

Abstract
The sixth transmembrane segment (TM6) of the CFTR chloride channel has been intensively investigated. The effects of amino acid substitutions and chemical modification of engineered cysteines (cysteine scanning) on channel properties strongly suggest that TM6 is a key component of the anion-conducting pore, but previous cysteine-scanning studies of TM6 have produced conflicting results. Our aim was to resolve these conflicts by combining a screening strategy based on multiple, thiol-directed probes with molecular modeling of the pore. CFTR constructs were screened for reactivity toward both channel-permeant and channel-impermeant thiol-directed reagents, and patterns of reactivity in TM6 were mapped onto two new, molecular models of the CFTR pore: one based on homology modeling using Sav1866 as the template and a second derived from the first by molecular dynamics simulation. Comparison of the pattern of cysteine reactivity with model predictions suggests that nonreactive sites are those where the TM6 side chains are occluded by other TMs. Reactive sites, in contrast, are generally situated such that the respective amino acid side chains either project into the predicted pore or lie within a predicted extracellular loop. Sites where engineered cysteines react with both channel-permeant and channel-impermeant probes occupy the outermost extent of TM6 or the predicted TM5−6 loop. Sites where cysteine reactivity is limited to channel-permeant probes occupy more cytoplasmic locations. The results provide an initial validation of two, new molecular models for CFTR and suggest that molecular dynamics simulation will be a useful tool for unraveling the structural basis of anion conduction by CFTR.
The CFTR (cystic fibrosis transmembrane conductance regulator)1 chloride channel is encoded by the gene that is mutated in the inherited disease, cystic fibrosis. CFTR is apparently unique in the ABC transporter family of proteins in that binding and hydrolysis of ATP, catalyzed by the cytoplasmic nucleotide binding domains, produce a conformational change that opens an anion conduction path through which chloride can move by electrodiffusion (1−4). Studies of channel function employing amino acid substitutions and cysteine scanning have implicated elements of the membrane-spanning domains in the anion conduction process, but the three-dimensional structure of the pore remains obscure. Homology models of CFTR based on the crystal structure of the prokaryotic transporter Sav1866, including those presented here, predict that the 12 transmembrane segments (TMs) fold so as to form an anion-selective channel (1−3). However, low homology between CFTR and Sav1866 in the membrane-spanning domains (<20%) results in uncertainty as to the structure of the pore. One approach to validating models of the pore domain is to evaluate experimentally the predicted reactivity toward thiol-directed reagents of cysteine-substituted CFTR constructs. Previous studies of TM6 by means of cysteine scanning leave little doubt that this segment contributes to the lining of the pore (5−10), but these studies have produced conflicting results with regard to both the reactivity of specific constructs and the nature of the functional effects attributed to chemical modification. Cysteines substituted for residues thought to lie within the “outer vestibule” of the pore (e.g., R334 and K335) have been consistently identified as reactive toward covalent labeling, but cysteines substituted for more cytoplasmic residues (e.g., F337, T338, S341) were reported to be reactive in some studies but not others (5,6,8−10).
The lack of consistent results from previous cysteine-scanning studies may be attributable in part to methodological limitations that we have tried to overcome in the present work. Earlier studies often employed thiol-reactive reagents like MTSET+ and MTSES− that, based on the results presented here, are unlikely to pass through the channel, so that their access to deeper lying (more cytoplasmic) residues is expected to be minimal. In addition, previous studies did not uniformly control for possible “spontaneous reactions” of the engineered cysteines that can block reactivity toward thiol-reactive probes (11), nor was the nature of the chemical reaction always confirmed, for example, by determining if a mixed disulfide reaction (as expected for MTS reagents) could be reversed by means of a reducing agent. Finally, none of the previous scanning studies have systematically compared the observed reactivity of substituted cysteines with the predictions of molecular model of the CFTR pore, although Mornon et al. (2) attempted this recently using published (albeit conflicting) data.
The aim of the present study was to scan the reactivity of cysteines substituted into TM6 (as defined by hydropathy, residues 330−353) using both channel-permeant (12) and channel-impermeant reagents and to map the observed reactivity onto molecular models of the CFTR pore. Reactive cysteines are presumed to be those for which the side chain, during at least some portion of the gating cycle, lies within a polar environment in which the thiol can ionize and participate in thiol−disulfide exchange (MTS compounds) or ligand exchange (metal dicyanates). Reactive cysteines, therefore, are expected to be those projecting into either the anion conduction path or some crevice that is water-accessible and probe-accessible during at least some phase of the gating cycle. Unreactive cysteines are presumed to be those located such that the thiol is occluded and either not water-accessible or not accessible to the thiol-reactive probe. We reasoned that the use of both channel-permeant and channel-impermeant probes would serve as a partial control for differences in thiol reactivity deriving from factors such as local differences in geometry or channel diameter and that molecular models would provide a framework for rationalizing patterns of reactivity in terms of underlying channel structure. Where possible, cysteines were engineered into a Cys-less background (13) to eliminate ambiguity as to the site of the thiol reaction, and care was taken to control for the potential effects of “spontaneous” reactions of the engineered cysteines (11).
We present here two new molecular models of the CFTR pore, one a homology model based on the crystal structure of the bacterial ABC transporter, Sav1866 (14), and the second based on 5 ns molecular dynamics simulation of the first. When the differential reactivity of substituted cysteines was mapped onto the 0 and 5 ns models, three regions could be clearly discerned: one comprising nonreactive sites and two comprising reactive sites. Nonreactive sites tended to cluster on the surface of TM6 that faces away from the predicted pore and toward the loop formed by TMs 3 and 4. The reactive regions comprised a more extracellular region where modification was relatively nonselective and a more cytoplasmic region where reactivity was restricted to the permeant, pseudohalide probes, [Au(CN)2]− or [Ag(CN)2]−. Side chains occupying reactive sites were predicted by the molecular models to face toward the pore. The striking correspondence between the predictions of the two models and the scanning results suggests that the two models provide a reasonably accurate depiction of the TM6 residues that form the “lining” of the CFTR pore.
Materials and Methods
Mutagenesis and In Vitro Transcription
The Cys-less CFTR construct (C76S, C126S, C225S, C276S, C343S, C491S, C524S, C590L, C592L, C657S, C832S, C866S, C1344S, C1355S, C1395S, C1400S, C1410S, C1458S) was a gift from Drs. Martin Mense and David Gadsby and was used in their pGEMHE vector previously described (13). The QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) was used to generate point mutations to both wild-type (wt) CFTR and the Cys-less CFTR constructs, as described previously (7,12,15). Mutations were confirmed by direct DNA sequencing. The CFTR cRNAs for Xenopus oocyte injection were synthesized using the Ambion mMessage mMachine T7 Ultra transcription kit as described previously (7,12,15).
Controls for the Effect of Cysteine Substitutions
Unless otherwise noted, the reactivity of engineered cysteines was first assayed on a wt background, and the values that appear in Table 1 reflect those obtained using these constructs. Cysteines that registered as reactive in one or more assays were then studied using a construct in which the single cysteine was placed on a Cys-less background to confirm the identity of the target. All of the reagents utilized in this study were tested extensively for reactivity with oocytes expressing either wt or Cys-less CFTR, at concentrations ranging from 1 μM to 1 mM and times of exposure ranging from minutes to hours.
