β, TNFα, and interferon γ), at a concentration of 50 ng/ml with and without inhibitors or the corresponding control medium for up to 24 h as a surrogate for the inflammatory edema fluid (21).
Acute Lung Injury Pulmonary Edema Fluid
Patients were categorized as having ALI by clinical criteria (bilateral infiltrates on chest radiograph, partial pressure of O2/fractional concentration of inspired O2 <300, and no evidence of left atrial hypertension with a pulmonary wedge pressure ≤18 mm Hg if measured) (22) and an edema fluid/plasma protein ratio >0.65 (Biuret method, (2)). The degree of lung injury was quantified by the lung injury score (23). To have a more uniform etiology of ALI for this study, we collected and combined samples only from patients with ALI due to sepsis. All human ALI pulmonary edema and plasma samples were collected from 1993 to 2005 at the University of California, San Francisco and Vanderbilt University Medical Centers (12, 13, 24). Briefly pulmonary edema fluid was obtained from patients meeting ALI criteria within 4 h of intubation through gentle luminal suction applied to a 14 French catheter passed into the distal airways. All ALI pulmonary edema fluid samples were centrifuged immediately at 14,000 rpm for 20 min to remove cells and debris, and the supernatant was stored at -80 °C until use. Plasma samples were drawn simultaneously and centrifuged (3000 × g for 10 min at 4 °C), and the supernatants were stored at -80 °C until further use. To have a sufficient quantity of edema fluid for all of the necessary experiments, the aliquots of ALI pulmonary edema fluid from all of the patients were combined as done previously (13, 24). The Committee for Human Research at the University of California, San Francisco and Vanderbilt Medical Center Institutional Review Board approved all human protocols.
Detection of Alveolar Epithelial Cell Apoptosis and Necrosis
Human alveolar epithelial type II cells were plated onto collagen I-coated 24-well plates and exposed to ALI pulmonary edema fluid or plasma for 24 h. Following the incubation, the cells were detached from the plate with trypsin (0.25%) and combined with the floating and detached cells in the supernatant. Total alveolar epithelial cells were double stained with annexin V-fluorescein isothiocyanate and propidium iodide (25) using the TACS annexin V-fluorescein isothiocyanate apoptosis detection kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The percentage of apoptotic cells was quantified by flow cytometry using FACScan (BD Biosciences), and data were analyzed using FlowJo software (version 6.4.3; Tree Star Inc.).
Cytokine Levels in ALI Pulmonary Edema Fluid
Total TNFα and IL-1β levels were measured in the ALI pulmonary edema fluid or simultaneously collected plasma prior to exposure to the human alveolar epithelial type II cells using the enzyme-linked immunosorbent assay Quantikine kit (R&D Systems) according to the manufacturer’s instructions.
Quantitative RT-PCR
Total RNA was isolated from the cells using the RNeasy minikit (Qiagen) according to the manufacturer’s instruction and incubated with RQ1 RNase-free DNase (Promega Corp.) and Superase RNA inhibitor (Ambion). Gene expression in human alveolar epithelial type II cells was measured using a two-step multiplex quantitative RT-PCR method described previously (26). Primer and probe sets for genes of interest were designed using “Primer Express” software (PerkinElmer Life Sciences) based on sequencing data from National Center for Biotechnology (NCBI) databases and purchased from Biosearch Technologies, Inc. (Novato, CA). The primer and probe sequences used are available upon request. Total RNA (10-20 ng) was first reverse transcribed using random hexamers, and the resulting cDNA product was amplified using a mixture of gene-specific primers and hot start PCR for 25 cycles. Transcript quantifications were run along with-RT cDNA controls in a 384-well format on an ABI Prism 7900 sequence detection system (Applied Biosystems, Inc., Foster City, CA). Data were collected as PCR cycle threshold values for each gene and then converted to relative gene copy number (GCN) based on linear regression (26). Transcript copy was normalized using 10 housekeeping genes. The geometric mean value of the housekeeping genes most stably expressed across the samples was used for normalization (GeNorm software).
Protein Isolation and Western Blotting
Total cellular protein was extracted from the cells using 0.15 ml of lysis buffer/well containing 1% Triton X-100, 20 mm Tris base (pH 8.0), 137 mm NaCl, 10% glycerol, 2 mm vanadate, 2 μg/ml aprotinin, 5 μg/ml leupeptin, and 1 mm Pefabloc and homogenized. Protein content was measured by the bicinchoninic acid method (Pierce). Each sample was first reduced and denatured with sample buffer and run on a 4-12% gradient bis-Tris gel (Invitrogen), 10-20 μg of proteins per lane, using a MOPS-SDS buffer (Invitrogen) at 100 V for roughly 2 h. The proteins were then transferred onto a nitrocellulose membrane and blocked with 5% milk in Tris-buffered saline with Tween 20 for 1 h. The nitrocellulose membrane was then exposed to the primary antibody overnight at 4 °C. Primary antibodies used were: αENaC (Calbiochem), α1- and β1Na,K-ATPase (Upstate Cell Signaling Solutions, Charlottesville, VA), and CFTR (R&D Systems). The protein bands were then visualized with a chemiluminescence agent, ECL+ (Amersham Biosciences), and quantitated with NIH ImageJ software.
Fluid Transport across Human Alveolar Epithelial Type II Cells
Net fluid transport was measured across human type II cells on Transwell plates (0.4-μm pore size and collagen I-coated, CoStar, Corning) in a humidified tent within a 37 °C, 5% CO2 incubator with 100% humidity. Measurement of fluid transport from the apical to basolateral membrane of the type II cell monolayers was done at 120 h following the initial isolation and 48 h after the air-liquid interface was achieved; previously the transmembrane electrical resistance peaked (1530 ohms × cm2) at 96 h with morphological evidence of tight junctions (9, 27). The cells were first exposed to 150 μl of ALI pulmonary edema fluid, plasma, or cytomix at 50 ng/ml (containing 0.3 μCi/ml 131I-albumin) in the apical chamber of the Transwell. For cAMP agonist studies, 10 μm forskolin and IBMX were added to the ALI pulmonary edema fluid prior to placement on the apical layer. After 5 min, 20 μl of the media was then aspirated as the initial sample. After 24 h, another 20 μl was aspirated from the upper compartment of the Transwell as the final sample. Each sample was weighed, and radioactivity was counted in a γ counter (Packard MINAXI 5000 series).
Net fluid transport (μl/cm2/h) was calculated as described previously (9): =[1 - (radioactivity in the initial sample/weight of the initial sample)/(radioactivity in the final sample/weight of the final sample)] × 100. At the end of the experiments, all the fluid in the lower compartment was also collected to determine the protein permeability by measuring the unidirectional flux of labeled 131I-albumin from the apical (upper compartment) to the basolateral side (lower compartment) (9, 27).
Confocal Microscopy with Immunofluorescence Labeling
Confocal microscopy was performed with immunofluorescence labeling of tight junction proteins to correlate the changes in protein levels with any redistribution of the protein around the tight junction. Briefly human type II cells, seeded on Transwell plates for 5 days, were exposed to plasma or pulmonary edema fluid with and without SB202190 from patients with ALI for 24 h and then fixed in 4% formaldehyde for 30 min. The cells were then permeabilized with 0.1% Triton X-100 for 5 min and blocked with 0.1% bovine serum albumin in PBS for 30 min. The cells were then exposed to the primary antibody for ZO-1 (1:250; Zymed Laboratories Inc.) for 1 h in 0.1% bovine serum albumin in PBS. After extensive washing with PBS, the cells were exposed to the secondary antibody (fluorescein isothiocyanate-goat anti-mouse or rhodamine red X-goat anti-rabbit and Texas Red-phalloidin, Molecular Probes) and 4′,6-diamidino-2-phenylindole for 45 min. The slides were mounted with Vectashield (Vector Laboratories), and images were obtained using the Bio-Rad MRC-1024 laser scanning confocal microscope.
