

Abstract
We investigated the renal responses to NO synthase (NOS) inhibition with N-mono-methyl-l-arginine (l-NMA; 30 mg/kg) in anesthetized rats in which renal perfusion pressure (RPP) to the left kidney was mechanically adjusted. Acute l-NMA increased blood pressure (BP, ∼20%) and renal vascular resistance (RVR) rose (∼50%) in the right kidneys that were always exposed to high RPP. In group 1, the left kidney was exposed to a transient increase (5 min) in RPP which was then normalized, and the rise in RVR was similar to the right kidney. In group 2 the left kidney was never exposed to high RPP, and the rise in RVR was attenuated relative to the right kidney. In group 3, rats were pretreated with the endothelin (ET) receptor antagonist Bosentan, immediately before exposure of the left kidney to a transient increase in RPP, and the rise in RVR was also attenuated relative to the right kidney. NOS inhibition resulted in a natriuresis and diuresis in the right kidneys, and ∼50% of the natriuresis persisted in the left kidney of group 2, in the absence of any rise in RPP. ET antagonism completely prevented the natriuresis and diuresis in response to acute l-NMA in both left and right kidneys. These data suggest that transient exposure to high RPP by NOS inhibition prevents an appropriate vasodilatory response when RPP is lowered, due to the intrarenal action of ET.
Keywords: nitric oxide, endothelin, blood pressure, renal plasma flow, natriuresis
Nitric Oxide (NO) has potent vasodilatory actions and maintains blood pressure (BP) and renal hemodynamics in the baseline state (4, 20, 22). The acute inhibition of endogenous NO synthesis with l-arginine analogs such as nitro-l-arginine methyl ester (NAME) or N-mono-methyl-l-arginine (l-NMA) causes large increases in systemic BP and renal vascular resistance (RVR) (4, 20, 22, 29). NO-induced vasodilation involves lowering vascular smooth muscle intracellular calcium concentration (1). Thus administration of the NO donor sodium nitroprusside (SNP) or calcium channel blocker verapamil should reverse the responses to acute NO synthase (NOS) inhibition. However, we previously reported that although the increased BP induced by NOS inhibition could be normalized completely by either SNP or verapamil, the increase in RVR was not acutely reversible, when the kidney had been exposed to a transient increase in renal perfusion pressure, RPP (6). This earlier study suggested that the renal vasculature was unable to vasodilate appropriately in response to a reduction in BP, after exposure to a transient rise in BP, when endogenous NOS was inhibited. In the present study in the anesthetized rat, we have investigated whether the persistent renal vasoconstriction after transient exposure to high RPP induced by acute systemic NOS inhibition could be prevented by mechanically controlling RPP, rather than by using vasodilatory drugs. In addition, there is increasing evidence suggesting that endothelin (ET) may mediate some of the vascular actions of acute NOS inhibition (14, 24, 32, 35), and accordingly, studies were also conducted to determine whether ET plays a role in this effect.
Methods
Studies were conducted in 22 male Sprague-Dawley rats (aged ∼4–5 mo) obtained from Harlan Sprague Dawley (Indianapolis, IN), maintained on ad libitum food (23% protein, 0.7% NaCl) and tap water. All aspects of these experiments were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals.”
On the day of the experiment, the rat was anesthetized with the thiobarbiturate general anesthetic, Inactin (120 mg/kg ip; supplemental doses 5–10 mg/kg ip as required), and transferred to a temperature-controlled table, and rectal temperature was maintained at 36–38°C throughout the experiment. Vascular catheters were placed in the left femoral vein and both jugular veins for the following infusions: artificial plasma (2.5% bovine serum albumin and 2.5% bovine globulin) to maintain the euvolemic preparation as described previously by us (11); tritiated inulin (5 μCi/ml; Dupont, NEN Research Products, Boston, MA) and p-aminohippurate (PAH, 1%; Merck, Sharp and Dohme, West Point, PA) in 0.9% NaCl at 1.6 ml/h after a 0.3-ml bolus. The left femoral artery and carotid artery were also catheterized for monitoring BP and for occasional blood sampling. A tracheotomy was performed and an oxygen-rich environment was provided by exposing the tracheal tube to 95% O2 and 5% CO2, throughout the experiment. The abdomen was opened through a ventral midline and a lateral incision and the intestines were reflected to expose the left kidney and the aorta. The left kidney was prevented from drying by covering with wet lens paper. Both ureters were catheterized for urine collection, and an adjustable Blalock clamp was placed on the abdominal aorta between the right and left renal arteries. This allowed independent control over RPP to the left and right kidneys. The carotid artery pressure was taken as right RPP and femoral artery pressure as left RPP. After the surgery, all the incisions were covered with Parafilm to prevent fluid loss.