Table 1. Percent Change in Oocyte Conductance in the Presence of Compounda.
| MTSET+ | MTSES− | [Ag(CN)2]− | [Au(CN)2]− | |
|---|---|---|---|---|
| G330C | ○ | ○ | ○ | ○ |
| I331C | −51.6 ± 6.3 | −28.9 ± 2.1 | −63.1 ± 8.8 | ○ |
| I332C | ○ | ○ | ○ | ○ |
| L333C | −58.5 ± 4.8 | −47.5 ± 7.6 | −83.1 ± 2.2 | ○ |
| R334C | +76.9 ± 11.3 | −84.4 ± 1.5 | −67.4 ± 7.4 | −41.4 ± 3.1 |
| K335C | +10.7 ± 2.4 | −37.3 ± 1.5 | −29.1 ± 6.4 | −54.6 ± 4.7 |
| I336C | −54.4 ± 7.9 | −75.0 ± 0.6 | −81.2 ± 10.5 | ○ |
| F337C | ○ | ○ | −89.6 ± 1.9 | −90.1 ± 1.3 |
| T338C | −37.1 ± 3.3 | −85.4 ± 2.5 | −75.0 ± 5.2 | −88.3 ± 1.6 |
| T339C | ○ | ○ | −24.5 ± 7.2 | ○ |
| I340C | ○ | ○ | −93.8 ± 1.0 | ○ |
| S341C | ○ | ○ | −49.3 ± 4.8 | ○ |
| F342C | ○ | ○ | −84.7 ± 1.8 | ○ |
| C343 | ○ | ○ | ○ | ○ |
| I344C | ○ | ○ | −66.9 ± 9.3 | −77.9 ± 2.1 |
| V345C | ○ | ○ | −49.1 ± 9.3 | ○ |
| L346C | ○ | ○ | ○ | ○ |
| R347C | ○ | ○ | ○ | ○ |
| M348C | ○ | ○ | −47.9 ± 8.8 | −50.1 ± 3.3 |
| A349C | ○ | ○ | −19.0 ± 2.0 | ○ |
| V350C | ○ | ○ | ○ | ○ |
| T351C | ○ | ○ | ○ | ○ |
| R352C | ○ | ○ | −77.5 ± 1.3 | ○ |
| Q353C | ○ | ○ | −72.6 ± 4.5 | −76.7 ± 2.8 |
Values are means ± SE of three or more oocytes. ○ = nonreactive.
Controlling for Spontaneous Reactions
All oocytes expressing cysteine-substituted CFTR constructs were screened for responses to 2-ME or DTT prior to any exposure to any of the thiol-reactive probes employed here. This protocol was based on an extensive documentation of “spontaneous” changes in the chemical state of a cysteine substituted at position 338 (11). We proposed that these spontaneous changes, that are not seen in either wt or Cys-less CFTR, reflect the coordination of trace amounts of copper (or some other, as yet unidentified, metal) by the Cys-substituted construct that can be reversed by mobile, metal ligands such as 2-ME, DTT, and CN−.
Homology Modeling
A homology model of the four “core” domains of the human CFTR protein was generated, consisting of two chains. Chain 1 is composed of transmembrane domain 1 (TMD1) and nucleotide binding domain 1 (NBD1; residues 68−664), and chain 2 is composed of TMD2 and NBD2 (residues 846−1455). The regulatory (R) domain (which lies between NBD1 and TMD2) was omitted, as there is no suitable template for this region. The template for all four domains was the 3D structure of the bacterial ABC transporter Sav1866 (14), which shares the “2 × 6” TM helix topology with CFTR. Each “half” of CFTR was aligned with Sav1866 using MUSCLE (16), yielding sequence identities of 16% at the TMDs and 26−27% at the NBDs. MODELER (17) was then used to build a set of models from the MUSCLE alignments, with the lowest energy structure (based on the MODELER energy term) selected for this study. A short energy minimization of the final model was performed using the GROMACS molecular dynamics package (18) as well as a check for stereochemical quality using PROCHECK (19).
Molecular Dynamics Simulation
To explore the short time scale conformational stability and dynamics of the Sav-based CFTR model, a 5 ns molecular dynamics (MD) simulation was performed in a phospholipid bilayer. The CFTR model was embedded in a dimyristoylphosphatidylcholine (DMPC) bilayer, containing 463 lipid molecules. Energy minimization of this complex was followed by solvation with 68921 explicit water molecules and 21 chloride counterions to preserve electroneutrality. This final system was further energy minimized and comprised a total of ca. 230000 atoms. Equilibration of the system was performed, with the protein heavy atoms (non-hydrogen) restrained, in order to allow a relaxation of the packing of the lipids around the protein. The production run was then carried out for a total of 5 ns, with all restraints removed. A snapshot of the simulation system is shown in the section (Figure S1).
MD simulation was carried out with GROMACS v3.2.1 (http://www.gromacs.org) (18,20,21), using the GROMOS96 force field (22). The CFTR model was positioned in the DMPC bilayer using a coarse-grained self-assembly simulation approach (23−26). Simulations were run in the NPT ensemble (constant number of molecules, temperature, and pressure) at 310 K and 1 atm. The system was equilibrated for 0.5 ns, with the non-H atoms of the protein harmonically restrained (force constant 1000 kJ mol−1 nm−2). During equilibration the Berendsen thermostat and Berendsen barostat algorithms (27) were employed, while the 5 ns production run employed the Nosé−Hoover thermostat (28,29) and Parrinello−Rahman barostat (30) algorithms. Long-range electrostatic interactions were calculated with the particle mesh Ewald (PME) method (31). All bonds were constrained using the LINCS algorithm (32), with the use of a 2 fs time step.
Preparation and Microinjection of Oocytes
The preparation and microinjection of Xenopus laevis oocytes were performed using methods previously described in detail (33,34,11). The follicular membranes were removed by mechanical agitation (1−2 h) in a Ca2+-free solution containing (mM) 82.5 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES-hemi Na, pH 7.5, with 0.2 Wünsch unit/mL Liberase Blendzyme 3 (Roche Molecular Biochemicals, Indianapolis, IN). Defolliculated oocytes were washed and maintained in a modified Barth’s solution containing (mM) 88 NaCl, 1 KCl, 0.82 MgSO4, 0.33 Ca(NO3)2 0.41 CaCl2, 2.4 NaHCO3, 10 HEPES-hemi Na, and 250 mg/L Amikacin at pH 7.5. Stage V−VI oocytes were injected with CFTR cRNA and cRNA encoding the human β2-adrenergic receptor. CFTR cRNA was diluted to yield 50−200 μS of stimulated conductance, that is, ∼0.2 ng/oocyte in a 50 nL volume for most constructs. Cys-less variants required 5−10 ng of cRNA per oocyte.
Whole-Cell Recordings
Whole-cell recording methods were similar to those described by Mansoura et al. (35). Briefly, individual oocytes were placed in the recording chamber and continuously superfused with frog Ringer’s solution. The Ringer’s solution contained (mM) 98 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES-hemi Na, at pH 7.4. The TEVC-200 amplifier (Dagan Corp., MN) and the pClamp 8 data acquisition program (Axon Instruments, Inc., CA) were used for data acquisition. Oocytes were maintained in the open circuit conditio, and the membrane potential was periodically ramped from −120 to +60 mV over 1.8 s to construct the whole-cell I−V plots.