Transmission Electron Microscopy
To examine the ultrastructure of the human alveolar epithelial type II cells exposed to ALI pulmonary edema fluid or plasma, treated human type II cell monolayers grown on Transwell membranes were fixed with 3% (w/v) Karnovsky fixative for 1 h at 0 °C, and the membranes were removed. The monolayers were postfixed for 1 h in 1% veronal-buffered osmic acid and then dehydrated in graded ethanols and/or propylene oxide. The cell preparations were then embedded in Epon or Araldite resins cured at 60 °C. Thin sections were contrasted with saturated aqueous uranyl acetate and Reynolds lead citrate. The sections were then imaged with a JEOL 1200 EX transmission electron microscope operating at 80 kV.
Inhibitor Studies
To test the mechanisms through which ALI edema fluid decreased gene and protein expression, human type II cells were exposed to five inhibitors individually 30 min prior to exposure to ALI pulmonary edema fluid or corresponding plasma: 1) anti-human TNFα/TNFSF1A antibody, 1 μg/ml (R&D Systems); IL-1RA, 5 μg/ml (R&D Systems); or anti-TGFβ1, -β2, -β3, 10 μg/ml (R&D Systems) and 2) MAPK inhibitors 10 μm PD98059, a MEK1 inhibitor (Cell Signaling Technology, Inc., Danvers, MA), or 10 μm SB202190, a p38 MAPK inhibitor (Calbiochem). Total RNA and protein were collected and analyzed as described above.
Total Nitric Oxide Level
The total nitric oxide level was measured using the Griess reaction (AssayDesigns, Ann Arbor, MI) following the manufacturer’s instructions.
Statistics
Each condition was run in triplicate and reported as the mean ± S.D. To determine significance, two-tailed Student’s t test and analysis of variance with posthoc Tukey test or Bonferroni correction were used when appropriate. p values less than or equal to 0.05 were determined to be significant.
RESULTS
Demographic and Hemodynamic Characteristics of the Patients with ALI from Sepsis
Undiluted pulmonary edema fluid from 19 patients with ALI due to sepsis was collected and pooled from the University of California, San Francisco and Vanderbilt Medical Centers. Base-line demographic and hemodynamic data for all the patients are listed in Table 1.
TABLE 1.
Clinical characteristics of the patients with acute pulmonary edema and acute lung injury from sepsis
| Clinical characteristics | Results |
|---|---|
| Age (years) | 39 ± 15a |
| Male | 79% |
| Caucasian | 79% |
| Shock | 74% |
| Lung injury scoreb | 3.1 ± 0.8a |
| SAPSIIc | 60 ± 16a |
| Mortality | 74% |
Data as mean ± S.D.
Out of four points.
Simplified acute physiology score II.
Effect of ALI Pulmonary Edema Fluid on Net Fluid Transport
Net fluid transport was essentially zero for human alveolar type II cells exposed to ALI pulmonary edema fluid compared with simultaneously collected plasma (0.02±0.05 versus 1.31±0.56 μl/cm2/h, mean ± S.D., p < 0.02). The significant decrease in net fluid transport caused by ALI pulmonary edema fluid was not altered by the addition of forskolin and IBMX (Fig. 1). The rate of net fluid transport for cells exposed to plasma derived from patients with ALI was equivalent to previously reported rates for human alveolar type II cells stimulated with forskolin and IBMX (1.35 ± 0.24 μl/cm2/h) over 24 h (9).
FIGURE 1. Net fluid transport across human alveolar epithelial type II cells exposed to plasma or ALI pulmonary edema fluid with or without forskolin + IBMX.
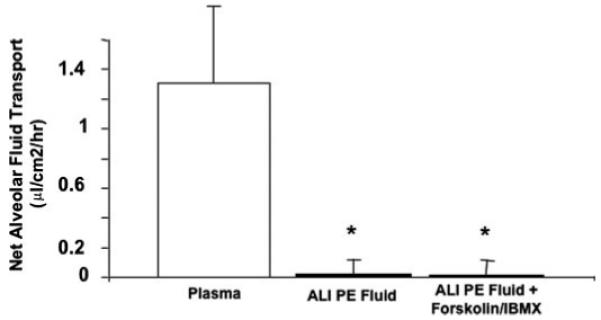
Human alveolar epithelial type II cells cultured for 5 days were exposed to plasma or ALI pulmonary edema fluid with or without forskolin + IBMX, 48 h after the air-liquid interface was achieved, for 24 h. Net fluid transport was calculated, as done previously, by the change in weight and radioactivity of 131I-albumin in the upper compartment of the Transwell plate and expressed as mean (μl/cm2/h) ± S.D. of each sample run in triplicates. *, p < 0.02 compared with plasma. PE, pulmonary edema.
The initial controls for the functional studies as well as the initial quantitative RT-PCR experiments included normal media, normal human plasma, and simultaneously collected plasma from the ALI patients. There was no significant difference in protein permeability, net fluid clearance, or gene expression between the controls (data not shown). ALI plasma was used as the control for all subsequent studies.
Effect of ALI Pulmonary Edema Fluid on Cell Death
There was no gross morphologic effect of ALI pulmonary edema fluid or plasma on the appearance of human alveolar epithelial type II cell monolayers. Total cell death as measured by propidium iodide staining increased among human alveolar type II cells exposed to ALI pulmonary edema fluid compared with plasma that was collected simultaneously (10 ± 1 versus 5 ± 1%, mean ± S.D., p < 0.01). The level of apoptosis, as measured by both annexin V-fluorescein isothiocyanate and propidium iodide staining, was not significantly different (6±1 versus 5±2%, mean ± S.D., p > 0.05; Fig. 2).
FIGURE 2. Effect of ALI pulmonary edema fluid on total cell death.
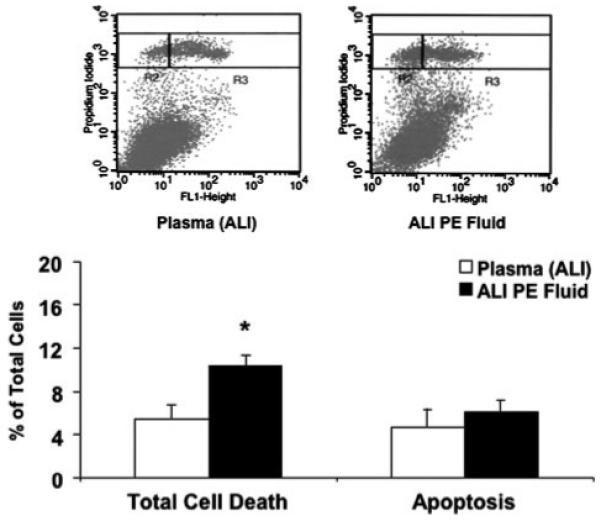
Human alveolar epithelial type II cells cultured for 3 days were exposed to ALI pulmonary edema fluid or plasma for 24 h. The percentage of apoptotic cells was quantified by flow cytometry after double staining with propidium iodide and annexin V-fluorescein isothiocyanate of both the floating and detached cells in the supernatant and the plated cells. Total cell death was measured by propidium iodide staining alone. Data represent mean±S.D. of each sample measured in triplicates. *, p < 0.01 compared with plasma controls. In the panels above each graph, total cell death = R2 + R3 where R2 represents necrotic cells and R3 represents the apoptotic portion. PE, pulmonary edema.