After a 60-min equilibration period at the end of surgery, two 15- to 20-min control urine collections were made with midpoint arterial blood samples (∼150 μl). The blood was centrifuged, plasma was removed for analysis (see below), and red blood cells were reconstituted with an equal volume of artificial plasma and restored to rats after completion of control measurements. Next, an intravenous bolus of l-NMA (30 mg/kg) was given, followed by a continuous infusion of l-NMA at the rate of 2 mg·kg−1·min−1. In group 1 experiments (n = 7), the Blalock clamp was manipulated 5 min after l-NMA infusion so that RPP to the left kidney was normalized after exposure of the left kidney to a transient increase in RPP. In group 2 experiments (n = 8), the Blalock clamp was manipulated during l-NMA administration to keep the RPP to the left kidney at the control level, with no exposure to the high BP. In group 3 experiments (n = 7), rats received the ET type A and B receptor antagonist, Bosentan (10 mg/kg; Hoffmann-La Roche), 5 min before l-NMA administration. In these rats, RPP to the left kidney was normalized 5 min after l-NMA infusion, as in group 1. In all groups, the right kidney was exposed to high perfusion pressure throughout the experimental period. Ten minutes after beginning l-NMA administration, two further urine collections were made with a midpoint femoral arterial blood sample.
The volume of urine samples was measured gravimetrically, and urine was analyzed for tritiated inulin activity and for PAH, sodium, and potassium concentrations. Arterial blood samples were analyzed for hematocrit, plasma tritiated inulin activity, PAH, sodium, and potassium concentrations. Tritiated inulin activity was measured in 10-μl aliquots of urine and plasma (in 0.3 ml of H2O + 3 ml of Scint A, XF; Packard, Meriden, CT) in a Packard scintillation counter. PAH was measured colorimetrically, and sodium and potassium concentrations were measured by flame photometry, as described previously (3, 6, 24).
These measurements allowed calculation of inulin clearance [equal to glomerular filtration rate (GFR)], PAH clearance [equal to renal plasma flow (RPF) when factored for renal extraction of PAH], RVR, filtration fraction (FF), urinary excretion of sodium and potassium (UNaV and UKV, respectively) and the fractional excretion of sodium (FENa). The calculations have been described previously (3, 5, 6, 24). Data are expressed throughout as means ± SE, and statistical significance (where P < 0.05) was determined by paired and unpaired t-tests.
Results
All data for renal function and BP are summarized in Table 1. There was no difference in renal function between the left and right kidney during control measurements in either group 1 or 2, nor were there any statistically significant differences in baseline variables between the two groups. Although BP and GFR were also similar in group 3, there were some differences in other baseline values, with group 3 rats generally showing more vasodilated kidneys and greater electrolyte excretions vs. groups 1 and 2 (Table 1). In all groups, acute NOS inhibition led to a sustained rise in BP of ∼25 mmHg. In group 3 rats, the ET antagonist led to a slight fall in BP 5 min after administration and immediately before the l-NMA was given (118 ± 5 vs. 123 ± 4 mmHg, P < 0.05). The right kidney, which was always exposed to the high BP, responded similarly in groups 1–3 with a large increase in RVR (Fig. 1). The renal vasoconstriction led to a fall in RPF but had no effect on GFR due to a compensatory increase in FF in groups 1 and 2. In group 3, the prior treatment with ET antagonist resulted in a fall in both RPF and GFR, since FF was not increased in response to acute NOS inhibition.
