

Abstract
Background
Shift of myocardial substrate preference has been observed in many chronic diseases such as diabetes and heart failure. This study was undertaken to elucidate the mechanisms underlying the chronic substrate switch in adult hearts and to determine the functional consequences of the switch.
Methods and Results
Transgenic mice with cardiac specific overexpression of the insulin-independent glucose transporter GLUT1 (TG) were used to increase intracellular glucose in cardiac myocytes. A high fat diet was used to increase the fatty acid supply to the heart. High fat diet induced a 40% increase in fatty acid oxidation in wild type hearts whereas glucose oxidation was decreased to 30% of the control. In contrast, glucose oxidation was more than 2-fold higher in TG hearts, and the high fat diet failed to up-regulate fatty acid oxidation in these hearts. Glucose induced changes in the expression of multiple metabolic genes, including PPARα (decreased by 51%), 3- oxoacid CoA transferase (decreased by 67%) and acetyl-CoA carboxylase (increased by 4-fold), resulting in a remodeling of the metabolic network to favor a shift of substrate preference toward glucose. Although TG mice on normal diet maintained normal cardiac energetics and function, inability to upregulate myocardial fatty acid oxidation in TG mice fed high fat diet resulted in increased oxidative stress in the heart, activation of p38 MAPK and contractile dysfunction.
Conclusions
We have demonstrated that chronic increases in myocardial glucose uptake and oxidation reduce the metabolic flexibility and render the heart susceptible to contractile dysfunction.
Keywords: fatty acids, glucose, metabolism, cardiomyopathy, contractility
Introduction
The heart requires constant and substantial energy supply for continuous pumping and thus has developed an elaborate metabolic network for utilizing all carbon substrates, including carbohydrates, fatty acids, ketones and amino acids. During development, the substrate preference of heart switches from primarily carbohydrate (fetal and neonatal stage) to predominately fatty acids (adult).1, 2 Although the adult heart utilizes fatty acids for over 50% of its energy supply the cardiac metabolic machinery is highly flexible allowing acute switch of substrate utilization in response to a variety of stresses, such as exercise, fasting and ischemia.2, 3 Chronic shift of myocardial substrate preference has also been noted in many diseases such as diabetes and heart failure.4, 5 However, the underlying mechanisms as well as the functional consequence of the shift are poorly understood.
We have previously shown that the adult mouse heart can adapt to sustained high intracellular glucose by switching to a fetal-like metabolic pattern for life with no adverse functional consequence.6-8 Here we demonstrate that chronic increases of intracellular glucose altered expressions and activities of key regulatory proteins in fatty acid and ketone metabolism pathways. Such a remodeling allows a long-term shift of substrate preference toward glucose while maintains cardiac energetic and function. However, in our mouse model of complete adaptation to high intracellular glucose milieu, the heart fails to up-regulate fatty acid oxidation during diet-induced obesity and suffers from increased oxidative stress and contractile dysfunction. Thus, the prevention of the high fatty acid oxidation during high fat diet induced obesity predisposes the heart to functional impairment.
Methods
Animal models
Transgenic mice overexpressing the insulin-independent glucose transporter GLUT1 in the heart (TG) were generated on FVB background as previously described.6 TG mice and their WT littermates (16 weeks old) were randomly assigned to high-fat diets (45% energy from fat, HF) and nutrient matched low-fat diet (12% energy from fat, LF, both from TestDiet, Richmond, IN) for 20 weeks. Mice were housed in a climate-controlled environment with a 12-h light/dark cycle and free access to food and water. Animal experimental protocols were approved by Harvard Medical Area Standing Committee on Animals. After 20 weeks of feeding, blood samples were drawn from mice for determinations of glucose (ONE TOUCH Glucose Monitor, Lifescan Inc.), free fatty acids (Wako Chemicals) and insulin (Crystal Chemical Inc.) levels using commercially available assay kits.