Cytokine Levels in ALI Pulmonary Edema Fluid and Plasma
Previously we found that the major biologically active inflammatory cytokines in human ALI pulmonary edema fluid are IL-1β and, to a lesser extent, TNFα (12). To confirm the proinflammatory nature of the ALI pulmonary edema fluid in this study, we measured the levels of both TNFα and IL-1β in the ALI pulmonary edema fluid prior to incubation with the human alveolar epithelial type II cells. As expected, the levels were markedly elevated compared with simultaneously collected plasma (Fig. 3).
FIGURE 3. Cytokine levels in ALI pulmonary edema fluid and plasma.
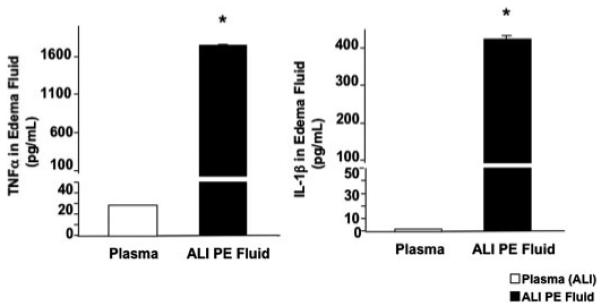
The levels of TNFα and IL-1β were measured by enzyme-linked immunosorbent assay from the combined ALI pulmonary edema fluid and the corresponding plasma of 19 patients with ALI from sepsis. Data represent mean ± S.D. of each sample measured in triplicates. *, p < 0.005 compared with plasma controls. The level of TNFα in ALI pulmonary edema fluid was 62× and the level of IL-1β was 220× the level found in the corresponding plasma. PE, pulmonary edema.
Effect of ALI Pulmonary Edema Fluid on Cytokine Gene Expression
The gene copy number for all the major inflammatory cytokines increased markedly following exposure to ALI pulmonary edema fluid compared with simultaneously collected plasma. The gene copy number of two major proinflammatory cytokines, TNFα and IL-1β, increased by 700 and by 900%, respectively, compared with cells exposed to plasma. The gene copy number of another major chemokine, IL-8, increased nearly 200 times above the levels induced by plasma (Fig. 4A).
FIGURE 4. A and B, effect of ALI pulmonary edema fluid on the gene expression of the major inflammatory cytokines and chemokines (A) and sodium and chloride transport proteins (B) by human alveolar epithelial type II cells exposed to ALI pulmonary edema fluid or plasma.
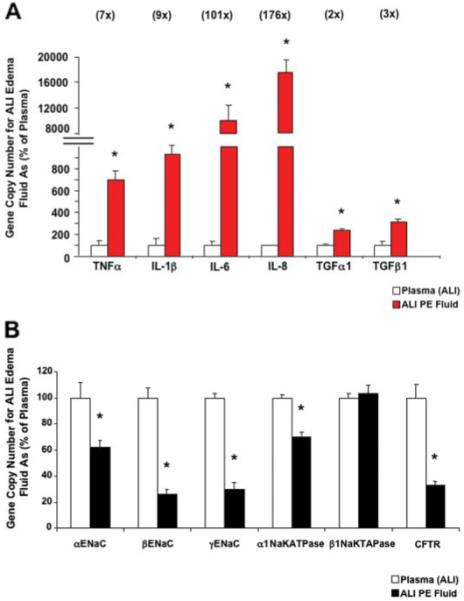
Human alveolar epithelial type II cells cultured for 3 days were exposed to ALI pulmonary edema fluid or plasma for 24 h. The gene expression was measured by two-step multiplex quantitative RT-PCR and expressed as the percent GCN of plasma ± S.D. of each sample measured in triplicates. *, p < 0.01 compared with plasma controls. The number above each cytokine/chemokine represents the multiple times the GCN of ALI pulmonary edema exposed cells exceeded the GCN of plasma exposed cells. PE, pulmonary edema.
Effect of ALI Pulmonary Edema Fluid on Sodium and Chloride Transport Gene Expression
In sharp contrast to the effect of the pulmonary edema fluid on proinflammatory cytokine gene expression, the gene copy number for the major sodium and chloride transport genes, except for β1Na,K-ATPase, decreased significantly with exposure to ALI pulmonary edema fluid. The gene copy number for each ENaC subunit, the major apical sodium transporter, decreased significantly: 39% for αENaC, 74% for βENaC, and 70% for γENaC. The gene copy number of the major basolateral sodium transporter, α1Na,K-ATPase, decreased by 30%, and the gene copy number of CFTR decreased by 67% (Fig. 4B).
Effect of ALI Pulmonary Edema Fluid on Sodium and Chloride Transport Protein Expression
By Western blot analyses, αENaC protein levels decreased significantly by 18%, CFTR protein levels decreased by 40%, and α1Na,K-ATPase protein levels decreased by 30%, whereas protein levels of β1Na,K-ATPase increased by 58% on exposure to ALI pulmonary edema fluid compared with plasma (Fig. 5, A and B). Western blot analyses were standardized using glyceraldehyde-3-phosphate dehydrogenase as the loading control.
FIGURE 5. A and B, effect of ALI pulmonary edema fluid on the protein expression of the major sodium and chloride transport proteins by human alveolar epithelial type II cells exposed to ALI pulmonary edema fluid or plasma.
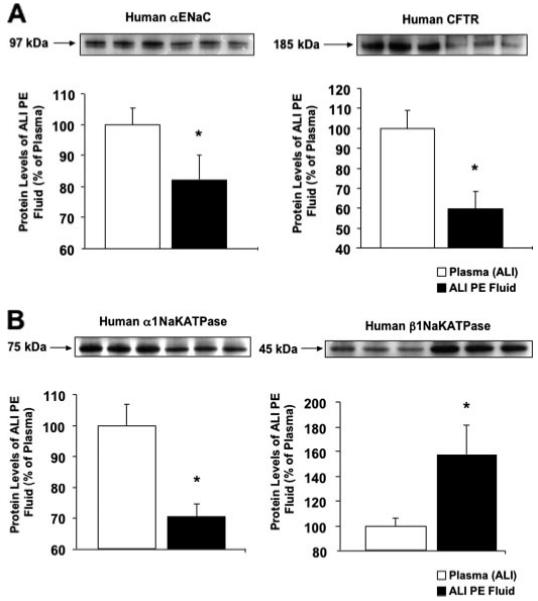
Human alveolar epithelial type II cells cultured for 3 days were exposed to ALI pulmonary edema fluid or plasma for 24 h. Protein levels were measured by Western blot analyses and expressed as the percent protein levels of plasma±S.D. of each sample measured in triplicates. *, p < 0.05 compared with plasma controls. A representative Western blot is depicted above each graph for the major sodium and chloride transport proteins. PE, pulmonary edema.
Effect of ALI Pulmonary Edema Fluid on Net Fluid Transport and Paracellular Protein Permeability
Although the human alveolar type II cell monolayer appeared intact by light microscopy, we measured the quantity of labeled albumin that crossed the human alveolar type II cell monolayer over 24 h to determine whether alterations in paracellular protein permeability were responsible for the net decrease in fluid transport in the presence of ALI pulmonary edema fluid. In the type II cells that were treated with ALI pulmonary edema fluid, there was a significant increase in paracellular protein permeability (28 ± 5 versus 1±1%, p < 0.005; Fig. 6, A and B). These results indicate that the decrease in net AFC was due to both an increase in paracellular protein permeability and a decrease in transcellular ion transport.
FIGURE 6. A-D, effect of ALI pulmonary edema fluid on the net fluid transport (A and C) and paracellular protein permeability (B and D) by human alveolar epithelial type II cells exposed to ALI pulmonary edema fluid or cytomix with and without pretreatment with a p38 MAPK inhibitor or plasma.