Reagents
The experiments presented here were conducted using 10 μM isoproterenol (Sigma, St. Louis, MO) and 1 mM IBMX (Sigma) as the stimulating cocktail (Isop + IBMX). Methanethiosulfonate reagents (MTSET+, MTSES−, MMTS) were purchased from Toronto Research Chemicals (Toronto, Canada). 2-Mercaptoethanol (2-ME), dithiothreitol (DTT), N-ethylmaleimide (NEM), iodoacetamide (IAM), K[Au(CN)2], and K[Ag(CN)2] were obtained from Sigma (St. Louis, MO). KCN was obtained from Fisher Chemicals (Fairlawn, NJ).
Results
Defining “Reactivity” for Different Classes of Reagents
In evaluating the impact of a panel of reagents on cysteine-substituted CFTRs, we found it necessary to employ several different criteria for reactivity. The simplest operational definition was the observation of a change in CFTR conductance that was “irreversible” in that it persisted after removing the reagent from the perfusion solution. In addition, we required that the reaction block reactivity toward other reagents was not seen in the Cys-less CFTR and could be reversed by exposure to a reducing agent (for the MTS compounds) or a competing metal ligand (for [Au(CN)2]− and [Ag(CN)2]−). The small thiols, 2-ME and DTT, served as both reducing agents and competing metal ligands, and CN− also served as a competing metal ligand. For several constructs we also determined if the reaction could be blocked by using either N-ethylmaleimide (NEM) or iodoacetamide (IAM) to alkylate the cysteine thiolate prior to exposure.
In the case of the pseudohalides, [Au(CN)2]− and [Ag(CN)2]−, we adopted a definition that allowed for reversible thiol modification, that is, reactions that reversed spontaneously when the pseudohalide was removed from the perfusate. In such instances it was possible to implicate a metal−thiolate reaction by showing that the inhibition could be reversed by adding excess KCN (1 mM) in the continued presence of the metal cyanide, thereby shifting the equilibrium of the reaction away from the mixed ligand product. The latter approach allowed us to distinguish a specific interaction of either [Ag(CN)2]− or [Au(CN)2]− with an engineered cysteine from the relatively nonspecific, “lyotropic” binding reported previously for [Au(CN)2]−36,37. In those instances in which none of the criteria for a reaction was met (Table 1), it is not possible in principle to eliminate with complete confidence the hypothesis that reactions occurred without functional effect, although the use of a panel of compounds reacting by different mechanisms renders this less likely.
A Cys-less CFTR Construct Was Not Reactive toward Channel-Permeant Reagents
We reported previously (12) that the Cys-less CFTR construct is not reactive toward MTS reagents and [Au(CN)2]−, but in the context of the present study it was important to test the Cys-less CFTR for reactivity toward [Ag(CN)2]−, as well as to characterize any lyotropic block by this compound. [Ag(CN)2]− is nearly identical to [Au(CN)2]− in size and hydration energy, but it is evident from the representative experiment depicted in Figure 1 that the blocking efficacy of [Ag(CN)2]− toward Cys-less CFTR channels (as well as wt channels) was substantially less than that seen previously with [Au(CN)2]−. Block of Cys-less CFTR by [Au(CN)2]− exhibited an apparent affinity of about 1 mM, similar to that seen with wt CFTR (12,37). In contrast, block of both wt and Cys-less CFTR by [Ag(CN)2]− exhibited an affinity that was at least 10-fold less, about 10 mM. Neither of these inhibitory effects was relieved by adding an excess (1 mM) of the competing ligand, CN−, as KCN. In fact, the addition of 1 mM KCN in the presence of 1 mM [Ag(CN)2]− actually produced a small additional decrease in conductance not seen with KCN alone . These results suggest that the Au(1) metal center interacts more favorably with non-thiol components of the CFTR pore than does the Ag(1) metal center but that neither of these blocking interactions involves ligand exchange or ligand addition. We speculate that the small exacerbation of block seen with [Ag(CN)2]− and KCN could reflect the formation of an unknown amount of the tricoordinate, Ag(1) species, [Ag(CN)3]2-, that may have increased affinity for the Cys-less CFTR channel or increased blocking efficacy due to the additional negative charge (38). We also confirmed that [Ag(CN)2]− is a permeant anion by demonstrating that an outward current seen in the presence of 30 mM external [Ag(CN)2]− ([Cl]o = 0) was blocked by GlyH-101, a specific CFTR, pore blocker (39) ( Figure S2).
Figure 1.

Differential effects of [Au(CN)2]− and [Ag(CN)2]− on Cys-less CFTR. Exposure to [Au(CN)2]− (1 mM) produced a “lyotropic” block that was not altered by exposure to KCN (1 mM). Exposure to [Ag(CN)2]− (1 mM) produced a substantially smaller block that was exacerbated by adding KCN in the continued presence of [Ag(CN)2]−.
The Overall Pattern: Selective and Nonselective Thiol Reactivity
The apparent reactivity of cysteines engineered into TM6 toward channel-permeant and channel-impermeant reagents is summarized in Table 1. Each of the four columns compares the apparent reactivity of cysteines substituted at different positions along TM6 toward channel-permeant and channel-impermeant, thiol-directed reagents. It is expected that scanning from the extracellular extent of TM6 toward the more cytoplasmic portion of TM6 will reveal selective reactivity related to changes in channel geometry as well as the differential properties of the thiol-reactive probes. The overall pattern of reactivity seen in Table 1 suggests that two regions can be delineated: one, more extracellular, exhibiting relatively nonselective reactivity, and another, more cytoplasmic, exhibiting selective reactivity toward channel-permeant reagents. In the following section we consider in more detail the thiol reactivity in these two regions of TM6.
Outer Region: Charge-Dependent and Charge-Independent Functional Effects
Cysteines at the more extracellular sites appeared to react with all (334, 335, and 338) or nearly all (331, 333, and 336) of the reagents utilized here, but there were distinct differences in the functional effects at different locations. In previous reports we described functional effects at positions 334, 335, and 338 that exhibited the charge dependence expected for a simple charged-vestibule model, that is, enhancement of conductance by deposition of positive charge and attenuation of conductance by deposition of negative charge, as well as predicted changes in the shape of the I−V curve (7,8). Charge dependence was most dramatic at position 334 where deposition of a positive charge (MTSET+ or MTSEA+) enhanced conductance, whereas deposition of a negative charge (MTSES−) attenuated conductance, a result confirmed by Fatehi and Linsdell (10) and Beck et al. (9). Furthermore, these charge-dependent changes in conductance could be described by a version of the Goldman equation, modified to include the effect of a charged vestibule. Charge-dependent changes could also be evoked by varying the bath pH between 6.0 and 9.0 so as to titrate the partial charge on the cysteine thiolate.2
We interpreted charge-dependent changes in conductance as indicating that residues at positions 334, 335, and 338 were positioned such that a charged side chain (native or chemically created) contributes to the electrostatic potential of the pore vestibule. In the present study, however, we identified three locations (331, 333, and 336) in the outer region of TM6 where the functional effects of MTS reagents, although they varied somewhat with charge, did not exhibit changes expected from a charged-vestibule model. Positions 331 and 333 had been previously identified as reactive sites by Cheung et al. (5,6) and Beck et al. (9). Figure 2 contains an example of results obtained with L333C CFTR on a wt background (L333C/wt). Here both MTSET+ and MTSES− produced an inhibitory effect on conductance, but at 331 inhibition was greater with the positive adduct and at 333 the functional effects of the positive and negative adducts were roughly equivalent. This result can be contrasted with that reported previously for a cysteine at 338 (8). Here, modification by either MTSET+ or MTSES− produced a decrease in conductance, but charge effects expected for a vestibule were, nevertheless, evident in the more profound inhibition by the negatively charged adduct. Reactions of MTSET+ and MTSES− with L333C/wt CFTR channels were reversed by exposure to DTT, but the rate of reversal was slower in the case of MTSES−-modified channels, as expected due to the unfavorable electrostatic interaction between the negatively charged adduct and the anionic DTT. At position 336 attenuation of conductance was greater for negatively charged adducts, but the rate of reaction was at least 10 times slower (t1/2 = 20 min; see Figure S9) than rates seen at 334, 335, and 338, as if access or reactivity of a cysteine at this site might be restricted. The CFTR models presented here suggest, in fact, that a cysteine at this position might be partially occluded by the TM3−4 loop (see below).
