

Abstract
Natural and non-natural cyclic peptides are a crucial component in drug discovery programs because of their considerable pharmaceutical properties. Cyclosporin, microcystins and nodularins all are notable pharmacologically important cyclic peptides. Because these biologically active peptides are often biosynthesized non-ribosomally, they often contain non-standard amino acids, thus increasing the complexity of the resulting tandem mass spectrometry data. In addition, due to the cyclic nature, the fragmentation patterns of many of these peptides showed much higher complexity when compared to related counterparts. Therefore, at the present time it is still difficult to annotate cyclic peptides MS/MS spectra. In this current work, an annotation program was developed for the annotation and characterization of tandem mass spectra obtained from cyclic peptides. This program, which we call MS-CPA is available as a web tool (http://lol.ucsd.edu/ms-cpa_v1/Input.py). Using this program, we have successfully annotated the sequence of representative cyclic peptides, such as seglitide, tyrothricin, desmethoxymajusculamide C, dudawalamide A and cyclomarins in a rapid manner, and also were able to provide the first-pass structure evidence of a newly discovered natural product base on predicted sequence. This compound is not available in sufficient quantities for structural elucidation by other means such as NMR.1 In addition to the development of this cyclic annotation program, it was observed that some cyclic peptides fragmented in unexpected ways resulting in the scrambling of sequences. In sum, MS-CPA not only provides a platform for rapid confirmation and annotation of tandem mass spectrometry data obtained with cyclic peptides but also enables quantitative analysis of the ion intensities. This program facilitates cyclic peptide analysis, sequencing, but also acts as a useful tool to investigate the uncommon fragmentation phenomena of cyclic peptides and aids the characterization of newly discovered cyclic peptides encountered in drug discovery programs.
Keywords: cyclic peptides, sequence-scrambling fragmentation pathway, mass spectrometry, natural products, non-ribosomal peptides
INTRODUCTION
Ribosomally as well as non-ribosomally derived cyclic peptides are an important group of compounds because of their wide range biological, toxic and pharmacological activities and they often exhibit unique chemical structures.2,3 For example the cyclic toxins microcystins and nodularins produced by cyanobacteria (Blue-green algae) can wipe out entire fisheries and can cause death in humans.4,5 In addition it is now becoming increasingly clear that these naturally occurring cyclic peptides have biological roles in quorem sensing,6,7 gliding,8,9 prevention of aerial growth,10 or cell adherence regulation,11 and that they can be used as a diagnostic markers for disease.12 In addition, many cyclic peptides are used in clinic. Well-known examples of cyclic natural products are cyclosporine, an immunosuppressant drug used to prevent organ rejection,13 seglitide, a potent growth factor release inhibitor,14 and ramoplanin, a novel antibiotic.15 Because of the importance of their therapeutic applications there is a continued development of strategies to generate cyclic libraries for drug screening programs.16–20 In fact, many cyclic natural products with potent therapeutic properties are discovered every week.1,21–24 Therefore it is important to continue developing methods not only for isolating or preparing such cyclic peptides but also to characterize such peptides.
Despite a lot of effort by mass spectrometrists25–37, we are still exploring the way cyclic peptides behave in a mass spectrometer, in particular during collision-induced dissociation (CID). Bioinformatics tools such as MASCOT, SEQUEST and InsPecT are capable of robust interpretation of tandem MS spectra and also enable protein identification with the equipped database search engines.38–40 However, few tools are designed for cyclic peptides with a user-friendly interface at a level accessible to non-mass spectrometrists. In addition most of the bioinformatics tools are based on somewhat refined fragmentation models i.e. they may only annotate b and y ions. Both of these are the likely reasons why most scientists that isolate cyclic natural products and that develop cyclic peptide libraries for drug screening programs ignore all but only annotate a small amount of the ions that are typically observed from cyclic peptides in their structural elucidation efforts, leaving tens to hundreds of ions unaccounted for.30 We became interested in this problem because when we attempted to annotate the tandem mass spectra of cyclic natural products isolated from marine organisms by manual means and discovered that a large proportion of the spectral intensity remained unaccounted for and that the annotation was very time consuming. Although a program that predict theoretical fragmentation patterns such as PIFA may assist in manually annotation of cyclic peptides by providing all possible b ions41, MS-CPA is capable of direct annotated the actual input cyclic peptide MS spectra and is also the first program that take into account of the fragments resulted in sequence-scrambling fragmentation pathways.