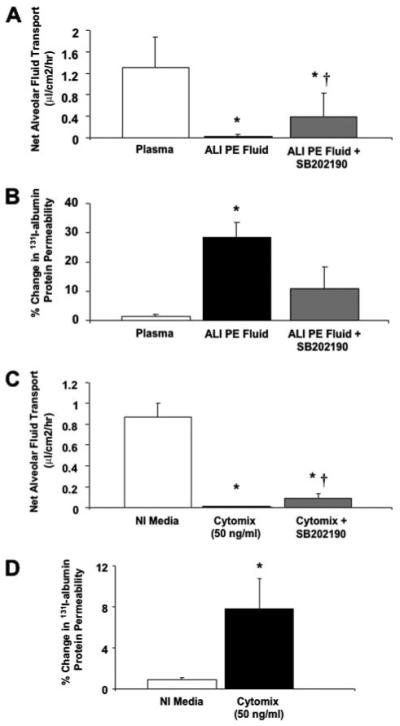
Human alveolar epithelial type II cells cultured for 5 days were exposed to ALI pulmonary edema fluid, plasma, or cytomix, 48 h after the air-liquid interface was achieved, for 24 h. Another set of human type II cells were pretreated with 10.0 μm SB202190 for 30 min prior to exposure with ALI pulmonary edema fluid or cytomix. Net fluid transport was calculated, as done previously, by the change in weight and radioactivity of 131I-albumin in the upper compartment of the Transwell plate and expressed as mean (μl/cm2/h) ± S.D. of each sample run in triplicates. Paracellular protein permeability was calculated, as done previously, by the passage of 131I-albumin from the upper compartment to the lower compartment of the Transwell plate and expressed as percent change in 131I-albumin permeability±S.D. of each sample run in triplicates. *, p < 0.02 compared with plasma controls; †, p < 0.01 compared with ALI pulmonary edema or cytomix-exposed cells. PE, pulmonary edema; NI, normal.
Effect of Cytomix on Net Fluid Transport and Paracellular Protein Permeability
To determine whether the increase in total cell death caused by ALI pulmonary edema fluid contributed to the changes in protein permeability or net fluid clearance, we exposed human alveolar epithelial type II cells to cytomix as a surrogate for the pulmonary edema fluid itself as we have done in prior studies (21). By flow cytometry, total cell death was not significantly different between human alveolar epithelial type II cells exposed to control media or cytomix (total cell death: 4 ± 1 versus 4 ± 0.7%, p = not significant; apoptosis level: 3 ± 0.2 versus 3 ± 0.5%, p = not significant). Similar to ALI pulmonary edema fluid, cytomix decreased net fluid clearance to zero while significantly increasing protein permeability (Fig. 6, C and D). Cytomix also increased the gene copy number of the major inflammatory cytokines while at the same time decreasing the gene copy number of the major sodium transport proteins in a dose-dependent manner (Fig. 7, A and B).
FIGURE 7. A and B, the dose response of human alveolar epithelial type II cells exposed to cytomix or normal human plasma on the expression of the major inflammatory cytokines and chemokines (A) and sodium transport proteins (B).
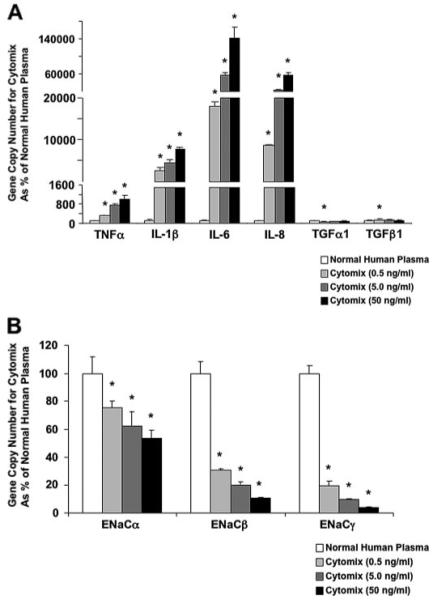
Human alveolar epithelial type II cells cultured for 3 days were exposed to different concentrations of cytomix (0.5-50 ng/ml) or plasma for 24 h. Gene expression was measured by two-step multiplex quantitative RT-PCR and expressed as the percent GCN of plasma ± S.D. of each sample measured in triplicates. *, p < 0.05 versus normal human plasma.
Confocal Microscopy of Tight Junctions
To study the morphologic appearance of the tight junction proteins, we used confocal microscopy to localize ZO-1 in the human alveolar type II cell monolayer. ZO-1 was localized to the cell boundaries in a discrete subapical region of the lateral membranes corresponding to the tight junction. The distribution of ZO-1 was not different between human alveolar type II cells exposed to plasma or ALI pulmonary edema fluid (Fig. 8).
FIGURE 8. Effect of ALI pulmonary edema fluid on ZO-1 distribution in primary cultures of human alveolar epithelial type II cells.
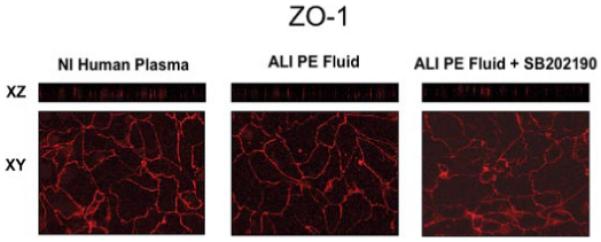
Human alveolar type II cells were grown on Transwell plates for 5 days and exposed to plasma, ALI pulmonary edema fluid, or ALI pulmonary edema fluid pretreated with 10 μm SB202190, a p38 MAPK inhibitor, for 24 h. The cells were fixed and stained with anti-ZO-1 antibodies and then analyzed by confocal microscopy. En face sections (x-y) were generated from selected planes in the vertical (x-z) sections. PE, pulmonary edema; NI, normal.
Transmission Electron Microscopy
Electron microscopy of the human epithelial type II cell monolayers demonstrated ultrastructural features characteristic of alveolar epithelial type II cells including microvillus, lamellar bodies, and cuboidal shape. No sign of apoptosis (condensation of the chromatin in tight apposition to the nuclear envelope, alteration of the nuclear envelope, or fragmentation of the nucleus) was present in type II cells exposed to ALI pulmonary edema fluid or simultaneously collected plasma. In addition, the tight junctions appeared intact as well (Fig. 9).
FIGURE 9. Effect of ALI pulmonary edema fluid on the ultrastructure of cultured human alveolar epithelial type II cells.
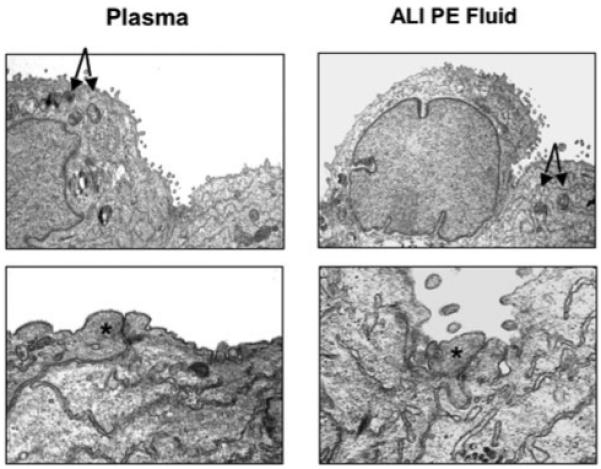
Transmission electron microscopy demonstrated no significant changes in the characteristic features of alveolar epithelial type II cells on exposure to ALI pulmonary edema fluid compared with plasma: the cuboidal shape or the presence of lamellar bodies and microvillus. More significantly, no signs of apoptosis were detected, and the tight junctions appeared intact. The arrows point toward lamellar bodies, and the asterisks depict the location of tight junctions. PE, pulmonary edema.