Isolated perfused heart experiments and NMR spectroscopy
Mice were heparinized (100 U, i.p.) and anesthetized by sodium pentobarbital (150mg/Kg, i.p). The heart was excised and perfused at a constant pressure of 80 mmHg at 37°C as previously described.7 The perfusate contained the following (in mmol/L) NaCl (118), NaHCO3 (25), KCl (5.3), CaCl2 (2), MgSO4 (1.2), EDTA (0.5), glucose (5.5), mixed long chain fatty acids (0.4, bound to 1% albumin), DL-β- hydroxybutyrate (0.38), lactate (1.0) and insulin (50 μU/ml), equilibrated with 95% O2 and 5% CO2 (pH 7.4). Hearts were paced at 7Hz throughout the protocol. Isovolumic contractile function was estimated by the product of LV developed pressure and heart rate (rate pressure product; RPP). Myocardial oxygen consumption (MVO2) was measured by determining the A-V differences in O2 saturation as previously described.8
After a 30-minute equilibration period, hearts were maintained at baseline workload or challenged with high workload by increasing CaCl2 concentration from 2 to 4 mM in the perfusate for 30 minutes. Dynamic changes in cardiac high energy phosphate content and intracellular pH (pHi) were monitored by 31P NMR spectroscopy simultaneously with a continuous recording of LV function. During baseline and high workload, the perfusion buffer contains 13C-labeled substrates for determination of the relative contribution of each substrate to the oxidative metabolism as previously described.8, 9 At the end of experiments, hearts were freeze-clamped with Wollenberger tongs pre-cooled in liquid nitrogen.
RNA extraction and quantitative RT-PCR
Total RNA extraction and quantitative RT-PCR of heart samples were performed as previously described.10 Primer and probe sequences for these assays are presented in Data Supplement Table I. Standard RNA was made for all assays by the T7 polymerase method (Ambion, Austin, TX) with the use of total RNA isolated from mouse hearts. Additional details are provided in the Data Supplement.
Biochemical assays
Myocardial lipids were extracted from unperfused hearts by chloroform/methanol and triglyceride content was assayed using a Sigma kit (TR0100) as described previously.11 Detection and quantification of myocardial malonyl-CoA level were performed by extracting CoA esters from powdered tissue into 6% perchloric acid and measuring with a modified HPLC as described.12 Cardiac glycogen content was determined by an alkaline extraction procedure to separate glycogen and exogenous glucose in the tissue. Glucose released from glycogen was measured with a glucose assay kit (GAHK-20; Sigma).
Immunoblotting
Western blotting was performed using cardiac tissue lysates. 40 μg of total protein extracts were resolved by SDS-PAGE and transferred to polyvinylidene fluoride membranes. Overnight incubation (4°C) was performed with the primary antibodies. Further details are provided in the Data Supplement.
Trans-thoracic echocardiography
Murine trans-thoracic echocardiography was conducted in conscious mice using an Acuson Sequoia C-256 echocardiograph machine (Siemens, Malvern, PA) and a 15 MHz probe as described previously.7
Measurements of cell shortening and intracellular Ca2+ transients
Freshly isolated cardiac myocytes were placed in a chamber mounted on the stage of a Nikon TS 100 microscope and superfused with a Tyrode buffer containing 1.8 mmol/l CaCl2, pH 7.4 at 37°C. Myocytes were field stimulated at 5 Hz, and cell shortening and relengthening were assessed using a video edge detector and specialized data acquisition software (SoftEdge Acquisition System and IonWizard, IonOptix). For simultaneous measurement of intracellular Ca2+ transient, myocytes were preloaded with 1 μmol/L fura-2/AM (Molecular Probes, Carlsbad, CA) for 15 min, and fluorescence intensity was recorded with a dual-excitation fluorescence photomultiplier system (IonOptix, Milton, MA) as previously described.13
Analysis of tissue ROS level
Tissue levels of lipid peroxides in heart homogenates were assessed by measuring the chromophore formed by the reaction of N-methyl-2-phenylindole and methanesulfonic acid as described.14 To prevent sample oxidation during the assay, 5mM of butylated hydroxytoluence was added prior to the homogenization. For histological assessments of tissue ROS level, cryosections of left ventricle were incubated for 1 h at 37°C with 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (DCFDA, 4 μM; Molecular Probes) as described.15 The positive DCFDA staining in the myocardium was quantified in Matlab (The Mathworks, Inc., Natick, MA). Data from each heart was normalized by background fluorescence intensity to calculate the DCFDA intensity.
Statistical Analysis
All data were presented as the mean ± SEM. ANOVA with Bonferroni post-hoc test was performed for multiple group comparisons and unpaired t-test was used for two group comparisons. Analyses were performed using GraphPad Prism 4.0 (GraphPad Software, San Diego, CA) and a value of p < 0.05 was considered to be significant.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
Animal models
We utilized transgenic mice with cardiac specific overexpression of the insulin-independent glucose transporter GLUT1 (TG) as a model of increased intracellular glucose in cardiac myocytes.6 High fat diet was used to increase the fatty acids supply to the heart. Compared with normal diet groups, mice fed HF for 20 weeks exhibited about 50% increase of body weight for both TG and their wildtype littermates (WT) (p < 0.05 for both, Figure 1A). There was no difference in heart weight normalized to body weight or to tibia length among all groups (Data Supplement Table II). Serum glucose and insulin levels were also significantly elevated but to the same extent in HFWT and HFTG mice (Figure 1B and 1C). These results suggest that the WT and TG mice responded similarly to the high fat diet, and both developed obesity, hyperglycemia and insulin resistance.