To improve our understanding of the fragmentation behavior of cyclic peptides we have developed a program that readily annotates a mass spectrum resulting from the collision-induced dissociation of cyclic peptides. In addition, we have created a user friendly web interface so that other scientist that are non-computer experts can easily use it to annotate their tandem mass spectra of cyclic peptides. Using this program we observed that much of the spectral intensity of an MS2 of a cyclic peptide could not be explained. Upon further analysis we realized that unanticipated fragmentation pathways involved in cyclic peptides when the standard fragmentation rules were applied. The data suggested those unanticipated fragments resulted in scrambling of sequence. These unusual fragments were first described by Harrisons et al., as nondirect sequence (NDS) ions based on the scrambling of original peptide sequence in contrast to the direct sequence (DS) ions derived from typical fragmentation pathways.30 While initially surprising to the authors that NDS are observed, the mechanistic details towards the formation of NDS ions have recently been described in detail.34 We have included NDS in our annotations. Therefore, our program, MS-CPA not only provides evidence for the existence of these NDS ions but also enables quantitative analysis of the spectral abundance that match to DS and NDS ions.
In order to demonstrate the utility of this program we have not only applied it to the representative testing peptides seglitide and the tyrocidines, but also used it to confirm the sequence of two newly discovered natural products, desmethoxymajusculamide C (DMMC) and dudawalamide A, both isolated from marine cyanobacteria Lyngbya majuscule (Figure 1). In addition, the program was used to verify the structure of desprenylcyclomarin C, a natural product isolated from a prenyltransferase mutant of the marine bacteria Salinispora arenicola CNS-205. This marine natural product could not be isolated in sufficient quantities to confirm its structure by NMR; therefore this program was critical in the confirmation of its structure. Finally, during these studies we discovered three additional dehydrated cyclomarin analogs and used our program to localize the site of dehydration.
Figure 1.
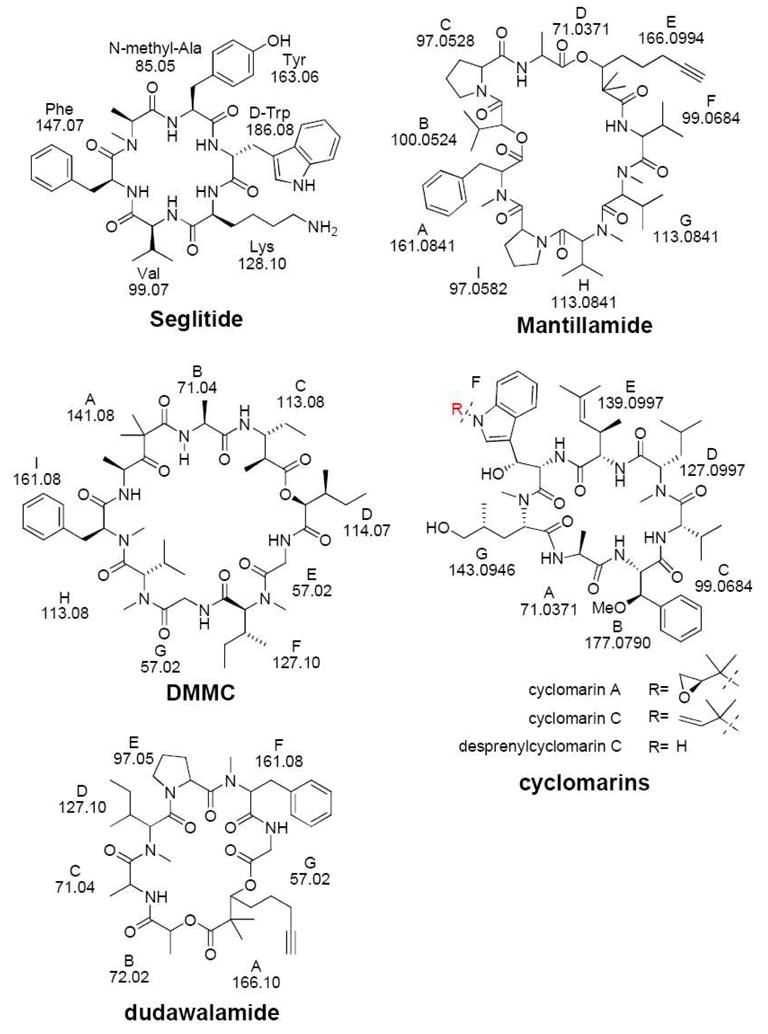
Structures of cyclic peptides discussed in this paper.