Effect of Inhibitor Pretreatment on Net Fluid Transport
To further investigate the mechanism by which ALI pulmonary edema fluid decreased net fluid transport, we exposed human alveolar type II cells to inhibitors of the major cytokines, TNFα, IL-1β, and TGFβ, and intracellular MAPKs, ERK1/2 and p38 MAPKs, both known to affect ENaC gene expression and protein levels as well as transepithelial current and sodium transport across type II cell monolayers (15-20). Inhibitors of TNFα, IL-1β, TGFβ, or phosphorylated ERK1/2 MAPK had no effect on restoring net fluid transport or paracellular protein permeability following exposure to ALI pulmonary edema fluid (data not shown). Only the inhibition of phosphorylated p38 MAPK with 10.0 μm SB202190 partially restored net fluid transport following exposure to ALI pulmonary edema fluid and attenuated the increase in paracellular protein permeability (Fig. 6, A and B). Pretreatment of the human alveolar type II cells with a p38 MAPK inhibitor, SB202190, also partially restored the decrease in net fluid clearance caused by cytomix (Fig. 6C). The dose of SB202190 was chosen based on our previous experiments with the effect of individual cytokines on vectorial sodium transport (18).
Pretreatment of human alveolar type II cells with SB202190 prior to ALI pulmonary edema fluid exposure also restored the gene copy numbers of the major cytokines/chemokines and the sodium/chloride transport proteins closer to the gene copy number observed after exposure to ALI plasma. SB202190 pretreatment significantly blunted a decrease in sodium/chloride transport genes (Fig. 10) induced by ALI pulmonary edema fluid (gene copy number levels of cells exposed to ALI pulmonary edema fluid with and without SB202190 pretreatment as percentage of plasma ± S.D.; *, p < 0.05 versus plasma; †, p < 0.02 versus ALI pulmonary edema (PE) fluid). By Western blot analyses, SB202190 pretreatment also significantly restored the protein levels of all the major sodium transport proteins from the effect of ALI pulmonary edema fluid (Fig. 11, A-D). SB202190 pretreatment had no effect on CFTR protein levels.
FIGURE 10. Changes in the gene expression of the major sodium and chloride transport proteins by human alveolar epithelial type II cells by pretreatment with a p38 MAPK inhibitor prior to exposure to ALI pulmonary edema fluid.
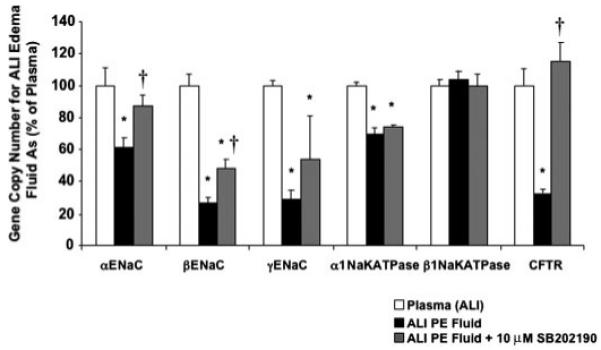
Human alveolar epithelial type II cells cultured for 3 days were exposed to ALI pulmonary edema fluid or plasma for 24 h. Another set of human type II cells were pretreated with 10.0 μm SB202190 for 30 min prior to exposure with ALI pulmonary edema fluid. The gene expression was measured by two-step multiplex quantitative RT-PCR and expressed as the percent GCN of plasma ± S.D. of each sample measured in triplicates. *, p < 0.05 compared with plasma controls; †, p is significant by analysis of variance with Bonferroni correction. PE, pulmonary edema.
FIGURE 11. A-D, changes in the protein levels of the major sodium and chloride transport proteins by human alveolar epithelial type II cells by pretreatment with a p38 MAPK inhibitor prior to exposure to ALI pulmonary edema fluid.
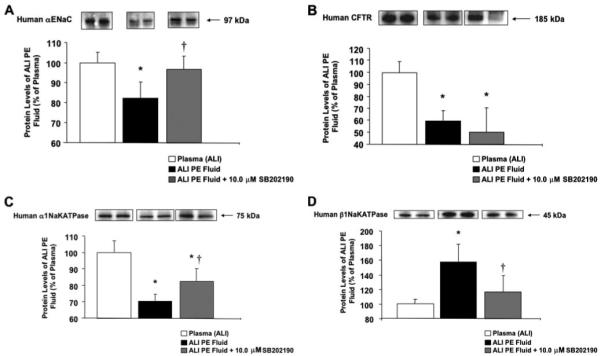
Human alveolar epithelial type II cells cultured for 3 days were exposed to ALI pulmonary edema fluid or plasma for 24 h. Another set of human type II cells were pretreated with 10.0 μm SB202190 for 30 min prior to exposure with ALI pulmonary edema fluid. Protein levels were measured by Western blot analyses and expressed as the percent protein levels of plasma±S.D. of each sample measured in triplicates. *, p < 0.05 compared with plasma controls; †, p < 0.02 compared with ALI pulmonary edema-exposed cells. A representative Western blot is depicted above each column for the major sodium and chloride transport proteins. PE, pulmonary edema.
Effect of ALI Pulmonary Edema Fluid on Total Nitric Oxide Level
The total nitric oxide level was elevated in the ALI pulmonary edema fluid compared with the corresponding plasma (224 ± 7 versus 180 ± 3 μmol/liter, p < 0.001). Incubating the ALI pulmonary edema fluid with human alveolar epithelial type II cells for 24 h further increased the total nitric oxide level compared with the control plasma (371 ± 9 versus 180 ± 3 μmol/liter, p < 0.0005) (Fig. 12).
FIGURE 12. Nitric oxide levels of the ALI pulmonary edema fluid before and after incubation with human alveolar epithelial type II cells for 24 h.
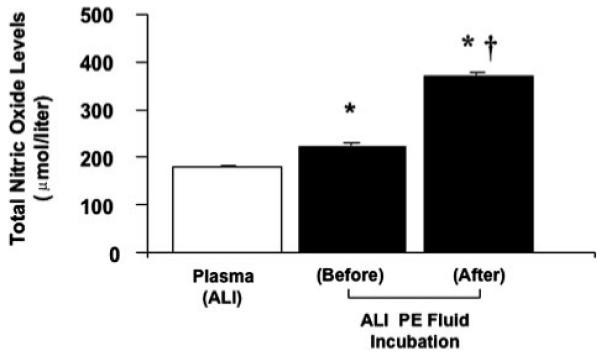
The total nitric oxide level was measured by the Griess reaction and expressed in μmol/liter±S.D. of each sample measured in triplicates. *, p < 0.001 compared with plasma controls; †,p<0.0005 compared with ALI pulmonary edema prior to exposure to alveolar epithelial type II cells. PE, pulmonary edema.
DISCUSSION
Impaired alveolar epithelial fluid transport is a characteristic feature in patients with ALI and has been associated with increased morbidity and mortality. Among ALI patients, we previously reported that patients with impaired AFC (<14%/h) exhibit reduced survival (62% hospital mortality versus 20% with AFC ≥ 14%/h) and a decreased median duration of unassisted ventilation (0 versus 23 days for AFC ≥ 14%/h) (2). Thus, the mechanisms responsible for the decrease in AFC are of major clinical and biological importance.
Several mechanisms can explain the decrease in AFC in the clinical setting. First alveolar epithelial barrier function in patients with ALI may be reduced because of severe injury from mechanical and inflammatory factors that results in epithelial cell death (apoptosis and necrosis) (3, 28, 29) as well as by hypoxia (30-32). Injury may also be mediated by activation of proinflammatory pathways that down-regulate sodium and chloride transporters that are responsible for vectorial fluid transport across the alveolar epithelium as we and others have hypothesized in prior studies (15-20). Therefore, in this study, we tested the direct effect of human ALI pulmonary edema fluid on cultured human alveolar epithelial type II cells to determine whether edema fluid, in contrast to simultaneously collected plasma, would alter net fluid transport.