Figure 1.

Body weight and blood metabolic parameters for WT and TG mice fed a high fat diet (HF) or control diet (LF) for 20 weeks. (A) Body weight (B) Blood glucose (C) Serum insulin and (D) Serum nonesterified fatty acid (NEFA) levels at fed state. Data are mean ± SEM from 5 to 6 mice per group. *p < 0.05 vs LFWT.
Enhanced myocardial glucose utilization in TG is resistant to modulation by high fat diet
To delineate the effects of increased glucose and fatty acids availability on the metabolic fate of each substrate, we examined the relative contributions of five classes of carbon substrates to the oxidative metabolism and MVO2 in isolated perfused hearts from the four groups of mice using 13C NMR isotopomer analysis (Figure 2). Compared to WT, the contribution of glucose to oxidative metabolism in TG hearts was increased by more than 2 fold in mice fed normal diet (p < 0.05). High fat diet increased the contribution of fatty acids to oxidative metabolism by 40% in WT, while the contribution from glucose decreased to 30% of LFWT suggesting an upregulation of myocardial fatty acid oxidation in mice with diet-induced obesity (p < 0.05 versus LFWT for both). Interestingly, high fat diet did not induce upregulation of myocardial fatty acid oxidation in TG mice, and the utilization of glucose remained high after 20 weeks of high fat diet. Of note, none of the relative contributions from other substrates, i.e. ketone, lactate and endogenous substrates in TG hearts was affected by high fat diet (Figure 2A) suggesting that TG hearts developed a metabolic pattern resistant to modulation by high fat diet. When the hearts were challenged with high workload (cardiac performance increased by 60%), the relative oxidation of glucose increased and that of fatty acids decreased in WT hearts regardless of diet (Figure 2B), consistent with the previous studies showing that the relative contribution of carbon substrates to oxidative metabolism shifts to glucose under stress.16,17 In contrast, when TG hearts were challenged with high workload, there was no change in the substrate utilization profile compared with baseline in either diet group, suggesting a loss of metabolic flexibility. Since the 13C NMR isotopomer analysis provided relative oxidation of each substrate, we determined MVO2 to assess total oxidative metabolism. MVO2 was similar among the four groups at baseline and increased ∼60% at high workload (Figure 2C and 2D). MVO2 was slightly lower in HFTG at high workload consistent with a lower contractile performance in the group (see below). MVO2 normalized to workload was similar for all groups at each workload (Figure 2E and 2F).
Figure 2.

Substrate utilization in the hearts of WT and TG mice fed a HF or LF diet. (A and B) Relative contributions of each substrate to the oxidative metabolism in isolated mouse hearts perfused with 13C-labeled substrates during baseline (A) and high workload (B) (n=5 to 7 per group). (C-F) Myocardial oxygen consumption and oxygen efficiency for contractile function during baseline (C and E) and high workload (D and F) (n=3 to 4 per group). Total glycogen (G) and triglyceride (H) content (n=5 to 7 per group) in the hearts of WT and TG mice fed a HF or LF diet for 20 weeks. Data are mean ± SEM. *p < 0.05 vs LFWT; #p < 0.05 vs LFTG (see text for statistical significance for figure 2A and 2B).
Consistent with our previous observation, we found that myocardial glycogen content increased by more than 3-fold in TG hearts (Figure 2G). Although the high fat diet increased glycogen content significantly (by 32%) in WT hearts, the glycogen content in TG hearts did not increase further by high fat diet. We also found a 2-fold increase of triglyceride content in TG hearts, which was in contrast to a modest increase (31%) caused by high fat diet in WT hearts (Figure 2H). This was consistent with the observation of suppressed fatty acids contribution to oxidation in TG hearts. Collectively, the data showed that glucose utilization was reduced and fatty acids oxidation increased in WT hearts by high fat diet but not in TG hearts even though diet-induced obesity and insulin resistance developed in both groups.