Table 1. Effect on renal function of acute NOS inhibition with l-NMA in rats with variable renal perfusion pressure.
| Hct, vol % | RPP, mmHg | GFR, ml/min | RPF, ml/min | FF | RVR, mmHg·ml−1·min | V, μl/min | UNaV, μeq/min | FENa, % | UKV, μeq/min | |
|---|---|---|---|---|---|---|---|---|---|---|
| Group 1 (n = 7) | ||||||||||
| Right kidney | ||||||||||
| Control | 49 ± 1 | 120 ± 5 | 1.57 ± 0.10 | 5.2 ± 0.4‡ | 0.31 ± 0.02‡ | 11.8 ± 0.9 | 5.6 ± 0.7 | 0.4 ± 0.1‡ | 0.17 ± 0.05‡ | 1.3 ± 0.1 |
| Exp | 51 ± 1 | 145 ± 6 | 1.49 ± 0.10 | 4.2 ± 0.3 | 0.37 ± 0.03 | 17.6 ± 1.1 | 45.2 ± 8.2 | 7.1 ± 1.5 | 3.57 ± 0.75 | 2.8 ± 0.3 |
| P value | <0.02 | <0.005 | NS | <0.05 | <0.02 | <0.0001 | <0.005 | <0.005 | <0.005 | <0.0005 |
| Left kidney | ||||||||||
| Control | 49 ± 1 | 119 ± 5 | 1.63 ± 0.09 | 5.3 ± 0.5‡ | 0.32 ± 0.02‡ | 11.4 ± 0.8 | 6.6 ± 1.1 | 0.6 ± 0.2‡ | 0.24 ± 0.09‡ | 1.4 ± 0.2 |
| Exp | 51 ± 1 | 119 ± 5 | 1.34 ± 0.06 | 3.7 ± 0.3* | 0.37 ± 0.02 | 16.1 ± 1.1 | 25.6 ± 4.9 | 3.3 ± 0.9 | 1.88 ± 0.52 | 2.2 ± 0.2 |
| P value | <0.02 | NS | <0.05 | <0.005 | <0.0005 | <0.001 | <0.005 | <0.01 | <0.05 | <0.05 |
| Group 2 (n = 8) | ||||||||||
| Right kidney | ||||||||||
| Control | 48 ± 1 | 123 ± 3 | 1.47 ± 0.11 | 4.4 ± 0.3‡ | 0.34 ± 0.02‡ | 14.4 ± 1.1‡ | 4.1 ± 0.3‡ | 0.3 ± 0.1‡ | 0.14 ± 0.04‡ | 1.0 ± 0.1‡ |
| Exp | 50 ± 1 | 147 ± 4 | 1.47 ± 0.11 | 3.7 ± 0.3 | 0.41 ± 0.03 | 20.5 ± 1.6 | 53.1 ± 9.6 | 8.6 ± 1.4 | 4.24 ± 0.67 | 2.8 ± 0.2 |
| P value | <0.0005 | <0.001 | NS | <0.05 | <0.02 | <0.001 | <0.002 | <0.001 | <0.001 | <0.0005 |
| Left kidney | ||||||||||
| Control | 48 ± 1 | 122 ± 3 | 1.52 ±0.07 | 4.7 ± 0.2‡ | 0.33 ± 0.02‡ | 13.2 ± 0.7‡ | 4.9 ± 0.6‡ | 0.4 ± 0.1‡ | 0.20 ± 0.06‡ | 1.1 ± 0.1‡ |
| Exp | 50 ± 1 | 120 ± 2 | 1.51 ± 0.11 | 3.8 ± 0.3 | 0.41 ± 0.03 | 15.9 ± 0.8† | 23.5 ± 2.2* | 3.5 ± 0.6* | 1.81 ± 0.35* | 2.5 ± 0.4 |
| P value | <0.0005 | NS | NS | <0.02 | <0.005 | <0.02 | <0.001 | <0.002 | <0.002 | <0.01 |
| Group 3 (n = 7) | ||||||||||
| Right kidney | ||||||||||
| Control | 46 ± 1 | 123 ± 5 | 1.46 ± 0.08 | 6.6 ± 0.5 | 0.22 ± 0.02 | 10.1 ± 0.8 | 6.8 ± 0.9 | 1.3 ± 0.3 | 0.67 ± 0.16 | 1.5 ± 0.1 |
| +Bos exp | 44 ± 1 | 144 ± 7 | 1.08 ± 0.07 | 4.3 ± 0.5 | 0.28 ± 0.3 | 20.1 ± 2.2 | 7.7 ± 1.8 | 1.0 ± 0.3 | 0.75 ± 0.24 | 1.3 ± 0.1 |
| P value | <0.01 | <0.001 | <0.02 | <0.001 | NS | <0.001 | NS | NS | NS | NS |
| Left kidney | ||||||||||
| Control | 46 ± 1 | 119 ± 6 | 1.43 ± 0.08 | 6.5 ± 0.5 | 0.23 ± 0.01 | 10.0 ± 1.0 | 9.9 ± 2.3 | 1.8 ± 0.5 | 0.95 ± 0.25 | 1.9 ± 0.2 |
| +Bos exp | 44 ± 1 | 116 ± 5 | 1.05 ± 0.06 | 4.3 ± 0.3 | 0.26 ± 0.02 | 15.3 ± 1.4 | 6.7 ± 1.4 | 0.8 ± 0.2 | 0.52 ± 0.14 | 1.3 ± 0.2 |
| P value | <0.01 | NS | <0.001 | <0.001 | NS | <0.005 | <0.05 | <0.05 | NS | <0.05 |
Values are means ± SE; n = no. of rats. Group 1 rats: left kidney was exposed to a transient increased (5 min) and then normalized blood pressure while right kidney was always exposed to high pressure. Group 2 rats: left kidney was never exposed to high pressure while right kidney was exposed to high pressure throughout. Group 3 rats: were pretreated with the endothelin antagonist Bosentan (Bos) 5 min before the left kidney was exposed to a transient increased (5 min) in blood pressure. Hct, hematocrit; RPP, renal perfusion pressure; GFR, glomerular filtration rate; RPF, renal plasma flow; FF, filtration fraction; RVR, renal vascular resistance; V, urine flow rate; UNaV, urinary sodium excretion; FENa, fractional excretion of sodium; UKV, urinary potassium excretion; Exp, experimental; NS, not significant.
P < 0.05 change were between right and left kidney (paired t-test).
P < 0.005 change between right and left kidney (paired t-test).
P < 0.05 difference in the control values vs. group 3 (1-way ANOVA).
Fig. 1.