Figure 2.

Functional impact of covalent labeling of L333C CFTR was charge-independent. After activation by isoproterenol and IBMX, exposure to either 1 mM MTSET+ or 1 mM MTSES− produced a substantial inhibition of conductance.
Inner Region: Selective Reactivity toward Channel-Permeant Reagents
The results compiled in Table 1 demonstrate that the cysteine-substituted CFTR constructs that exhibited reactivity toward MTS reagents also reacted with at least one of the two, metal-based, ligand exchange reagents. These latter reagents, however, also appeared to react with 11 additional cysteines, 1 at position 337 and 10 placed cytoplasmic to position 338 (apparently the site of the deepest, or most cytoplasmic, reactivity of MTSET+ and MTSES−). This is the result expected if channel-permeant probes like [Au(CN)2]− and [Ag(CN)2]− can reach cysteines that are inaccessible to, or unreactive toward, the bulkier MTS reagents.
Lack of Reactivity of F337C CFTR toward MTSET+ and MTSES−
As indicated in the introduction, previous attempts to assess the reactivity of cysteines placed at position 337 have not produced consonant results. Beck et al. (9) reported no reactivity of F337C on a wt CFTR background (F337/wt CFTR) toward MTSEA+ or MTSES−, but Cheung and Akabas (5,6) and Fatehi and Linsdell (10) reported reactivity toward both MTSET+ and MTSES−. Accordingly, we examined the reactivity toward MTS reagents of F337C CFTR (wt and Cys-less backgrounds) carefully to ensure that any change in conductance observed in the presence of an MTS reagent met our criteria for a thiol−disulfide exchange reaction. In some experiments we observed that exposure of oocytes expressing F337C/wt CFTR to MTSET+ or MTSES− produced decreases in conductance. Some of these changes were reversed by simply washing off the compound while others persisted to a variable extent after washing. We determined, however, that these variable effects of exposure to MTSET− or MTSES− were not due to thiol−disulfide exchange reactions.3
Reactivity of F337C CFTR toward Channel-Permeant Probes
The reactivity of F337C/wt CFTR toward the channel-permeant probes, although similar to that seen previously with T338C/wt CFTR (12), differed significantly in detail. Exposure of oocytes expressing F337C/wt CFTR to 1 mM [Au(CN)2]− produced a profound inhibition that was not reversed by superfusing the oocytes with a [Au(CN)2]−-free solution (Figure 3). After inhibition of F337C conductance by [Au(CN)2]−, exposure of oocytes to a competing thiol, 2-ME, did not reverse the inhibition of conductance as previously seen with T338C/wt CFTR (12), but the inhibition was relieved by exposing the oocyte to a solution containing 1 mM KCN as expected from the high-affinity liganding of Au(1) by the cyanide anion (12). Similar results were obtained with oocytes expressing F337C/Cys-less CFTR, confirming that the cysteine at 337 is the site of the reaction with [Au(CN)2]− ( Figure S3). F337C/Cys-less CFTR was also reactive toward the second, channel-permeant probe, [Ag(CN)2]−. In the latter case, the reaction was more likely to reverse spontaneously upon washout of the reagent, but reactivity was confirmed by exposing the oocyte to KCN as described in Materials and Methods (not shown). The reaction of oocytes expressing F337/wt CFTR with [Au(CN)2]− was blocked by prior exposure to an alkylating agent, NEM, or the smallest MTS compound, MMTS ( Figure S4).
Figure 3.

Selective reactivity of F337C CFTR. Exposure of an oocyte to 1 mM MTSET+ or 1 mM MTSES− resulted in small, reversible reductions of conductance. In some experiments, no change in conductance was seen after exposure to these reagents. Subsequent exposure of the oocyte to [Au(CN)2]− (1 mM) produced a profound inhibition of conductance that was only slightly reversed by removing [Au(CN)2]− from the perfusate. Inhibition was not reversed by 1 mM 2-ME but was reversed by exposure to 1 mM KCN.
These results are consistent with the hypothesis that a cysteine at 337 is positioned within the CFTR pore such that it can react with the channel-permeant, thiol-reactive compounds, [Au(CN)2]− and [Ag(CN)2]−, as well as the small mixed disulfide, MMTS, but the location or orientation of the side chain, and perhaps other environmental constraints, precludes reaction with MTSET+ and MTSES−. Likewise, the reversal of the [Au(CN)2]− ligand exchange product (protein-S-[AuCN]−) and the MMTS-protein mixed disulfide product by small thiols like 2-ME or DTT was not observed despite the fact that these small thiols can reverse the inhibition seen with [Ag(CN)2]− (not shown).
Narrowing of the Pore Cytoplasmic to Position 338
The distinctive pattern of reactivity seen with the two channel-permeant reagents, [Au(CN)2]− and [Ag(CN)2]− (Table 1, columns 3 and 4), points to a possible narrowing of the pore after residue 338. For example, the permeant reagents reacted with a cysteine at 341, but contrary to the reports of Cheung and Akabas (5,6) and Fatehi and Linsdell (10) we found no evidence for a reaction of a cysteine at this locus with either MTSET+ or MTSES−. That this could reflect the size and/or polarity of the latter reagents is suggested by the observation that S341C, like F337C CFTR, clearly reacts with a smaller MTS compound, MMTS (not shown).
The results depicted in Figure 4 show that exposure of S341C/Cys-less CFTR to 100 μM [Ag(CN)2]− produced about 70% inhibition of conductance (gCl), substantially greater than that seen in wt or Cys-less CFTR. The inhibition was readily and completely reversed by washing (not shown), but adding KCN to the superfusion solution (in the continued presence of [Ag(CN)2]−) produced a stepwise reversal of inhibition consistent with reversal of a reaction between [Ag(CN)2]− and the cysteine thiolate. In contrast, exposure of S341C CFTR to [Au(CN)2]− produced inhibition that was similar to that seen in the wt or Cys-less CFTR and was not altered by KCN, indicating the absence of a ligand exchange reaction. Thus, although the cysteine at 341 met our criteria for a reactive site, its reactivity toward two channel-permeant pseudohalides differed significantly from that of a cysteine at 337.
Figure 4.

Differential reactivity of S341C/Cys-less CFTR toward [Ag(CN)2]− and [Au(CN)2]−. Inhibition of conductance by [Ag(CN)2]− (100 μM) was reversed by adding KCN in the continued presence of [Ag(CN)2]−, indicative of a ligand exchange or ligand addition reaction. In contrast, inhibition by [Au(CN)2]− (1 mM) was unaffected by adding KCN as expected for lyotropic block.