Chronic increases in substrate availability remodeled the metabolic network at molecular level
To determine the molecular mechanisms responsible for the changes of substrate preference caused by long-term alterations of substrate availability to the heart, we measured the expression of a subset of genes known to play key roles in myocardial substrate utilization in the heart (Figure 3 and Data Supplement Figures I). PPARα, a key player in transcriptional control of cardiac fatty acid metabolism, was significantly downregulated in hearts with increased glucose or fatty acid availability (Figure 3A). The downregulation was more pronounced in heart with high glucose supply (TG) than in hearts with high fatty acids supply (high fat diet). The expression of PPARδ, another important regulator of fatty acid oxidation in cardiac tissue, decreased modestly only in HFWT group (Figures IA in the Data Supplement). The expression of RXRα, the obligate partner of PPARs, was not different among the groups (Figures IB in the Data Supplement). Despite the marked decrease of PPARα expression, the expression of its target genes involved in fatty acid oxidation were mostly unsuppressed (Figures IC-IH in the Data Supplement). However, one of the PPARα target genes, the fatty acid transporter CD36, was induced by high fat diet in WT but not in TG hearts (Figure 3B).
Figure 3.

Expression of genes involved in myocardial substrate utilization and myocardial malonyl CoA level in the hearts of WT and TG mice fed a HF or LF diet. (A-E) PPAR α, CD36, SCOT, ACC α and ACC β mRNA expression levels determined by real-time PCR (n=6 per group). (F) ACC protein level in the hearts determined by Western blot (n=4-6 per group). (G) Myocardial malonyl CoA content in hearts of WT and TG fed a HF or LF diet (n=5 to 7 per group). Data are mean ± SEM. *p < 0.05 vs LFWT; #p < 0.05 vs HFWT.
We found significant changes in the expression of 3-oxoacid CoA transferase (SCOT) and acetyl-CoA carboxylase (ACC); neither of them was PPARα target gene but played important roles in ketone and fatty acid metabolism. SCOT expression was downregulated by 67% in TG hearts and the change was independent of the diet regimen (Figure 3C). SCOT catalyzes the rate-limiting step in the formation of acetyl-CoA from acetoacetate by transferring the CoA from succinyl-CoA, and is therefore, essential for ketone oxidation. The marked downregulation of SCOT predicts decreased ketone oxidation in TG hearts, which has been demonstrated by the 13C NMR study (Figure 2).
The expressions of ACCα, mainly expressed in the liver, and ACCβ, mainly expressed in the heart, were both significantly induced in the LFTG and HFTG hearts (Figure 3D and 3E). The fact that ACC was not induced in HFWT and was not further induced by high fat diet in TG hearts suggested that this change, similar to the downregulation of SCOT, was a glucose-dependent event. Consistent with the gene expression changes, western blotting revealed significant increases of ACC protein in the hearts of TG mice on either diet regimen, and no change of ACC protein amount was found in the HFWT hearts (Figure 3F).
ACC catalyzes the synthesis of malonyl-CoA, a key metabolite in fatty acid metabolism that is degraded by malonyl-CoA decarboxylase (MCD). MCD expression was not altered in all groups (data not shown). Cardiac content of malonyl-CoA increased by 32% in the TG hearts (Figure 3G) suggesting that the total ACC activity was increased in these hearts. Similar to the pattern of ACC upregulation, cardiac malonyl-CoA content increased only in TG hearts and was not altered by high fat diet in either WT or TG group. Since malonyl CoA is a potent inhibitor of mCPT-1 in oxidative tissue, increased malonyl-CoA in TG hearts inhibits the rate-limiting step of fatty acids entering mitochondria thus limiting the upregulation of fatty acid oxidation and increasing acyl-CoA content (Figure 4). In addition, malonyl CoA produced by ACCα in lipogenic tissue is an essential substrate for fatty acid synthesis raising the possibility that the substantial upregulation of ACCα in TG hearts results in increased fatty acid synthesis and triglyceride accumulation although fatty acid synthesis activity is considered very low in the heart.
Figure 4.
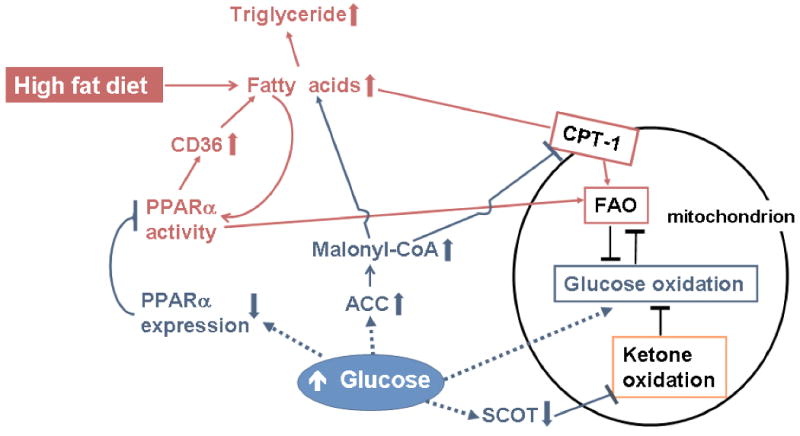
Mechanisms by which high intracellular glucose remodel metabolic network to perpetuate glucose oxidation and to resist the stimulation of fatty acids during high fat diet feeding. Increased intracellular glucose downregulates the expression of 3-oxoacid CoA transferase (SCOT), a key enzyme in ketone metabolism, resulting in decreased oxidation of ketones. High glucose also downregulates PPARα expression, which limits the stimulatory effects of fatty acids on the expression of array of genes involved in fatty acid uptake and fatty acid oxidation (FAO) such as CD36 and CPT-1. Independent of PPARα mechanisms, glucose induced expression of acetyl-CoA carboxylase (ACC) that leads to increased synthesis of malonyl-CoA which inhibits fatty acid entering mitochondria via CPT-1 and promotes lipid synthesis and storage in the cytosol.