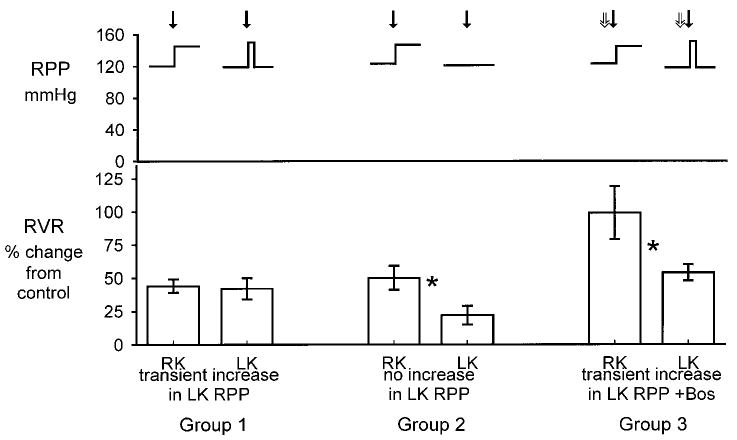
Top: change in mean renal perfusion pressure (RPP) to right kidney (RK) and left kidney (LK) in groups 1–3. Bottom: percentage change from control of renal vascular resistance (RVR) in RK and LK after systemic NO synthase (NOS) inhibition with l-NMA (solid arrows). Right kidney was always exposed to increased RPP, whereas measurements were made on the left kidney at control RPP. In groups 1 and 3, left kidney was transiently exposed to increased RPP (5 min), whereas in group 2 the left kidney was never exposed to increased RPP. In group 3, Bosentan (Bos) was given 5 min before l-NMA (open arrows). *Significant difference between right and left kidney, P < 0.05 by paired t-test.
In group 1 rats (413 ± 12 g body wt), the left kidney was exposed to a transiently increased (5 min) RPP, which was then lowered to control values. Despite rapid reversal of the increase in RPP, RVR remained high and RPF fell, similar to the right kidney. As shown in Fig. 1, the magnitude of the rise in RVR (expressed as percent change from control) was similar in left and right kidneys of group 1. The absolute change in RVR after l-NMA was also similar (4.6 ± 0.7 and 5.8 ± 0.5 mmHg·ml−1·min, left and right kidneys, respectively; P = not significant). In contrast, the GFR fell moderately in the left kidney, by ∼15% vs. the control value, presumably because reduction of RPP meant that a lower pressure was transmitted to the glomerulus. In group 2 rats (402 ± 11 g body wt), the left kidney was never exposed to increased RPP during acute NOS inhibition. In this case, the RVR increased, but the magnitude of the rise was smaller than in the right kidney, both by absolute change (+6.1 ± 1.1 vs. +2.7 ± 0.9 mmHg·ml−1·min; P < 0.05) and as percent change (Fig. 1). In group 2 rats, GFR was similar between the kidneys and was not affected by NOSI. In group 3 rats (body wt, 433 ± 21 g), prior ET antagonism prevented the exaggerated rise in left kidney RVR in response to transient exposure to high RPP after l-NMA. Therefore, although RVR increased in both left and right kidneys with l-NMA, the magnitude of the rise was blunted in the left vs. right kidney both by absolute change (+5.3 ± 0.7 vs. +10.0 ± 1.9 mmHg·ml−1·min; P = 0.05) and as percent change (Fig. 1). In group 3 rats, GFR fell similarly in both left and right kidneys after l-NMA as did RPF, whereas FF did not change.
In response to acute NOSI, urine flow rate (V), UNaV, and FENa rose significantly and a smaller increase occurred in UKV, in the right kidneys, subjected to high RPP in both group 1 and group 2 rats. Although not shown, plasma sodium and potassium concentrations were unaffected by any of the experimental maneuvers. Compared with the right kidney, the magnitude of the increases of V, UNaV, and FENa in left kidneys was less, by ∼50% in both groups 1 and 2 (Fig. 2), whereas the kaliuresis was similar. It is interesting that acute NOS inhibition still exerted natriuretic and diuretic effects in the left kidneys of both groups 1 and 2, despite the fact that RPP was not elevated during the urine collections and, in the case of group 2, had never been exposed to increased RPP during the course of the experiment. Prior Bosentan eradicated the natriuresis, diuresis, and kaliuresis in response to l-NMA in both kidneys, despite the fact that the right kidney was exposed to a high RPP.
Fig. 2.
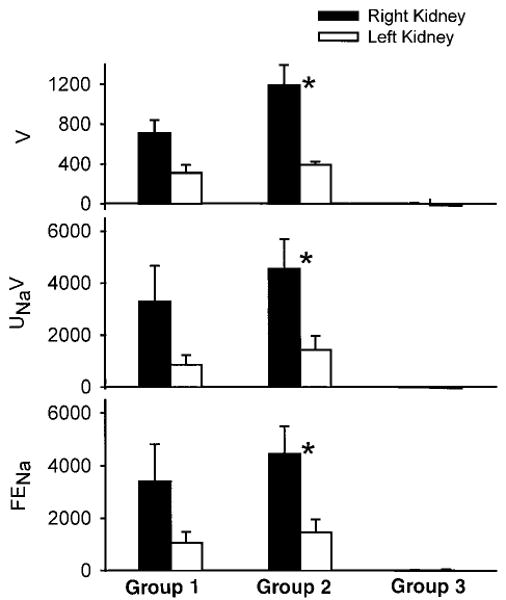
Percentage change from control of urine flow rate (V), urinary excretion of sodium (UNaV), and fractional excretion of sodium (FENa) of right and left kidney after acute NOS inhibition with l-NMA in groups 1–3. Right kidney was always exposed to increased RPP in both groups, whereas measurements were made on the left kidney at control RPP. In groups 1 and 3, left kidney was transiently exposed to increased RPP (5 min), whereas in group 2 the left kidney was never exposed to increased RPP. Bosentan was given 5 min before l-NMA in group 3. *Significant difference between right and left kidney, P < 0.05 by paired t-test.