Cysteines placed at 339, 340, 341, 342, 344, 345, 348, 349, 352, and 353 all exhibited selective reactivity toward the channel-permeant reagents, [Ag(CN)2]− and [Au(CN)2]− (Table 1). Representative records appear in Figure 5 and Figures S5, S6, S7, and S8. We had previously reported that R352C/wt CFTR was not reactive toward MTSET+ and MTSES−(7), a result consistent with the findings of Beck et al. (9). We also reported, however, that R352C/wt CFTR displayed an unusual, spontaneously reversible reaction with MTSEA+(7). In those experiments, however, qualitatively identical, reversible reactivity toward MTSEA+ was also seen using either R352Q/wt or R352H/wt CFTR constructs, suggesting that the target of MTSEA+ was 1 of the 18 endogenous cysteines in the R352C/wt protein (7). It was essential, therefore, that we explore reactivity toward channel-permeant probes at position 352 using both wt and Cys-less backgrounds. The experiment depicted in Figure 5 demonstrates that R352C/Cys-less CFTR exhibited reactivity toward [Ag(CN)2]−. Reactivity toward [Au(CN)2]− was not seen, and experiments with R352C/wt CFTR yielded identical results (not shown). We also confirmed that neither R352C/wt nor R352C/Cys-less exhibits reactivity toward either MTSET+ or MTSES− (not shown), contrary to the reports of Chueng and Akabas (5,6).
Figure 5.

R352C/Cys-less CFTR was reactive toward [Ag(CN)2]− (1 mM) as judged by reversal of inhibition by adding KCN (1 mM) in the continued presence of [Ag(CN)2]−.
Nonreactive Sites
Table 1 lists seven locations in TM6 where cysteines were apparently unreactive across the panel of reagents used for these experiments. We cannot eliminate completely the possibility that reactions occurred that were without a discernible functional consequence, but the molecular models presented below suggest that the majority of these residues are positioned in such a way as to preclude thiol−disulfide exchange or ligand exchange reactions with a substituted cysteine, perhaps due to close apposition to other helices or the lipid bilayer.
Mapping Reactive and Nonreactive Sites onto a Model of the CFTR Pore
Any attempt to formulate a structural interpretation of the reactivity of cysteines substituted into CFTR must begin by considering two potential issues. First, constructing a Sav1866-based model of CFTR required the alignment of sequences that were less than 20% similar, so that it was essential to apply an experimental method like a cysteine scan to begin to validate structural predictions. Second, the crystal structure of Sav1866 represents a single state of that protein. Accordingly, the resulting CFTR homology model must be taken to represent one potential structure of CFTR, of several that the protein could potentially visit during its gating cycle. In an attempt to address the latter issue, we conducted a 5 ns molecular dynamics simulation using our Sav-based model as a starting point. The procedure allowed us to explore the stability of the homology model, refine it, and possibly sample other potential conformational states of the protein that lie nearby to the starting point on the energy landscape.
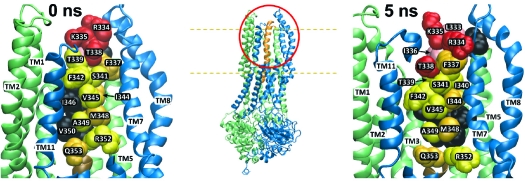
Figure 6 contains representations of both the 0 and 5 ns models of CFTR based on the crystal structure of Sav1866 (0 ns) and the MD simulation (5 ns). The overall body plan of the predicted CFTR channel ( Figure S1 and movies M1 and M2) is similar to that presented in models developed by Gadsby et al. (4), Serojijos et al. (1), Mornon et al. (2), and Jordan et al. (3), in that the 12 TMs are predicted to surround a pore-like cavity that is visible from the extracellular side. The two NBDs form a gondola-like structure on the cytoplasmic side that is joined to the TMs via intracellular loops. We did not attempt to model the R-domain as Sav1866 lacks this structural element.
Figure 6.

Three views of two models of the CFTR pore based on Sav1866 and a 5 ns molecular dynamics simulation. Insets show the portion of the protein that is enlarged in each panel, and dashed lines indicate the approximate boundaries of the lipid bilayer. Also, the contains two movies that offer an overall view of the two models. Cytoplasmic domains have been removed from the enlarged views. (A) Top view looking “down” into the pore from the extracellular side. (B) Side view with TMs 9, 10, and 12 partially removed to reveal the “pore-lining” face of TM6. (C) Side view from behind the TM3−4 loop. Residues indicated in black are those where substituted cysteines were unreactive with any of the probes employed in this study. Residues colored in red and pink represent sites where thiol reactivity was detected toward both channel-permeant and channel-impermeant, thiol-directed reagents. The pink residues are sites where the resulting functional effects were charge-independent, whereas those in red represent sites where functional effects were distinctly charge-dependent. Residues colored in yellow and orange represent those sites where cysteines reacted exclusively with permeant reagents, [Ag(CN)2]− and [Au(CN)2]−. [Ag(CN)2]− reacted at all of these sites while [Au(CN)2]− reacted only at those labeled in orange. All molecular representations were generated using “Visual Molecular Dynamics” (vmd) (54).
The six panels of Figure 6 illustrate the reactivity of cysteines substituted in TM6 toward channel-permeant and channel-impermeant reagents. The insets indicate the locations that are enlarged in each pair of panels. Each pair compares the 0 and the 5 ns models from three views: From the extracellular end looking “down” into the pore (Figure 6A), from the side looking through TMs 9, 11, and 12, which have been partially removed to create a window onto the “pore-lining” face of TM6 (Figure 6B), and from the side behind the TM3−4 loop (Figure 6C). The pattern of reactivity exhibited by the substituted cysteines suggests three, more or less distinct groups: those sites where the cysteine was unreactive, those where the cysteine was nonselectively reactive, and those where the cysteine was selectively reactive toward channel-permeant reagents. Residues colored in black represent those sites where substituted cysteines were not reactive toward any of the reagents used in this study. The majority of these residues (C343, L346, R347, V350, T351) tend to be located on the side of TM6 that is predicted by both the 0 and 5 ns models to face “away” from the pore, as if cysteines at these positions might be occluded by apposition to the extracellular loop connecting TMs 3 and 4 (Figure 6C). This apparent occlusion is more obvious in the 5 ns model. Position 332, predicted to reside in the TM5−TM6 loop, is not obviously occluded in the 0 ns model but in the 5 ns model appears to be occluded by TMs 7 and 8. Position 330 is not obviously occluded in the 5 ns model but could be occluded by relatively minor motions of the TM7−8 and TM3−4 loops.
Residues colored in red and pink represent sites where cysteine reactivity that was nonselective, that is, sites where reactions were seen with both channel-permeant and channel-impermeant, thiol-directed reagents. Those in red represent sites where functional effects were charge-dependent in the manner expected for a simple, charged-vestibule model of the pore (7). The pink residues are sites where reactivity was nonselective, but the resulting functional effects did not exhibit the charge dependence predicted for a charged vestibule. Accordingly, the model locates the red residues (R334, K335, T338) in positions that line the outermost extent of the predicted pore entrance, whereas the pink residues (I331, L333, I336) are displaced from the pore lumen toward the surrounding helices (Figure 6A).