The initial studies demonstrated that ALI edema fluid markedly reduced net fluid transport compared with plasma (Fig. 1). This was a remarkable finding that provides direct translation of our clinical studies of impaired alveolar epithelial fluid transport in patients with ALI (1, 2) to our laboratory model of cultured human alveolar type II cells. To study the mechanisms, we carried out further studies to evaluate the effects of ALI pulmonary edema fluid on cell death and the expression of the major known alveolar epithelial ion transporters on cultured human alveolar epithelial type II cells. The primary goal was to understand some of the mechanisms by which undiluted pulmonary edema fluid from patients with ALI reduced net fluid transport (i.e. resolution of pulmonary edema or AFC). We collected and combined pulmonary edema samples from 19 patients with ALI. We chose sepsis as the source of ALI due to the high mortality associated with this etiology of ALI (33) and combined the fluid due to the limited volume of pulmonary edema fluid available from each individual patient as in our prior studies (12, 13, 24).
Total cell death was modest for human alveolar epithelial type II cells exposed to ALI pulmonary edema fluid (10 ± 1%) compared with plasma (5 ± 1%) by 24 h. The level of necrosis was elevated in type II cells exposed to ALI pulmonary edema fluid (4 ± 1 versus 1 ± 0.3%, p < 0.01), whereas the level of apoptosis was not significantly different (Fig. 2). However, by confocal and transmission electron microscopy, the vast majority of the cells appeared intact, retaining features characteristic of alveolar epithelial type II cells without evidence of apoptosis or injury when exposed to either the ALI pulmonary edema fluid or plasma (Figs. 8 and 9). The modest increase in necrosis but not apoptosis among type II cells in these in vitro studies was in general agreement with several prior studies of lung injury (29, 34, 35).
To further determine whether the modest increase in total cell death caused by ALI pulmonary edema fluid contributed to the changes in protein permeability or net fluid clearance, we exposed human alveolar epithelial type II cells to cytomix, a mixture of the major biologically active cytokines found in ALI pulmonary edema fluid, as a surrogate for the pulmonary edema fluid itself (21). We previously found by flow cytometry that total cell death was not significantly different between human alveolar type II cells exposed to control media or cytomix, <5%. Similar to ALI pulmonary edema fluid, cytomix decreased net fluid clearance to zero while increasing protein permeability significantly above the level of the control media (Fig. 6, C and D). Cytomix also increased the gene expression of the major inflammatory cytokines and decreased the expression of the major sodium transport proteins in a dose-dependent manner (Fig. 7, A and B). Combined with the relatively low level of cell death in these current studies, the changes in net fluid transport were not likely to be primarily explained by necrosis or apoptosis.
In an earlier study, we reported that IL-1β and, to a lesser extent, TNFα are the predominant cytokines responsible for the proinflammatory activity in pulmonary edema fluid in patients with ALI (12, 13, 24). In this study, we confirmed that both TNFα and IL-1β were markedly elevated in the ALI pulmonary edema fluid compared with simultaneously collected plasma (Fig. 3).
In cultured human alveolar epithelial type II cells, ALI pulmonary edema fluid markedly increased the gene expression of all the major inflammatory cytokines and chemokines compared with plasma (Fig. 4A). The changes in gene copy numbers suggested that human alveolar epithelial type II cells were capable of propagating an inflammatory response once stimulated in an autocrine manner. In sharp contrast, ALI pulmonary edema fluid decreased the gene expression of all the major sodium and chloride transport proteins (Fig. 4B) that are essential for net fluid transport or AFC. By Western blot analysis (Fig. 5, A and B), the protein levels of αENaC, CFTR, and α1Na,K-ATPase decreased significantly. The decrease in protein levels of the major sodium and chloride transport proteins involved in AFC corroborates previous studies of the effect of individual cytokines (TNFα (17), IL-1β (18), and TGFβ1 (19, 20)) on the mRNA and protein levels of αENaC and CFTR and fluid transport.
In contrast to the other transport proteins, the gene copy number of β1Na,K-ATPase increased by 3% (p = not significant), and the protein levels increased by 58% on exposure to ALI pulmonary edema fluid compared with plasma (Fig. 5B). The β1 subunit, expressed in significantly lower amounts than the α1 subunit, is the regulatory and rate-limiting element in the assembly of the functional αβNa,K-ATPase enzyme complex whose expression is controlled at the transcriptional, posttranscriptional, and translational levels (32, 36, 37). In a state where Na,K-ATPase enzyme activity is decreased such as during exposure of LLC-PK1 cells, a pig kidney cell line, to low extracellular potassium, Lescale-Matys et al. (38) found that the gene expression of β1 subunit is increased compared with the α1 subunit, leading to an actual increase in the αβ active complex delivered to the membrane. The decrease in the sodium transport proteins, αENaC and α1Na,K-ATPase, as well as the likely consequent changes in the gradient and membrane potential due to ALI pulmonary edema fluid in these studies may stimulate an environment in which Na,K-ATPase enzyme activity is decreased, leading to an increase in β1Na,K-ATPase protein expression.
In parallel with the reduced gene and protein expression of the key alveolar epithelial transport proteins, net alveolar type II fluid transport was significantly decreased (Fig. 6A) whereas paracellular protein permeability was increased (Fig. 6B) for human alveolar type II cells exposed to ALI pulmonary edema fluid compared with plasma. Interestingly net fluid transport for human type II cells exposed to plasma from ALI patients was similar to that of human type II cells stimulated with forskolin and IBMX, suggesting that plasma from ALI patients can activate cAMP perhaps due to a modest increase in epinephrine levels as reported previously (39). The lack of improvement in net fluid transport for cells treated with forskolin and IBMX following treatment with ALI pulmonary edema fluid (Fig. 1) is consistent with our finding that CFTR mRNA and protein levels were significantly decreased following ALI pulmonary edema exposure; the CFTR protein is important in mediating the cAMP up-regulation of AFC especially in β-adrenergic receptor-driven alveolar Na+ transport (7, 9, 10, 40).
Despite the changes in the sodium and chloride transport proteins reported previously with individual cytokine exposure (15-20), pretreatment of the human alveolar type II cells with inhibitors of TNFα or TGFβ or with IL-1RA prior to exposure to ALI pulmonary edema fluid had no impact on net fluid transport or paracellular protein permeability perhaps because neutralization of individual cytokines has limited value in the context of the actual ALI edema fluid that contains several proinflammatory cytokines. Because of a limitation of the quantity of edema fluid available for study, we could not test the potential additive effect of combining different neutralization strategies such as IL-1RA plus an anti-TGFβ inhibitor.
Because neutralization of individual cytokines did not have any effect on reversing fluid transport, we tested inhibitors of intracellular signaling pathways that we and other investigators have found to be linked to vectorial sodium transport in prior studies of alveolar epithelial type II cells (18, 19, 32). Although inhibition of the ERK1/2 MAPK pathway had no effect, pretreatment of ALI edema fluid with SB202190, a p38 MAPK inhibitor (10 μm), improved net fluid transport (Fig. 6A). Pretreatment with SB202190 also attenuated the increase in paracellular protein permeability from 28% for cells treated with ALI pulmonary edema fluid alone to 11% for cells pretreated prior to exposure to ALI pulmonary edema, although this difference did not quite reach statistical significance (p = 0.06; Fig. 6B).
In addition to these functional effects, pretreatment with a p38 MAPK inhibitor prior to ALI pulmonary edema fluid exposure partially restored the gene copy number of the inflammatory cytokines/chemokines and sodium/chloride transport proteins closer to those of the plasma control (Fig. 10). In addition, Western blot analysis showed that αENaC, α1Na,K-ATPase, and β1Na,K-ATPase protein levels of human alveolar type II cells pretreated with SB202190 prior to ALI pulmonary edema fluid were partially restored to the levels of type II cells exposed to the plasma (Fig. 11, A-E).