These data collectively suggest that high intracellular glucose induced changes of the regulatory enzymes at the level of transcription in favor of glucose oxidation at the expense of fatty acids and ketone oxidation (Figure 4). Furthermore, the induction of ACC, together with downregulation of PPARα and consequently failure to upregulate CD36, prevented the upregulation of fatty acid oxidation despite diet-induced obesity.
Impaired contractile function but normal energetics in TG hearts during diet-induced obesity
To determine whether the altered substrate utilization profile had any functional significance, we assessed in vivo cardiac function by echocardiography in all mice at the end of 20-week diet feeding (age of mice: 36-40 weeks). Compared with WT mice, LV fractional shortening was modestly but significantly decreased in TG mice fed high fat diet (57 ± 2 vs. 65 ± 1, p < 0.05). HFTG mice also showed modest increases in LV end-diastolic and end-systolic dimensions with no changes in wall thickness (Figure 5A-5D). Thus, inability to shift myocardial substrate utilization in TG mice fed high fat diet was associated with mild cardiac dysfunction in vivo.
Figure 5.

Cardiac function assessed by trans-thoracic echocardiography. (A) LV factional shortening, (B) LV end systolic diameter, (C) LV end diastolic diameter and (D) LV posterior wall thickness in WT and TG fed a HF or LF diet for 20 weeks. Data are mean ± SEM (n=8 to10 per group). *p < 0.05 vs LFWT.
We also assessed isovolumic contractile function of isolated perfused hearts from all groups in order to evaluate myocardial contractile function independent of neurohormonal regulation. At the baseline workload, all hearts were able to achieve and sustain a RPP of ∼50,000 mmHg/min (Figure 6A). When subjected to high workload by doubling the Ca2+ concentration in the perfusion buffer, the RPP increased ∼60% in all groups. Importantly, all hearts except the HFTG group could sustain the high contractile performance. At the end of the high-workload period, the RPP of HFTG was decreased by 37% (Figure 6A) suggesting an impaired contractile reserve. This finding corroborated the in vivo function study and suggested that TG hearts, although protected from high fatty acid oxidation, were maladaptive to high fat diet-induced obesity.
Figure 6.

Contractile function and myocardial high-energy phosphate of isolated perfused mouse hearts measured by 31P NMR spectroscopy. (A) Cardiac contractile function estimated as RPP (n= 6 to 12 per group). (B-D) Myocardial concentrations of PCr, ATP and intracellular pH (n=4 to 5 per group) in the hearts of WT and TG fed a HF or LF diet during baseline and high workload. Dotted vertical lines represent the switch from baseline to high workload. Data are mean ± SEM. *p < 0.05 vs LFWT.
Since TG mice on high fat diet failed to increase myocardial fatty acid oxidation, we tested whether the inability to sustain high workload could be attributed to impaired myocardial energetic status due to inadequate ATP synthesis. Using 31P NMR spectroscopy we measured dynamic changes of high-energy phosphate content in isolated perfused hearts at baseline and high workload. The concentrations of ATP and PCr, the energy reserve compound, as well as the intracellular pH were not significantly different in all groups during both baseline and high workload challenge (Figure 6B-6D); suggesting myocardial energetic status was not impaired in these models. Thus, the contractile dysfunction associated with the failure to increase fatty acid oxidation in HFTG hearts was unlikely due to insufficient energy supply.