Residues colored in yellow and orange represent those sites where cysteines reacted exclusively with the channel-permeant reagents, [Ag(CN)2]− and [Au(CN)2]−. With the exception of 337, these sites lie cytoplasmic to 338, apparently “deeper” in the pore (Figure 6B). Interestingly, each of the permeant reagents exhibited a distinct pattern of reactivity within these deeper lying sites, a result that may reflect the differing coordination preferences of Au(1) and Ag(1) (see text T1 for discussion). [Ag(CN)2]− reacted with cysteines substituted at all of the yellow- and orange-labeled sites, whereas [Au(CN)2]− reacted only with cysteines at those sites indicated in orange. Those sites where cysteines reacted with [Au(CN)2]− (337, 344, 348, 353) tended to form a stripe along TM6 that would appear to be partially occluded by the TM7−8 hairpin in the 0 ns model but is oriented toward the pore in the 5 ns model. Those sites where cysteines were reactive only toward [Ag(CN)2]− are predicted to face directly into the pore, with the possible exception of R352, which could be partially occluded by TM7 in the 0 ns model but moves toward the pore lumen in the 5 ns model (see also below and discussion).
Among the deeper lying residues, a notable exception in terms of side-chain orientation is position 340. A cysteine substituted here reacted exclusively with [Ag(CN)2]−, but the models place it in a location that would appear to be occluded in both the 0 and the 5 ns conformational states. The I340 side chain, rather than projecting into the pore, is oriented toward TMs 4, 5, 7, and 8. This apparent contradiction may be resolvable by additional refinement of the homology models and may also reflect the impact of the cysteine substitution on the conformation of the channel.
R352C/wt CFTR is not reactive toward MTSET+ and MTSES−, but St. Aubin and Linsdell (40), Cui et al. (41), and Jordan et al. (3) identified R352 as critical for the maintenance of normal anion conduction and gating in CFTR channels. On the basis of amino acid substitutions for R352, St. Aubin and Linsdell (40) proposed that this residue provided positive charge at the intracellular mouth of the CFTR pore, while Cui et al. (41) and Jordan et al. (3) proposed that the effects of mutations at this position were due to interference with an electrostatic “salt bridge” interaction between the arginine and aspartic acid 993 in TM9. They speculated that, although these two residues appear quite separated in the Sav-based model of Serohijos et al. (1), they might be brought into proximity due to the relative motion of the TMs during gating. In Figure 7A it can be seen that, in the model presented here, the side chain of D993 is predicted to face toward the pore lumen where its negative charge might be expected to impede anion conduction. However, comparing panels B and C of Figure 7 reveals that the relative motion of A352 and D993 postulated by Cui et al. (41) and Jordan et al. (3) is in fact predicted by the 5 ns MD simulation. Proximity of the two oppositely charged side chains predicted by the 5 ns model might be expected to produce a partial cancellation of their net electrical impact. In contrast, residues R347 and D924, predicted by Cotten and Welsh (42) to form a salt bridge, are not predicted to be in proximity by our model. Neither, however, is R347 predicted by our model to be “pore-lining” as suggested by an earlier study (43). Rather this Arg is predicted to face away from the pore, toward TM7 (not shown).
Figure 7.

Views of R352 and its relation to D993 in the 1 and 5 ns molecular models. Atom coloring is carbon = cyan, nitrogen = blue, white = hydrogen, and red = oxygen. (A) View from the top of a cross section of the pore (0 ns model) showing that both R352 and D993 side chains are predicted to project into the pore. (B, C) Side views showing the change in relative position of the two residues in the 0 and 5 ns models. It can be seen that the nitrogen on the arginine side chain and the oxygen on the aspartate side chain move closer in the 5 ns model.
Three positions where cysteines were reactive toward [Ag(CN)2]−, 331, 336, and 339, stood out because the rates of reaction were slow compared to the remaining [Ag(CN)2]−-reactive sites. The half-times for these reactions were greater than 5 min whereas <1 min was typical for the remaining [Ag(CN2]−-reactive sites. It was, therefore, intriguing to find that, in the 5 ns model (Figure 6), these residues are grouped together in a manner suggesting that cysteines substituted at these positions could be partially occluded by the TM3−4 loop.
Discussion
Figure 8 summarizes the reactivity of cysteine-substituted CFTR constructs as determined in this study and compares our findings to those obtained in previous cysteine-scanning studies of TM6. It can be seen that there is a reasonable consensus on the reactivity of cysteines placed at positions 330−335 and 338, whereas there are significant disparities at 336, 337, and the remaining residues in TM6. The most obvious differences relate to the use in the present study of the channel-permeant, thiol-reactive probes, [Au(CN)2]− and [Ag(CN)2]−. It is apparent that these reagents are able to access and react with cysteines that, in the present study as well as that of Beck et al. (9), were not reactive toward the bulkier MTS reagents. The fact that the majority of these sites lie cytoplasmic to position 338 suggests that the lack of reactivity reflects the inability of the bulkier MTS reagents to pass through the CFTR channel. A more detailed discussion of the individual discrepancies can be found in text T2.
Figure 8.

Summary of the results of previous cysteine-scanning studies of TM6 of the CFTR chloride channel compared to the present study. Note the lack of consistent results reported for F337C, S341C, I344C, R347C, T351C, R352C, and Q353C (shaded). ● = sites where reactivity with channel-impermeant probes was inferred. ◆ = sites where reactivity with [Ag(CN)2]− was inferred. ◇ = sites where reactivity with [Au(CN)2]− was inferred. ○ = sites scored as nonreactive. Superscripts refer to bibliographic references.
Implications for Pore Structure
Mapping patterns of cysteine reactivity onto models of the CFTR pore produced results remarkably consistent with general expectations for CFTR pore structure, despite the limitations of the modeling process due to low sequence similarity and the availability of only a single state of the Sav1866 protein. Nonreactive sites appear to be largely those occluded by virtue of their placement with respect to other elements of the protein, whereas reactive sites exhibit a gradient of selective reactivity that runs from more extracellular sites exhibiting nonselective reactivity to the more cytoplasmic sites reacting exclusively with channel-permeant reagents, as expected for a pore that narrows after position 338.
The Outer Vestibule of the Pore
Cysteines at 334, 335, and 338 reacted with all of the probes utilized here. This property, taken together with the highly charge-dependent nature of the functional effects previously described and the fact that the partial negative charge on thiolates at 334, 335, and 338 can be titrated by varying bath pH (7,8), suggests that they occupy a relatively wider portion of the pore presumably occupied by anions, cations, and water. Accordingly, in both the 0 and 5 ns models these residues reside near the apparent “entrance” to the pore where the positive charges of R334 and K335 would be expected to create an electrostatic potential favorable to the entry of anions (Figure 6A,B). The location of these residues is consonant with the conclusion of previous studies in which we modeled anion conduction using a charged vestibule scheme (7,8) as well of the more recent findings of Fatehi et al. (10) and Beck et al. (9).
The most outward-lying reactive sites, 331 and 333, where the impact of reactions was charge-independent, mapped to the TM5−6 loop (Figure 6A,B). Beck et al. (9) reported that modification of I331C or L333C CFTR channels results in a profound reduction in open probability, and the authors suggested that these sites may experience significant movement during the gating cycle. These observations are consistent with the notion that I331 and L333, despite their predicted outward location, are not situated within the pore vestibule as defined electrostatically (7,44). On the other hand, they may be important for maintaining the structure of the anion-conducting pore or facilitating the movement of TM6 that is associated with gating as suggested by Beck et al. (9). The 5 ns MD simulation presented here is also consistent with the notion that during channel gating there could be significant relative motion of these sites with respect to the TM3−4 loop and the TM7−8 loop.