Discussion
In the present study, acute systemic NOS inhibition in the anesthetized, volume-replete rat led to hypertension and marked renal vasoconstriction. The renal vasoconstrictor responses of the right kidney (always exposed to high RPP) were similar to those previously reported by us (3, 4, 6, 24) and others (22, 30) in both conscious and anesthetized, volume-replete rats. An increase in RPP leads to an autoregulatory rise in RVR separate from any direct actions of NOS inhibition within the renal vasculature. Accordingly, local intrarenal NOS inhibition, without a rise in RPP, has been reported to evoke a smaller renal vasoconstriction than seen with a pressor dose of systemic NOS inhibitor (11, 15), reflecting the selective removal of local, tonically produced NO. As predicted by these earlier findings, in the present study an attenuated rise in RVR occurred in the left kidney of group 2 rats (never exposed to high RPP) vs. the right. This observation is qualitatively similar to recent reports in which only ∼50% of the rise in RVR in response to systemic NOS inhibition occurred when RPP was held constant (10, 31). Much of this residual rise in RVR could be prevented by inhibition of tubuloglomerular feedback (TGF) in a setting where the ANG II, cyclooxygenase, and sympathetic nervous systems were also blocked (10). These observations suggest that removal of directly acting endothelium-derived NO plays only a minor role in the renal vasoconstrictor response to acute NOS inhibition. A less predictable finding in the present study was the amplified increase in RVR seen in the left kidney of group 1 rats. Although these kidneys were exposed to a transient (5 min) increase in RPP, the measurements of RVR were made up to 30–40 min later, during which time the left kidney had been exposed to control levels of RPP. Nevertheless, the left kidney “remembered” the transient increase in RPP and responded with a persistent, amplified elevation in RVR that was indistinguishable from that seen in the right kidney, exposed to high RPP throughout.
In a previous study performed in conscious chronically catheterized rats, normalization of BP with either the NO donor SNP or the calcium channel blocker, verapamil (6) after transient (5 min) hypertension failed to lower the RVR to the expected value. Thus transient exposure of the kidney to high RPP leads to prolonged renal vasoconstriction which cannot be reversed by either pharmacological or mechanical reduction of RPP. This effect is specific to a pressor dose of NOS inhibitor, because when an equivalent, transient rise in RPP was produced by ANG II, the RVR fell appropriately upon reduction in RPP (6).
One explanation for the prolonged increase in RVR after a transient rise in RPP due to NOS inhibition is that renal autoregulatory mechanisms may become deranged. However, several reports indicate that autoregulation of RPF was unattenuated in both dog and rat given high, pressor doses of l-NAME (2, 7, 13, 27). In fact, NOS inhibition should improve renal autoregulation since macula densa NO provides a vasodilatory “brake” to TGF-induced increases in afferent arteriolar tone (17, 36), NOS inhibition improves TGF efficiency (8, 34), and autoregulation of glomerular BP is enhanced by NOS inhibition in the “in vitro” juxtamedullary nephron preparation (16). However, some observations have suggested that NO might contribute to the autoregulatory vasodilation that occurs when RPP is lowered (18, 21, 28). In these studies relatively low levels of NOS inhibitors were given so that intrarenal NOS was inhibited with minimal increases in BP. In fact, studies in the rat have shown that autoregulation of cortical blood flow is improved with high-dose l-NAME but impaired with a lower dose (13).
It is not clear what role, if any, NO plays in autoregulatory vasodilation when RPP is lowered and the present studies were not designed to address this issue. Nevertheless, it is possible that a component of the persistent, exaggerated increase in RVR seen with acute systemic NOS inhibition, after normalization of BP, results from an autoregulatory response. If NO is required for an appropriate fall in RVR when RPP is reduced, then the increased RVR secondary to a pressor dose of NOS inhibitor will be “stuck,” and RVR will be unresponsive to subsequent normalization of BP. However, renal autoregulatory adjustments are confined to the preglomerular vessels (23), and we have both direct and indirect evidence to suggest that efferent arterioles are also involved; thus other mechanisms may also be operating. In a previous micropuncture study, we showed that acute systemic NOS inhibition led to large increases in both afferent and efferent arteriolar resistance, whereas local intrarenal inhibition of NO caused a small increase in afferent arteriolar resistance without any effect on efferent arteriolar resistance (11). The increased efferent resistance was a result of a pressor dose of systemic NOS inhibitor, and we subsequently showed that this was largely prevented by ET receptor antagonism, which also blunted the increase in afferent resistance (25). We have also reported previously that ET inhibition reduces the rise in RVR seen in the conscious chronically catheterized rat during acute NOS inhibition (24).