Intracellular calcium homeostasis is maintained in cardiac myocytes of HFTG mice
Impaired calcium handling has been shown in diabetic cardiomyopathy.18 We thus determined whether abnormal intracellular calcium handling contributes to the impaired contractile function in HFTG. We found a significant decrease of sarcoplasmic reticulum calcium ATPase (SERCA) protein in HFTG hearts (Figure 7A). Phospholamban (PLN), the inhibitory regulator of SERCA, was unchanged in total amount but its phosphorylation was increased significantly (Figure 7B). Phosphorylation of PLN would decrease the inhibition of SERCA, thus, serving as a compensatory mechanism for maintaining the activity of SERCA protein. The sodium-calcium exchanger protein 1 (NCX1) was unchanged in all groups (Figure 7C). To directly assess the intracellular calcium cycling and its effect on cardiac myocyte contraction, we isolated cardiac myocytes from the four groups of hearts and measured intracellular calcium simultaneously with cell shortening (Figure 7D and 7E). The amplitude of cell shortening was decreased in myocytes isolated from HFTG hearts but the cytosolic calcium level was unaltered either at resting state or at the peak of contraction. Thus, the myocyte study reproduced the contractile dysfunction observed both in isolated hearts and in vivo. Furthermore, it suggested that mechanisms independent of intracellular calcium handling were responsible for the impaired contraction of HFTG hearts.
Figure 7.

Assessment of intracellular Ca2+ handling and cardiomyocyte contractility. (A-C) Protein expression of calcium cycling proteins, SERCA2a, PLN, p-PLN and NCX1. (D) Percentage shortening and (E) intracellular calcium levels in freshly isolated cardiomyocyte. Data are expressed as mean ± SEM from 4-5 hearts for each group. *p < 0.05 vs LFWT.
Increased oxidative stress and activation of p38 MAPK in HFTG hearts
Previous studies have shown that activation of p38 MAPK exerts negative inotropic effects on cardiac myocytes independent of cytosolic calcium cycling.19-21 We examined MAP kinase pathway and found increased phosphorylation of p38 MAPK in HFTG hearts (Figure 8A). The activation of p38 MAPK appeared to be unique since phosphorylations of other MAPKs, such as ERK or JNK, were increased only slightly and non-discriminatorily by high fat diet in WT and TG hearts. Using two independent methods to evaluate ROS level in the heart we found increased oxidative stress in HFTG group. Cardiac malondialdehyde (MDA) and 4-hydroxyalkenals (HAE) levels, indicators of lipid peroxide content, were increased by over 2.5-fold in HFTG group (Figure 8B). Increased tissue ROS load was also indicated by a higher intensity of DCFDA fluorescence in frozen tissue sections from HFTG hearts (Figure 8C). Concurrent increases of oxidative stress and p38 MAPK activation in HFTG suggest an important role of this pathway in the pathogenesis of contractile dysfunction during chronic metabolic derangement.
Figure 8.

p38 MAPK activation and increased ROS production in the heart of TG mice fed high fat diet. (A) Protein levels of MAP kinases and their phosphorylation in the hearts of WT and TG fed a HF or LF diet for 20 weeks. Since p-P38 MAPK was nearly undetectable in LF diet group, no quantitative comparisons among the groups were made. (B) Lipid peroxide content and (C) Frozen heart sections stained with green fluorescence dye DCFDA. Scale bar represents 20 μm. Data are expressed as mean ± SEM from 4-5 hearts for each group. *p < 0.05 vs LFWT.
Discussion
Fatty acid oxidation supplies more than 50% of energy for a normal adult heart while glucose and lactate are the main energy source for fetal hearts.1-3, 5 Using genetic manipulations, we have generated an adult mouse heart (TG) that utilizes carbohydrates (glucose and lactate) for 50% of its energy supply whereas the utilization of fatty acids and ketone is suppressed, thus mimicking the fetal metabolic profile.8 The TG hearts have normal morphology and function and the TG mice live a normal life span, suggesting a complete adaptation to the fetal-like cardiac metabolism.7 However, unlike the neonatal hearts that switch to fatty acid oxidation when circulating levels of fatty acids and oxygen supply increase after birth, glucose oxidation is sustained and high fatty acid oxidation is prevented in the adult TG hearts despite increased fatty acids availability during high fat diet induced obesity. We have identified a significant upregulation of ACC protein and activity in TG hearts that led to increased malonyl-CoA production resulting in decreased fatty acid entry into mitochondria and increased lipid storage in the cytosol. Furthermore, the induction of ACC is glucose-specific and is not affected by fatty acid availability thus represents a unique mechanism that cross-communicates glucose and fatty acid metabolism in the heart. A similar observation is made for SCOT that accounts for inhibition of ketone oxidation in TG hearts.