Narrowing of the Pore beyond 338
The differential reactivity toward channel-permeant and channel-impermeant reagents describes an out-to-in gradient suggestive of a narrowing of the anion conduction pathway cytoplasmic to residue 338. Reactivity toward MTSET+ and MTSES− disappears beyond this point. This conclusion is consistent with the earlier predictions of McCarty and Zhang (45) and Linsdell et al. (46) that this point might be the site of a narrowing of the pore. In the structure represented by the 0 ns model, the channel is wider at the mouth (>8 Å) than positions just cytoplasmic to 338 (5.5−6 Å), and in the 5 ns model this diameter narrows to 5.2−5.5 Å. These dimensions might exclude MTSES−, estimated by Karlin and Akabas (47) to fit into a 6 Å cylinder. It may be, however, that further refinement of the model will be necessary to accurately predict channel diameter. McCarty et al. (45,48) and Linsdell et al. (36,46,49) reported that amino acid substitutions at 338 altered anion selectivity and conductance. Similarly, at position 337, Linsdell et al. (49) reported changes in permeation with amino acid substitutions and suggested that this residue might contribute to the selectivity filter. Whatever the roles of these two residues may be in the permeation process, the present results suggest that they occupy distinctly different physical environments that are reflected in the differential reactivity of cysteines at the two positions. In this regard it is important to note that we do not know the extent to which the chemical modification results were affected by structural changes resulting from the cysteine substitution itself.
Moving Parts of the Conduction Pathway
Three previous studies have presented evidence for changes in the reactivity of cysteines engineered into CFTR that are correlated with the gating cycle of the channel (9,10,50). In these studies the reactivity of open channels was seen to differ from that of closed channels, suggesting that comparing the rate of cysteine reactivity can provide information about the movements of residues on the protein that are associated with the conformational changes that underlie the gating process (51). It is expected that CFTR gating involves significant motion within the protein in view of the “alternating access” conformational changes that are thought to occur in CFTR’s ABC transporter relatives (4,14,52). The MD simulation presented here provides an initial glimpse of how protein dynamics might alter the positions of residues that make up the anion conduction path of CFTR. The simulation also illustrates how a consideration of potential states of the protein can be used to clarify the functional significance of specific residues. The reactivity of R352C CFTR toward the channel-permeant probe, [Ag(CN)2]−, clearly implicates R352 as a “pore-lining” residue, as suggested by the effect of amino acid substitutions at this position on single-channel conductance, envisioned by St. Aubin and Linsdell (40) as contributing a positive electrostatic potential to a cytoplasmic vestibule for the pore. Modeling results, however, also provide support for the speculation of Cui et al. (42) and Jordan et al. (3) that R352 and D993 could move together during the gating cycle and experience a Coulombic attraction that has important functional consequences.
These two views of the function of R352 may not be mutually exclusive. The positive charge on the arginine could facilitate anion conduction, as suggested by St. Aubin and Linsdell (40), and also contribute to the stability of one or more of the conformational states of the pore that are visited during the gating cycle (42). A unifying hypothesis would hold that the “functional role” of the positive charge on Arg 352 is to partially “neutralize” the negative charge on the Asp 993 side chain which, according to the model presented here, would project into the pore and, absent Arg 352, perhaps electrostatically impede anion conduction. Single-channel records presented in St. Aubin and Linsdell (40) and Cui et al. (41) provide some support for this speculation. In both studies, placing a negative charge at position 352, by substituting a glutamic acid for R352, reduced the single-channel conductance for inward current (outward Cl− flow) when compared to wt CFTR. Conversely, Cui et al. (41) reported that substitution of a positive charge at position 993 (D993R) increased single-channel conductance for inward current above that of wt CFTR.
The molecular models of the CFTR chloride channel developed in the course of these studies appear to depict the orientation of TM6 in the CFTR pore in a way that is remarkably consistent with the results of cysteine scanning. Both the “hits” (the “lining” of the pore) and the “misses” (occluded residues) are largely predicted by the models. The use of both channel-permeant and channel-impermeant probes provides an empirical approach to defining the topology of the CFTR pore which predicts a narrowing cytoplasmic to T338. The present results also suggest that the use of molecular dynamics simulations in conjunction with homology modeling can yield additional insights into CFTR pore structure. It remains to be seen if models derived in this way can predict conduction properties of the pore such as anion selectivity and single-channel conductance.
Acknowledgments
We are grateful to Drs. David Gadsby and Nael McCarty for insightful comments on the manuscript.
Supporting Information Available
Additional discussion of previous conflicting results and nine figures and two movies, the movies providing an enhanced perspective on the overall body plan of the two models as well as the locations of reactive and nonreactive sites. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the National Institute for Diabetes, Digestive and Kidney Diseases, NIH (DK45880 to D.C.D. and DK60312 and DK070755 to X.L.), and the Cystic Fibrosis Foundation (DAWSON08G0 and Dawson05X0 to D.C.D. and SERRAN04F0 to J.R.S). Dr. Sansom’s laboratory was supported by the Wellcome Trust and the BBSRC.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; TM6, transmembrane segment six; wt, wild type; ISOP, isoproterenol; IBMX, isobutylmethylxanthine; MTSET+, [2-(trimethylammonium)ethyl] methanethiosulfonate bromide; MTSES−, sodium (2-sulfonatoethyl)methanethiosulfonate; MMTS, methyl methanethiosulfonate; 2-ME, 2-mercaptoethanol; DTT, dithiothreitol; NEM, N-ethylmaleimide; IAM, iodoacetamide; K[Au(CN)2], potassium dicyanoaurate(I); K[Ag(CN)2], potassium dicyanoargentate(I); KCN, potassium cyanide.
We pointed out previously (7,8) that the reaction of a cysteine thiolate with an uncharged reagent can also produce a change in charge by eliminating the partial charge on the thiolate anion, the magnitude of which depends on the pH of the solution and pKa of the cysteine thiolate in question.
Rapid reversal (t1/2 < 30 s) of MTS reagent effects by 1 mM KCN (a concentration that does not readily reverse mixed disulfide bonds) suggested that the inhibition represented the binding of a metal compound that was displaced from the superfusion tubing, rather than a thiol−disulfide exchange reaction with the target cysteine. Effects of MTSET+ and MTSES− were seen only when Tygon tubing used to supply the recording chamber with fluid had been used previously for solutions containing metallic compounds such as [Ag(CN)2]−. No effects of either mixed disulfide were seen if the Tygon superfusion tubing was washed extensively with 5% nitric acid.