The extensive interactions between NO and ET suggest that ET may mediate some of the renal vasoconstrictor responses to acute NOS inhibition. ET stimulates endothelial NO release, via the ETB receptor (32, 35), whereas tonically produced NO inhibits endothelial ET release (8). NO blunts the vasoconstrictor response to ET, both by its general vasodilatory action and because NO abbreviates ET signaling at its receptor (14). In addition, the prolonged vasoconstrictor action of ET following binding to its vascular receptor (19) could provide a mechanism for the prolonged rise in RVR seen after transient exposure of the left kidney to high RPP in the present study. We therefore conducted an additional experiment to determine whether combined ETA and ETB receptor antagonism could prevent the exaggerated, prolonged renal vasoconstriction due to l-NMA, associated with transient exposure of the left kidney to high RPP. As shown, blockade of ETA and ETB receptors reduces the rise in left kidney RVR by ∼50% compared with that seen in the right kidney, so that the pattern of change in RVR resembles that of group 2 rats, where the left kidney is never exposed to high RPP. These observations suggest that an intact renal NO system is necessary for an appropriate fall to occur in RVR due to reduction in RPP. In the absence of NO even a transient rise in RPP leads to a persistent, ET-mediated renal vasoconstriction. We did not separately assess the renal actions of ET blockade alone, in the present study, although we previously reported that Bosentan produces no change in total RVR, RPF, or GFR, despite a small fall in BP and a small constriction of the outer cortical afferent arterioles (26). It is therefore likely that the Bosentan-induced attenuation of the rise in left RVR seen in group 3 reflects the inhibition of an effect secondary to NOS inhibition-induced stimulation of ET.
One limitation of the present study is that group 3 studies (performed ∼1 yr after groups 1 and 2) showed some differences in baseline function, suggestive of either less surgical stress or greater volume expansion (both of which are likely to impact the NOS system). Therefore there may be some differences in the baseline level of activity of NOS between group 3 and groups 1 and 2. Nevertheless, the important comparison is the response to acute NOS inhibition between the left and right kidneys within each group. ET blockade clearly restores the attenuated response of the left vs. right kidney exposed to transient increases in RPP during acute NOS inhibition (group 3), to that of the NOS-inhibited left kidney never exposed to increased RPP (group 2). This strongly suggests that ET mediates the exaggerated, prolonged renal vasoconstriction associated with acute NOS inhibition and transient rise in RPP.
In addition to the hypertensive and renal hemodynamic effects, systemic NOS inhibition produced a significant natriuresis and diuresis. We suggested earlier that this might result from a pressure natriuresis/diuresis due to an abrupt increase in arterial BP (4). We note, however, in the present study ∼50% of the natriuresis persisted in the left kidney despite either rapid normalization of RPP (group 1) or absence of any rise in RPP (group 2), demonstrating that the natriuresis observed due to systemic NOS inhibition in this experiment is not entirely the result of a pressure effect. De Nicola and colleagues (12) previously reported that l-NMA has a direct ANG II-dependent inhibitory action on proximal reabsorption that is independent of RPP (12). We have previously observed that prior renal denervation attenuates the natriuretic response to a pressor dose of NOS inhibitor (5), despite persistence of an unblunted rise in BP, in the conscious rat. Similar findings were reported for the anesthetized rat (33). We recognize that there is considerable evidence supporting a primary natriuretic role for NO by inhibition of epithelial sodium transport (20). It is therefore likely that a component of the natriuretic response to widespread NOS inhibition results from some secondary event related to the rise in BP but separate from a pressure natriuresis. Based on our group 3 studies, it is clear that ET plays a pivotal role, since Bosentan prevents the natriuresis and diuresis due to NOS inhibition in both the left kidney and the right kidney exposed to prolonged high RPP. The present observation agrees with our previous report that ET receptor blockade, or ET converting enzyme inhibition, leads to suppression of the natriuretic response to acute systemic NOS inhibition in the conscious rat (24). In addition, an α2-adrenoceptor natriuretic effect, mediated via the renal nerves, has also been implicated (33). Exactly how these various systems interact and influence sodium excretion in response to acute NOS inhibition is not clear at this time.
In summary, the present study confirms and extends our finding that once the kidney is exposed to a transient increase in BP due to acute systemic inhibition of NO, the RVR remains high despite subsequent normalization of BP. It seems that the renal vasculature can remember a short-term exposure to high RPP, when the pressor effect is due to NOS inhibition, and that this exaggerated renal vasoconstriction is due to an action of ET. In addition to blunting the vasoconstrictor actions of ET and reducing ET release, NO also shortens the interaction time between ET and its receptor (8, 14). It is possible that a transient rise in RPP leads to intrarenal ET release, which is normally buffered by endogenous NO. As a result, in the absence of NO, a prolonged ET-dependent renal vasoconstriction occurs even after RPP has been normalized. This has potential clinical relevance, since an abrupt reduction in BP in NO-deficient individuals could lead to prolonged high RVR and low RPP; a combination that would seriously compromise renal function. These studies also support recent findings that the natriuresis of acute systemic NOS inhibition can be partially dissociated from the rise in BP.
Acknowledgments
The technical assistance of Lennie Samsell and Kevin Engels is gratefully acknowledged.
These studies were supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-45517.