Despite the restoration of the gene copy number of CFTR with SB202190 pretreatment, CFTR protein levels remained depressed following exposure to SB202190 and ALI pulmonary edema fluid. Regulation of CFTR-mediated chloride transport in the alveolar epithelium may occur at multiple levels: phosphorylation leading to channel opening, trafficking of CFTR to the apical membrane, CFTR endocytosis and degradation, and CFTR transcription (41). In this study, ALI pulmonary edema fluid decreased the gene expression and protein levels of CFTR compared with plasma. Our results were similar to previous studies that demonstrated that inflammatory cytokines (42) or mediators such as nitric oxide (NO) (43) are capable of suppressing CFTR cAMP-stimulated activity. In human colonic epithelial cells in culture, Howe et al. (44) demonstrated that TGFβ suppresses CFTR gene expression, intracellular protein levels, and apical membrane channel number. They also found that the inhibition of cAMP-mediated Cl- secretion by TGFβ is reversed with p38 MAPK inhibition not c-Jun NH2-terminal kinase (JNK) or ERK1/2 inhibition (42). In our current study, pretreatment with a p38 MAPK antagonist prior to ALI pulmonary edema fluid exposure fully restored the gene copy number of CFTR, but protein levels remained depressed. In the future, to understand the role of CFTR in net fluid transport on exposure to ALI pulmonary edema fluid, we may need to perform 1) biotinylation studies to determine the changes in the number of CFTRs at the apical membrane and 2) short circuit studies to determine any changes in CFTR activity. The persistent decrease in CFTR protein levels correlated with the lack of improvement in net fluid transport with cAMP agonist treatment following ALI pulmonary edema fluid exposure.
There are other possible mechanisms that could contribute to the decrease in alveolar epithelial fluid transport that we measured. Although we primarily studied the role of the inflammatory cytokines in this study, ALI pulmonary edema fluid contains other biologically active substances such as proteolytic enzymes, lipids, and reactive oxygen species, particularly the NO metabolite peroxynitrite, which can affect not only fluid clearance but other alveolar epithelial type II cell activity such as surfactant homeostasis. In a similar model in the rat, O’Brodovich and co-workers (45-48) found that endotoxin-stimulated alveolar macrophages or the corresponding supernatant decreases ENaC mRNA levels and activity as well as amiloride-sensitive sodium channel activity among distal lung epithelial cells. The inhibitory effect is prevented by the addition of a nitric-oxide synthase inhibitor or an antioxidant. The role of NO or reactive oxygen species in the activity of the ALI pulmonary edema fluid is complex and will require further extensive analyses. Interestingly we found that human ALI pulmonary edema significantly increased the level of NO induced from human alveolar epithelial type II cells following 24 h of exposure (Fig. 12).
Another potential contributing mechanism includes the trafficking of ENaC, CFTR, or Na,K-ATPase channels to the apical or basolateral membranes, although the failure of cAMP agonists to increase alveolar fluid clearance makes this possibility less likely (31). Because we studied type II cells alone, we cannot comment on what effects the edema fluid might have on alveolar epithelial type I cells. Recent work has shown that alveolar epithelial type I cells contain functional sodium and chloride transport proteins, which are essential for AFC (40). We also did not measure the electrophysiologic characteristics of these human type II cells, although these studies were beyond the scope of this work (49).
In conclusion, ALI pulmonary edema reduced the gene expression of major ion transporters in human alveolar type II cells. There was a substantial decrease in all of the major sodium and chloride transport gene and protein levels. These changes were associated with a decrease in net vectorial fluid transport as well as an increase in paracellular protein permeability across cultured human type II cell monolayers. Inhibition of p38 MAPK phosphorylation partially reversed the effect of the ALI pulmonary edema fluid on protein levels, net fluid transport, and paracellular permeability. These results demonstrate that ALI pulmonary edema fluid contains soluble factors that are capable of adversely affecting the resolution of pulmonary edema.
Footnotes
This work was supported by NHLBI, National Institutes of Health Grants HL-51856 and HL-51854 (to M. A. M.) and HL081332 (to L. B. W.) and by the John Severinghaus Fellowship (to J. W. L.). The abstract was presented in part at the American Thoracic Society Meeting, May 19-24, 2006 in San Diego, CA.
- AFC
- alveolar fluid clearance
- ALI
- acute lung injury
- CFTR
- cystic fibrosis transmembrane conductance regulator
- MAPK
- mitogen-activated protein kinase
- IL
- interleukin
- TNF
- tumor necrosis factor
- TGF
- transforming growth factor
- ERK
- extracellular signal-regulated kinase
- PBS
- phosphate-buffered saline
- RT
- reverse transcription
- GCN
- gene copy number
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MOPS
- 4-morpholinepropanesulfonic acid
- IBMX
- 3-isobutyl-1-methylxanthine
- ENaC
- epithelial sodium channel
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- IL-1RA
- interleukin-1 receptor antagonist
REFERENCES
- 1.Matthay MA, Wiener-Kronish JP. Am. Rev. Respir. Dis. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. Am. J. Respir. Crit. Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, Folkesson HG, Clerici C. Physiol. Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 4.Folkesson HG, Matthay MA. Am. J. Respir. Cell Mol. Biol. 2006;35:10–19. doi: 10.1165/rcmb.2006-0080SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matalon S, O’Brodovich H. Annu. Rev. Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 6.Eaton DC, Chen J, Ramosevac S, Matalon S, Jain L. Proc. Am. Thorac. Soc. 2004;1:10–16. doi: 10.1513/pats.2306008. [DOI] [PubMed] [Google Scholar]
- 7.Mutlu GM, Sznajder JI. Am. J. Physiol. 2005;289:L685–L695. doi: 10.1152/ajplung.00247.2005. [DOI] [PubMed] [Google Scholar]
- 8.Matalon S, Lazrak A, Jain L, Eaton DC. J. Appl. Physiol. 2002;93:1852–1859. doi: 10.1152/japplphysiol.01241.2001. [DOI] [PubMed] [Google Scholar]
- 9.Fang X, Song Y, Hirsch J, Galietta LJ, Pedemonte N, Zemans RL, Dolganov G, Verkman AS, Matthay MA. Am. J. Physiol. 2006;290:L242–L249. doi: 10.1152/ajplung.00178.2005. [DOI] [PubMed] [Google Scholar]
- 10.Brochiero E, Dagenais A, Prive A, Berthiaume Y, Grygorczyk R. Am. J. Physiol. 2004;287:L382–L392. doi: 10.1152/ajplung.00320.2002. [DOI] [PubMed] [Google Scholar]