The transcriptional program mediated by PPARα and PPAR δ plays a central role in maintaining fatty acid oxidation in adult hearts.8,11,22 It is also responsible for the upregulation of fatty acid oxidation during increased fatty acid supply such as fasting or high fat diet.23, 24 Interestingly, chronic increases in the availability of either glucose or fatty acids result in a decrease of PPARα expression although the expression of many PPARα target genes are not suppressed. The dissociation of PPARα expression and its activity in these models suggest that PPARα expression level alone may not be an appropriate marker of cardiac substrate metabolism in vivo. We speculate that increased fatty acids supply to the heart during high fat diet presents a higher amount of PPARα ligands that stimulate the transcriptional activity. It is possible that increased lipid storage in the TG hearts also stimulate PPARα activity. Similar observation has been made previously in hearts with suppressed fatty acid oxidation and increased lipid content.12, 25 These observations indicate a very important role of feedback circuits in the network response to metabolic stress that would not have been predicted by the overexpression or deletion of PPARα. In addition, the substantial down regulation of PPARα expression (51% lower) in TG hearts may have played a key role in counteracting the stimulatory effects of fatty acids, e.g. upregulation of fatty acid transporter CD36, during high fat diet. We suggest that this mechanism adds to the ACC upregulation and renders the TG hearts resistance to high fat diet-induced shift to fatty acid oxidation (Figure 4).
Despite the striking differences in substrate utilization pattern, cardiac energetics and function in WT mice subjected to high fat diet or in TG mice on regular diet are well sustained supporting the notion that a normal heart is capable of utilizing a wide range of energy substrates without compromising its function. These observations suggest that a simple shift of substrate preference towards carbohydrates or fatty acids is not sufficient to cause cardiac “glucotoxicity” or “lipotoxiciy”. However, the adjustments of cardiac metabolic network to chronic changes of substrate availability deplete the system of its flexibility at molecular level in such that the reestablished homeostasis is vulnerable to further stresses. When the TG mice were subjected to high fat diet and developed obesity and insulin resistance, the absence of increased fatty acid oxidation was not protective but associated with cardiac dysfunction. Thus, although high fatty acid oxidation in the heart has been associated with a number of pathological conditions our findings suggest that maneuvers to sustain myocardial glucose oxidation and/or to prevent high fatty acid oxidation in obesity should be evaluated with great caution.
Altered substrate metabolism in the heart has been observed in multiple disease conditions, but its role in the pathogenesis of cardiac dysfunction has been elusive. It has been shown that a shift toward glucose preference is favorable for sustaining energy production under conditions of impaired ATP generation from fatty acid oxidation such as cardiac hypertrophy.6, 8 However, here we show that glucose-centered metabolic network in adult heart increases the susceptibility to fatty acid overload despite a complete functional adaptation in the absence of additional metabolic perturbation.7 In diet-induced obesity, we find that a higher level of reactive oxygen species in HFTG hearts when increased fatty acids availability failed to upregulate fatty acid oxidation. Activation p38 MAPK, observed in HFTG hearts, is a known consequence of increased oxidative stress.20, 26, 27 Interestingly, chronic activation of p38 MAPK has also been shown to impair contractile function in the absence of abnormal calcium handling,19-21 thus a likely mechanism for cardiac dysfunction observed in HFTG.
The present study has also identified molecular responses in E-C coupling mechanisms during chronic changes of substrate availability. For example, increased oxidative stress in HFTG hearts is associated with a downregulation of SERCA protein at post-transcriptional level. Interestingly, decreased SERCA protein is compensated by inactivation of phospholamban in HFTG yielding normal calcium transient. These mechanisms partially compensate for the impairment of cardiac function in chronic metabolic derangement. Indeed, the in vivo cardiac dysfunction observed in the present study is rather mild. Nevertheless, the study demonstrated that the underlying molecular remodeling in response to altered metabolism form a basis for accelerated deterioration when other pathological conditions superimpose.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jun Yoshioka for his advice on measurement of tissue ROS, Dr. Benbo Gao for his help with biochemical assays, Drs. Jianru Shi and Bo Wang for their help in cardiac myocyte experiments.
Sources of Funding: This study is supported by NIH fund R01 HL59246 (to Dr Tian). Dr. Tian is an Established Investigator of the American Heart Association.
Footnotes
Disclosures: None.