References
- 1.Anderson TJ, Meredith IT, Ganz P, Selwyn AP, Yeung AC. Nitric oxide and nitrovasodilators: similarities, differences and potential interactions. J Am Coll Cardiol. 1994;24:555–566. doi: 10.1016/0735-1097(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 2.Baumann JE, Persson PB, Ehmke H, Nafz B, Kirchheim HR. Role of endothelium-derived relaxing factor in renal autoregulation in conscious dogs. Am J Physiol. 263 doi: 10.1152/ajprenal.1992.263.2.F208. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1992;32:F208–F213. [Google Scholar]
- 3.Baylis C, Engels K, Samsell L, Harton P. Renal effects of acute endothelium-derived relaxing factor blockade are not mediated by angiotensin II. Am J Physiol. 264 doi: 10.1152/ajprenal.1993.264.1.F74. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1993;33:F74–F78. [Google Scholar]
- 4.Baylis C, Harton P, Engels K. Endothelial-derived relaxing factor (EDRF) controls renal hemodynamics in the normal rat kidney. J Am Soc Nephrol. 1990;1:875–881. doi: 10.1681/ASN.V16875. [DOI] [PubMed] [Google Scholar]
- 5.Baylis C, Braith R, Santmyire BR, Engels K. Renal nerves do not mediate vasoconstrictor response to acute nitric oxide synthesis inhibition in conscious rats. J Am Soc Nephrol. 1997;8:887–892. doi: 10.1681/ASN.V86887. [DOI] [PubMed] [Google Scholar]
- 6.Baylis C, Masilamani S, Losonczy G, Samsell L, Harton P, Engels K. Blood pressure (BP) and renal vasoconstrictor responses to acute blockade of nitric oxide: persistence of renal vasoconstriction despite normalization of BP with either verapamil or sodium nitroprusside. J Pharmacol Exp Ther. 1995;274:1135–1141. [PubMed] [Google Scholar]
- 7.Beierwaltes WH, Sigmon DH, Carretero OA. Endothelium modulates renal blood flow but not autoregulation. Am J Physiol. 262 doi: 10.1152/ajprenal.1992.262.6.F943. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1992;31:F943–F949. [Google Scholar]
- 8.Boulanger C, Luscher TF. Release of endothelin from porcine aorta. Inhibition by endothelium-derived nitric oxide. J Clin Invest. 1990;85:587–590. doi: 10.1172/JCI114477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braam B, Koomans HA. Reabsorption of nitro-l-arginine infused into the late proximal tubule participates in modulation of TGF responsiveness. Kidney Int. 1995;47:1252–1257. doi: 10.1038/ki.1995.179. [DOI] [PubMed] [Google Scholar]
- 10.Brand-Schieber E, Pucci M, Nasjletti A. Determinants of renal vasoconstriction after systemic inhibition of nitric oxide synthesis in rats. Am J Physiol. 270 doi: 10.1152/ajpregu.1996.270.6.R1203. [DOI] [PubMed] [Google Scholar]; Regulatory Integrative Comp Physiol. 1996;39:R1203–R1207. [Google Scholar]
- 11.Deng A, Baylis C. Locally produced EDRF control preglomerular resistance and the ultrafiltration coefficient. Am J Physiol. 264 doi: 10.1152/ajprenal.1993.264.2.F212. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1993;33:F212–F215. [Google Scholar]
- 12.De Nicola L, Blantz RC, Gabbai FB. Nitric oxide and angiotensin. Glomerular and tubular interaction in the rat. J Clin Invest. 1992;89:1248–1256. doi: 10.1172/JCI115709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenoy FJ, Ferrer P, Carbonell L, Salom MG. Role of nitric oxide on papillary blood flow and pressure natriuresis. Hypertension. 1995;25:408–414. doi: 10.1161/01.hyp.25.3.408. [DOI] [PubMed] [Google Scholar]
- 14.Goligorsky MS, Tsukahara H, Magazine H, Anderson TT, Malik AB, Bahou WF. Termination of endothelin signaling: role of nitric oxide. J Cell Physiol. 1994;158:485–494. doi: 10.1002/jcp.1041580313. [DOI] [PubMed] [Google Scholar]
- 15.Granger JP, Alberola AM, Salazar FJ, Nakamura T. Control of renal hemodynamics during intrarenal and systemic blockade of NO synthesis in conscious dogs. J Cardiovasc Pharmacol. 1992;20:S160–S162. doi: 10.1097/00005344-199204002-00045. [DOI] [PubMed] [Google Scholar]
- 16.Imig JD, Roman RJ. Nitric oxide modulated vascular tone in preglomerular arterioles. Hypertension. 1992;19:770–774. doi: 10.1161/01.hyp.19.6.770. [DOI] [PubMed] [Google Scholar]
- 17.Ito S, Ren Y. Evidence for the role of nitric oxide in macula densa control of glomerular hemodynamics. J Clin Invest. 1993;92:1093–1098. doi: 10.1172/JCI116615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiyomoto H, Matsuo H, Tamaki T, Aki Y, Iwai H, Abe Y. Effect of l-NG-nitro-arginine, inhibitor of nitric oxide synthesis, on autoregulation of renal blood flow in dogs. Jpn J Pharmacol. 1992;58:147–155. doi: 10.1254/jjp.58.147. [DOI] [PubMed] [Google Scholar]