Supplementary Material
References
- Serohijos A. W.; Hegedus T.; Aleksandrov A. A.; He L.; Cui L.; Dokholyan N. V.; Riordan J. R. (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105, 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon J. P.; Lehn P.; Callebaut I. (2008) Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell. Mol. Life Sci. 65, 2594–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan I. K.; Kota K. C.; Cui G.; Thompson C. H.; McCarty N. A. (2008) Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 18865–18870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby D. C.; Vergani P.; Csanady L. (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M.; Akabas M. H. (1996) Identification of cystic fibrosis transmembrane conductance regulator channel-lining residues in and flanking the M6 membrane-spanning segment. Biophys. J. 70, 2688–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M.; Akabas M. H. (1997) Locating the anion-selectivity filter of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel. J. Gen. Physiol. 109, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. S.; Liu X.; Zhang Z. R.; Sun F.; Kriewall T. E.; McCarty N. A.; Dawson D. C. (2001) CFTR. Covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J. Gen. Physiol. 118, 407–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Zhang Z. R.; Fuller M. D.; Billingsley J.; McCarty N. A.; Dawson D. C. (2004) CFTR: a cysteine at position 338 in TM6 senses a positive electrostatic potential in the pore. Biophys. J. 87, 3826–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck E. J.; Yang Y.; Yaemsiri S.; Raghuram V. (2008) Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J. Biol. Chem. 283, 4957–4966. [DOI] [PubMed] [Google Scholar]
- Fatehi M.; Linsdell P. (2008) State-dependent access of anions to the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 283, 6102–6109. [DOI] [PubMed] [Google Scholar]
- Liu X.; Alexander C.; Serrano J.; Borg E.; Dawson D. C. (2006) Variable reactivity of an engineered cysteine at position 338 in cystic fibrosis transmembrane conductance regulator reflects different chemical states of the thiol. J. Biol. Chem. 281, 8275–8285. [DOI] [PubMed] [Google Scholar]
- Serrano J. R.; Liu X.; Borg E. R.; Alexander C. S.; Shaw C. F. III; Dawson D. C. (2006) CFTR: ligand exchange between a permeant anion ([Au(CN)2]−) and an engineered cysteine (T338C) blocks the pore. Biophys. J. 91, 1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense M.; Vergani P.; White D. M.; Altberg G.; Nairn A. C.; Gadsby D. C. (2006) In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 25, 4728–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R. J.; Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185. [DOI] [PubMed] [Google Scholar]
- Liu X.; Smith S. S.; Sun F.; Dawson D. C. (2001) CFTR. Covalent modification of cysteine-substituted channels expressed in xenopus oocytes shows that activation is due to the opening of channels resident in the plasma membrane. J. Gen. Physiol. 118, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Berendsen H.; van der Spoel D.; van Drunen R. (1995) GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56. [Google Scholar]
- Laskowski R. A.; Moss D. S.; Thornton J. M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231, 1049–1067. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess B.; van der Spoel D. (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model 7, 306–317. [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Scott W. R. P.; Hunenberger P. H.; Tironi I. G.; Mark A. E.; Billeter S. R.; Fennen J.; Torda A. E.; Huber T.; Kruger P.; van Gunsteren W. F. (1999) The GROMOS biomolecular simulation program package. J. Phys. Chem. A 103, 3596–3607. [Google Scholar]
- Bond P. J.; Holyoake J.; Ivetac A.; Khalid S.; Sansom M. S. (2007) Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 157, 593–605. [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Sansom M. S. (2006) Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 128, 2697–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S.; Scott K. A.; Bond P. J. (2008) Coarse-grained simulation: a high-throughput computational approach to membrane proteins. Biochem. Soc. Trans. 36, 27–32. [DOI] [PubMed] [Google Scholar]
- Scott K. A.; Bond P. J.; Ivetac A.; Chetwynd A. P.; Khalid S.; Sansom M. S. (2008) Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure 16, 621–630. [DOI] [PubMed] [Google Scholar]
- Berendsen H.; Postma J.; van Gunsteren W.; DiNola A.; Haak J. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Hoover W. (1985) Canonical dynamics: equilibrium phase−space distributions. Phys. Rev. A 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- Nose S. (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268. [Google Scholar]
- Parrinello M.; Rahman A. (1981) Polymorphic transitions in single-crystals—a new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald—an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H.; Fraaije J. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- Smit L. S.; Wilkinson D. J.; Mansoura M. K.; Collins F. S.; Dawson D. C. (1993) Functional roles of the nucleotide-binding folds in the activation of the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. U.S.A. 90, 9963–9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D. J.; Mansoura M. K.; Watson P. Y.; Smit L. S.; Collins F. S.; Dawson D. C. (1996) CFTR: the nucleotide binding folds regulate the accessibility and stability of the activated state. J. Gen. Physiol. 107, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoura M. K.; Smith S. S.; Choi A. D.; Richards N. W.; Strong T. V.; Drumm M. L.; Collins F. S.; Dawson D. C. (1998) CFTR: anion binding as a probe of the pore. Biophys. J. 74, 1320–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X.; Burbridge S. M.; Cowley E. A.; Linsdell P. (2002) Molecular determinants of Au(CN)(2)(−) binding and permeability within the cystic fibrosis transmembrane conductance regulator Cl(−) channel pore. J. Physiol. 540, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. S.; Steinle E. D.; Meyerhoff M. E.; Dawson D. C. (1999) Cystic fibrosis transmembrane conductance regulator. Physical basis for lyotropic anion selectivity patterns. J. Gen. Physiol. 114, 799–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal M. A.; Novoa J. J.; Alvarez S. (2004) Choice of coordination number in d10 complexes of group 11 metals. J. Am. Chem. Soc. 126, 1465–1477. [DOI] [PubMed] [Google Scholar]
- Muanprasat C.; Sonawane N. D.; Salinas D.; Taddei A.; Galietta L. J.; Verkman A. S. (2004) Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J. Gen. Physiol. 124, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Aubin C. N.; Linsdell P. (2006) Positive charges at the intracellular mouth of the pore regulate anion conduction in the CFTR chloride channel. J. Gen. Physiol. 128, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G.; Zhang Z. R.; O'Brien A. R.; Song B.; McCarty N. A. (2008) Mutations at arginine 352 alter the pore architecture of CFTR. J. Membr. Biol. 222, 91–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotten J. F.; Welsh M. J. (1999) Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR—Evidence for disruption of a salt bridge. J. Biol. Chem. 274, 5429–5435. [DOI] [PubMed] [Google Scholar]
- Tabcharani J. A.; Rommens J. M.; Hou Y. X.; Chang X. B.; Tsui L. C.; Riordan J. R.; Hanrahan J. W. (1993) Multi-ion pore behaviour in the CFTR chloride channel. Nature 366, 79–82. [DOI] [PubMed] [Google Scholar]
- Allen T. W.; Andersen O. S.; Roux B. (2004) Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. U.S.A. 101, 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty N. A.; Zhang Z. R. (2001) Identification of a region of strong discrimination in the pore of CFTR. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L852–L867. [DOI] [PubMed] [Google Scholar]
- Linsdell P.; Zheng S. X.; Hanrahan J. W. (1998) Non-pore lining amino acid side chains influence anion selectivity of the human CFTR Cl− channel expressed in mammalian cell lines. J. Physiol. 512, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A.; Akabas M. H. (1998) Substituted-cysteine accessibility method. Methods Enzymol. 293, 123–145. [DOI] [PubMed] [Google Scholar]
- McDonough S.; Davidson N.; Lester H. A.; McCarty N. A. (1994) Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron 13, 623–634. [DOI] [PubMed] [Google Scholar]
- Linsdell P.; Tabcharani J. A.; Rommens J. M.; Hou Y.-X.; Chang X.-B.; Tsui L.-C.; Riordan J. R.; Hanrahan J. W. (1997) Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J. Gen. Physiol. 110, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Cui G.; Liu X.; Song B.; Dawson D.; McCarty N. (2005) Determination of the functional unit of the cystic fibrosis transmembrane conductance regulator chloride channel. One polypeptide forms one pore. J. Biol. Chem. 280, 458–468. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Jurman M. E.; Yellen G. (1996) Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron 16, 859–867. [DOI] [PubMed] [Google Scholar]
- Ward A.; Reyes C. L.; Yu J.; Roth C. B.; Chang G. (2007) Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinamard R.; Akabas M. H. (1999) Arg352 is a major determinant of charge selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel. Biochemistry 38, 5528–5537. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD—Visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.