References
- 1.Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol. 1991;261:H1698–1705. doi: 10.1152/ajpheart.1991.261.6.H1698. [DOI] [PubMed] [Google Scholar]
- 2.Opie LH. Metabolism of the heart in health and disease I. Am Heart J. 1968;76:685–698. doi: 10.1016/0002-8703(68)90168-3. [DOI] [PubMed] [Google Scholar]
- 3.Opie LH. The heart: physiology, from cell to circulation. Philadelphia: Lippincott-Raven Publishers; 1998. pp. 295–342. [Google Scholar]
- 4.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 5.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 6.Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002;106:2125–2131. doi: 10.1161/01.cir.0000034049.61181.f3. [DOI] [PubMed] [Google Scholar]
- 7.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116:901–909. doi: 10.1161/CIRCULATIONAHA.107.691253. [DOI] [PubMed] [Google Scholar]
- 8.Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112:2339–2346. doi: 10.1161/CIRCULATIONAHA.105.534594. [DOI] [PubMed] [Google Scholar]
- 9.Malloy CR, Sherry AD, Jeffrey FM. Analysis of tricarboxylic acid cycle of the heart using 13C isotope isomers. Am J Physiol. 1990;259:H987–995. doi: 10.1152/ajpheart.1990.259.3.H987. [DOI] [PubMed] [Google Scholar]
- 10.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 11.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 12.Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, Kawase Y, Jishage K, Lopaschuk GD. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. 2006;114:1721–1728. doi: 10.1161/CIRCULATIONAHA.106.642009. [DOI] [PubMed] [Google Scholar]
- 13.Nagata K, Ye C, Jain M, Milstone DS, Liao R, Mortensen RM. Galpha(i2) but not galpha(i3) is required for muscarinic inhibition of contractility and calcium currents in adult cardiomyocytes. Circ Res. 2000;87:903–909. doi: 10.1161/01.res.87.10.903. [DOI] [PubMed] [Google Scholar]
- 14.Yoshioka J, Imahashi K, Gabel SA, Chutkow WA, Burds AA, Gannon J, Schulze PC, MacGillivray C, London RE, Murphy E, Lee RT. Targeted Deletion of Thioredoxin-Interacting Protein Regulates Cardiac Dysfunction in Response to Pressure Overload. Circ Res. 2007;101:1328–1338. doi: 10.1161/CIRCRESAHA.106.160515. [DOI] [PubMed] [Google Scholar]
- 15.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A. 2006;103:7432–7437. doi: 10.1073/pnas.0510444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 17.Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. 1998;273:29530–29539. doi: 10.1074/jbc.273.45.29530. [DOI] [PubMed] [Google Scholar]
- 18.Trost SU, Belke DD, Bluhm WF, Meyer M, Swanson E, Dillmann WH. Overexpression of the sarcoplasmic reticulum Ca(2+)-ATPase improves myocardial contractility in diabetic cardiomyopahy. Diabetes. 2002;51:1166–71. doi: 10.2337/diabetes.51.4.1166. [DOI] [PubMed] [Google Scholar]
- 19.Liao P, Wang SQ, Wang S, Zheng M, Zheng M, Zhang SJ, Cheng H, Wang Y, Xiao RP. p38 Mitogen-activated protein kinase mediates a negative inotropic effect in cardiac myocytes. Circ Res. 2002;90:190–196. doi: 10.1161/hh0202.104220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palomeque J, Sapia L, Hajjar RJ, Mattiazzi A, Vila Petroff M. Angiotensin II-induced negative inotropy in rat ventricular myocytes: role of reactive oxygen species and p38 MAPK. Am J Physiol. 2006;290:H96–106. doi: 10.1152/ajpheart.00324.2005. [DOI] [PubMed] [Google Scholar]
- 21.Vahebi S, Ota A, Li M, Warren CM, de Tombe PP, Wang Y, Solaro RJ. p38-MAPK induced dephosphorylation of alpha-tropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circ Res. 2007;100:408–415. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- 22.Tian R. Transcriptional regulation of energy substrate metabolism in normal and hypertrophied heart. Curr Hypertens Rep. 2003;5:454–458. doi: 10.1007/s11906-003-0052-7. [DOI] [PubMed] [Google Scholar]
- 23.Finck BN. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res. 2007;73:269–277. doi: 10.1016/j.cardiores.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 24.Young ME, Patil S, Ying J, Depre C, Ahuja HS, Shipley GL, Stepkowski SM, Davies PJ, Taegtmeyer H. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor (alpha) in the adult rodent heart. Faseb J. 2001;15:833–845. doi: 10.1096/fj.00-0351com. [DOI] [PubMed] [Google Scholar]
- 25.Okere IC, Chandler MP, McElfresh TA, Rennison JH, Kung TA, Hoit BD, Ernsberger P, Young ME, Stanley WC. Carnitine palmitoyl transferase-I inhibition is not associated with cardiac hypertrophy in rats fed a high-fat diet. Clin Exp Pharmacol Physiol. 2007;34:113–119. doi: 10.1111/j.1440-1681.2007.04545.x. [DOI] [PubMed] [Google Scholar]
- 26.Clerk A, Fuller SJ, Michael A, Sugden PH. Stimulation of “stress-regulated” mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J Biol Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- 27.Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol. 2002;282:L1324–1329. doi: 10.1152/ajplung.00326.2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.