- 11.Mutlu GM, Adir Y, Jameel M, Akhmedov AT, Welch L, Dumasius V, Meng FJ, Zabner J, Koenig C, Lewis ER, Balagani R, Traver G, Sznajder JI, Factor P. Circ. Res. 2005;96:999–1005. doi: 10.1161/01.RES.0000164554.21993.AC. [DOI] [PubMed] [Google Scholar]
- 12.Pugin J, Verghese G, Widmer MC, Matthay MA. Crit. Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- 13.Olman MA, White KE, Ware LB, Simmons WL, Benveniste EN, Zhu S, Pugin J, Matthay MA. J. Immunol. 2004;172:2668–2677. doi: 10.4049/jimmunol.172.4.2668. [DOI] [PubMed] [Google Scholar]
- 14.Ware LB, Matthay MA. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, Matthay MA. Am. J. Physiol. 2001;280:L1258–L1265. doi: 10.1152/ajplung.2001.280.6.L1258. [DOI] [PubMed] [Google Scholar]
- 16.Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, Brundler MA, Totsch M, De Baetselier P, Fransen L, Fukuda N, Morel DR, Lucas R. Am. J. Respir. Crit. Care Med. 2003;168:1043–1050. doi: 10.1164/rccm.200206-618OC. [DOI] [PubMed] [Google Scholar]
- 17.Dagenais A, Frechette R, Yamagata Y, Yamagata T, Carmel JF, Clermont ME, Brochiero E, Masse C, Berthiaume Y. Am. J. Physiol. 2004;286:L301–L311. doi: 10.1152/ajplung.00326.2002. [DOI] [PubMed] [Google Scholar]
- 18.Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, Canessa CM, Pittet JF. J. Biol. Chem. 2005;280:18579–18589. doi: 10.1074/jbc.M410561200. [DOI] [PubMed] [Google Scholar]
- 19.Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, Howard M, Canessa CM, Fang X, Sheppard D, Matthay MA, Pittet JF. J. Biol. Chem. 2003;278:43939–43950. doi: 10.1074/jbc.M304882200. [DOI] [PubMed] [Google Scholar]
- 20.Pruliere-Escabasse V, Fanen P, Dazy AC, Lechapt-Zalcman E, Rideau D, Edelman A, Escudier E, Coste A. Am. J. Physiol. 2005;288:L77–L83. doi: 10.1152/ajplung.00048.2004. [DOI] [PubMed] [Google Scholar]
- 21.Pittet JF, Lu LN, Morris DG, Modelska K, Welch WJ, Carey HV, Roux J, Matthay MA. J. Immunol. 2001;166:6301–6310. doi: 10.4049/jimmunol.166.10.6301. [DOI] [PubMed] [Google Scholar]
- 22.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. Am. J. Respir. Crit. Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 23.Murray JF, Matthay MA, Luce JM, Flick MR. Am. Rev. Respir. Dis. 1988;138:720–723. doi: 10.1164/ajrccm/138.3.720. [DOI] [PubMed] [Google Scholar]
- 24.Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, Matthay MA. Am. J. Respir. Crit. Care Med. 2001;163:1384–1388. doi: 10.1164/ajrccm.163.6.2006131. [DOI] [PubMed] [Google Scholar]
- 25.Geiser T, Ishigaki M, van Leer C, Matthay MA, Broaddus VC. Am. J. Physiol. 2004;287:L448–L453. doi: 10.1152/ajplung.00177.2003. [DOI] [PubMed] [Google Scholar]
- 26.Dolganov GM, Woodruff PG, Novikov AA, Zhang Y, Ferrando RE, Szubin R, Fahy JV. Genome Res. 2001;11:1473–1483. doi: 10.1101/gr.191301. [DOI] [PubMed] [Google Scholar]
- 27.Fang X, Song Y, Zemans R, Hirsch J, Matthay MA. Am. J. Physiol. 2004;287:L104–L110. doi: 10.1152/ajplung.00176.2003. [DOI] [PubMed] [Google Scholar]
- 28.Frank JA, Gutierrez JA, Jones KD, Allen L, Dobbs L, Matthay MA. Am. J. Respir. Crit. Care Med. 2002;165:242–249. doi: 10.1164/ajrccm.165.2.2108087. [DOI] [PubMed] [Google Scholar]
- 29.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, Zimmerman GA, Matthay MA, Ware LB. Am. J. Pathol. 2002;161:1783–1796. doi: 10.1016/S0002-9440(10)64455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivona ML, Matthay M, Chabaud MB, Friedlander G, Clerici C. Am. J. Respir. Cell Mol. Biol. 2001;25:554–561. doi: 10.1165/ajrcmb.25.5.4420. [DOI] [PubMed] [Google Scholar]
- 31.Planes C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. J. Biol. Chem. 2002;277:47318–47324. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- 32.Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. J. Clin. Investig. 2003;111:1057–1064. doi: 10.1172/JCI16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. N. Engl. J. Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 34.Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, Cutz E, Liu M, Keshavjee S, Martin TR, Marshall JC, Ranieri VM, Slutsky AS. J. Am. Med. Assoc. 2003;289:2104–2112. doi: 10.1001/jama.289.16.2104. [DOI] [PubMed] [Google Scholar]
- 35.Matute-Bello G, Liles WC, Steinberg KP, Kiener PA, Mongovin S, Chi EY, Jonas M, Martin TR. J. Immunol. 1999;163:2217–2225. [PubMed] [Google Scholar]
- 36.Shao Y, Ismail-Beigi F. Am. J. Physiol. 2004;286:C580–C585. doi: 10.1152/ajpcell.00117.2003. [DOI] [PubMed] [Google Scholar]
- 37.Vadasz I, Morty RE, Olschewski A, Konigshoff M, Kohstall MG, Ghofrani HA, Grimminger F, Seeger W. Am. J. Respir. Cell Mol. Biol. 2005;33:343–354. doi: 10.1165/rcmb.2004-0407OC. [DOI] [PubMed] [Google Scholar]
- 38.Lescale-Matys L, Hensley CB, Crnkovic-Markovic R, Putnam DS, McDonough AA. J. Biol. Chem. 1990;265:17935–17940. [PubMed] [Google Scholar]
- 39.Sakuma T, Gu X, Wang Z, Maeda S, Sugita M, Sagawa M, Osanai K, Toga H, Ware LB, Folkesson G, Matthay MA. Crit. Care Med. 2006;34:676–681. doi: 10.1097/01.CCM.0000201403.70636.0F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, Jain L, Dobbs LG, Eaton DC. Proc. Natl. Acad. Sci. U. S. A. 2006;103:4964–4969. doi: 10.1073/pnas.0600855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertrand CA, Frizzell RA. Am. J. Physiol. 2003;285:C1–C18. doi: 10.1152/ajpcell.00554.2002. [DOI] [PubMed] [Google Scholar]
- 42.Howe K, Gauldie J, McKay DM. Am. J. Physiol. 2002;283:C1667–C1674. doi: 10.1152/ajpcell.00414.2001. [DOI] [PubMed] [Google Scholar]
- 43.Skinn AC, MacNaughton WK. Am. J. Physiol. 2005;289:G739–G744. doi: 10.1152/ajpgi.00425.2004. [DOI] [PubMed] [Google Scholar]
- 44.Howe KL, Wang A, Hunter MM, Stanton BA, McKay DM. Exp. Cell Res. 2004;298:473–484. doi: 10.1016/j.yexcr.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 45.Rafii B, Gillie DJ, Sulowski C, Hannam V, Cheung T, Otulakowski G, Barker PM, O’Brodovich H. J. Physiol. 2002;544:537–548. doi: 10.1113/jphysiol.2002.024612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding JW, Dickie J, O’Brodovich H, Shintani Y, Rafii B, Hackam D, Marunaka Y, Rotstein OD. Am. J. Physiol. 1998;274:L378–L387. doi: 10.1152/ajplung.1998.274.3.L378. [DOI] [PubMed] [Google Scholar]
- 47.Dickie AJ, Rafii B, Piovesan J, Davreux C, Ding J, Tanswell AK, Rotstein O, O’Brodovich H. Pediatr. Res. 2000;48:304–310. doi: 10.1203/00006450-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 48.Elias N, Rafii B, Rahman M, Otulakowski G, Cutz E, O’Brodovich H. Am. J. Physiol. 2007 doi: 10.1152/ajplung.00373.2006. in press. [DOI] [PubMed] [Google Scholar]
- 49.Jain L, Chen XJ, Ramosevac S, Brown LA, Eaton DC. Am. J. Physiol. 2001;280:L646–L658. doi: 10.1152/ajplung.2001.280.4.L646. [DOI] [PubMed] [Google Scholar]