- 19.Kohan DE. Endothelins in the normal and diseased kidney. Am J Kidney Dis. 1997;29:2–26. doi: 10.1016/s0272-6386(97)90004-4. [DOI] [PubMed] [Google Scholar]
- 20.Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the kidney. Am J Physiol. 272 doi: 10.1152/ajprenal.1997.272.5.F561. [DOI] [PubMed] [Google Scholar]; Renal Physiol. 1997;41:F561–F578. [Google Scholar]
- 21.Majid DSA, Navar LG. Suppression of blood flow autoregulation plateau during nitric oxide blockade in canine kidney. Am J Physiol. 262 doi: 10.1152/ajprenal.1992.262.1.F40. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1992;31:F40–F46. [Google Scholar]
- 22.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 23.Navar LG. Renal autoregulation: perspective from whole kidney and single nephron studies. Am J Physiol. 234 doi: 10.1152/ajprenal.1978.234.5.F357. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1978;3:F357–F370. [Google Scholar]
- 24.Qiu C, Engels K, Baylis C. Endothelin modulates the pressor actions of acute systemic nitric oxide blockade. J Am Soc Nephrol. 1995;6:1476–1481. doi: 10.1681/ASN.V651476. [DOI] [PubMed] [Google Scholar]
- 25.Qiu C, Samsell L, Baylis C. Endothelin (ET) and angiotensin II (AII) modulate glomerular hemodynamic responses to acute NO blockade (NOB) J Am Soc Nephrol. 1995;6:668. Abstract. [Google Scholar]
- 26.Qiu C, Samsell L, Baylis C. Actions of endogenous endothelin on glomerular hemodynamics in the rat. Am J Physiol. 269 doi: 10.1152/ajpregu.1995.269.2.R469. [DOI] [PubMed] [Google Scholar]; Regulatory Integrative Comp Physiol. 1995;38:R469–R473. [Google Scholar]
- 27.Salom MG, Lahera V, Miranda-Guardiola F, Romero JC. Blockade of pressure natriuresis induced by inhibition of renal synthesis of nitric oxide in dogs. Am J Physiol. 262 doi: 10.1152/ajprenal.1992.262.5.F718. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1992;31:F718–F722. [Google Scholar]
- 28.Schultz MF, Robinette JB, Conger JD. Nitric oxide is a factor in the lower range of renal autoregulation in the rat. J Am Soc Nephrol. 1992;3:551. Abstract. [Google Scholar]
- 29.Tolins JP, Palmer PMJ, Moncada S, Raij L. Role of endothelium-derived relaxing factor in regulation of renal hemodynamic responses. Am J Physiol. 258 doi: 10.1152/ajpheart.1990.258.3.H655. [DOI] [PubMed] [Google Scholar]; Heart Circ Physiol. 1990;27:H655–H662. [Google Scholar]
- 30.Tolins JP, Shultz PJ, Raij L, Brown DM, Mauer SM. Abnormal renal hemodynamic response to reduced renal perfusion pressure in diabetic rats: role of NO. Am J Physiol. 265 doi: 10.1152/ajprenal.1993.265.6.F886. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1993;34:F886–F895. [Google Scholar]
- 31.Treeck B, Aukland K. Effect of l-NAME on glomerular filtration rate in deep and superficial layers of rat kidney. Am J Physiol. 272 doi: 10.1152/ajprenal.1997.272.3.F312. [DOI] [PubMed] [Google Scholar]; Renal Physiol. 1997;41:F312–F318. [Google Scholar]
- 32.Tsukahara H, Ende H, Magazine HI, Bahou WF, Goligorsky MS. Molecular and functional characterization of the non-isopeptide-selective ETB receptor in endothelial cells: receptor coupling to nitric oxide synthase. J Biol Chem. 1994;269:21778–21785. [PubMed] [Google Scholar]
- 33.Vallon V, Peterson OW, Gabbai FB, Blantz RC, Thomson SC. Interactive control of renal function by alpha 2-adrenergic system and nitric oxide: role of angiotensin II. J Cardiovasc Pharmacol. 1995;26:916–922. doi: 10.1097/00005344-199512000-00010. [DOI] [PubMed] [Google Scholar]
- 34.Vallon V, Thomson SC. Inhibition of local nitric oxide synthase increases homeostatic efficiency of tubuloglomerular feedback. Am J Physiol. 269 doi: 10.1152/ajprenal.1995.269.6.F892. [DOI] [PubMed] [Google Scholar]; Renal Fluid Electrolyte Physiol. 1995;38:F892–F899. [Google Scholar]
- 35.Verhaar MC, Strachan FE, Newby DE, Cruden NL, Koomans HA, Rabelink TJ, Webb DJ. Endothelin-A receptor antagonist-mediated vasodilatation is attenuated by inhibition of nitric oxide synthesis and by endothelin-B receptor blockade. Circulation. 1998;97:752–756. doi: 10.1161/01.cir.97.8.752. [DOI] [PubMed] [Google Scholar]
- 36.Wilcox CS, Welch WJ, Murad F, Gross SS, Taylor G, Levi R, Schmidt HH. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci USA. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]