RESULTS AND DISSCUSSION
Complexity of cyclic peptide fragmentation
Because so many researchers work with cyclic peptides, the annotation of tandem mass spectra from cyclic peptides is important. The annotation, however, of tandem mass spectra of cyclic peptides is often difficult for mass spectrometrists and natural product scientists alike. The difficulty in the annotation of cyclic peptides arises from the nature of cyclic peptides itself. A cyclic peptide with n amino acid residues, theoretically will yield n series of b-ions but not any y-ions32. If there are other ions such as a-ions, internal fragments, and small neutral losses such as H2O and NH3, this complexity increases significantly. Therefore, it is difficult to annotate each and every ion in the spectrum of cyclic peptides and thus becomes an informatics problem. To overcome some of the complexity in the annotation of these peptides we have developed a program that assists in the annotation of tandem mass spectrometry data based on input amino acid values and an experimental tandem mass spectrometric data set in .dta and .mzXML formats.
While we have presented, at a conference that de novo sequencing of these non-ribosomal peptides can be accomplished with near “perfect” mass spectral data sets using spectral alignments and a combination of de novo and database searching algorithms42, it quickly became clear that when we applied our first generation de novo sequencing algorithms to “non-perfect” mass spectrometry data sets typically encountered with more complex non-ribosomally encoded peptides or symmetric cyclic peptides that these algorithms often identified a slightly different sequence. To improve the de novo sequencing algorithms that can be used confirm the structures of isolated natural products we need to improve our understanding of the resulting ions from a tandem mass spectrometry experiment. This is in particular important when it comes to complex cyclic peptides.
Cyclic peptide annotation program
To aid in the sequencing as well as to improve our understanding of the fragmentation behavior of cyclic peptides of non-ribosomal origin, we developed a program named the MS-Cyclic Peptide Annotation program (MS-CPA) that readily annotates a mass spectrum resulting from the CID of a cyclic peptide. In particular, this program annotates b ions, a ions (losses of CO), and b0 ions (losses of H2O). However, y ions are not included, because cyclic peptides do not yield such ions.32 The annotation program started as a Python script to mark b, a, and b0 ions given a mass spectrum. The current implementation is capable of handling .dta and .mzXML file formats as this data format is becoming the standard format for reporting or depositing mass spectra and/or proteomic data-sets43,44 as spectrum inputs. For the reason that many cyclic peptides contain unusual or modified amino acids, we leave the freedom for users to input the amino acid masses manually. There is no size limitation to the mass of the amino acid that can be manually imported. Additionally, default standard amino acids masses are provided. Finally, the amino acid sequence is specified by the user in the order that they are encountered in the peptide. For example, seglitide has a methylation on the nitrogen of alanine. This is a nonstandard amino acid therefore we can input 85.05280 for methyl-alanine rather than the alanine mass 71.03711. In addition, once it was recognized that even for mass spectrometrically well behaved peptides, a large proportion of the ion intensity remained unexplained, the capabilities of this program was expanded to consider neutral amino acid losses from the b-ion ladder, as well as, evaluation of possible rearrangements based on the series of masses initially given. The current program has thousands of lines of code to annotate a spectrum, and for the generation of a graphical and tabular output on a web server.
We have made the MS-CPA program publicly available as a web tool at the UCSD center for computational mass spectrometry (http://lol.ucsd.edu/ms-cpa_v1/Input.py) and have also included a tutorial in supporting information. In this paper, we demonstrate the utility of MS-CPA for the characterization of the cyclic peptides shown in Figure 1. The cyclic peptides in Figure 1 are representative of the type of cyclic peptides encountered in drug screening programs.
Pre-analysis data processing of the tandem mass spectrometry input file
While the main code for this program is thousands of lines, the main challenge in the annotation process is actually the generation of a spectrum in which most peaks can be interpreted. Because of the great variance of experimental settings, instrumentation, and fragmentation properties of the compounds, pre-processing steps of the data that is required for each compound and experiment can vary a lot. To this end, we implemented a series of filters to enhance the signal to noise ratio of the experimental spectrum. Our current implementation regarding pre-processing includes centroid filtering, rank filtering, water filtering, isotope filtering, peak tolerance, and symmetrization. These preprocessing steps are detailed described in the supporting information but the user can also choose not to carry out any pre-processing. Given that noise peaks are unavoidable in a real mass spectrometry experiment, the main goal of the filters is to eliminate ions that are likely noise, or ions that are uninformative without losing the important data. In addition this gives the users of this program the flexibility to annotate their spectra in a manner they prefer. For example the user may only want to annotate the top 10 ions in the spectrum. This is possible with this interface. In addition it is possible to annotate unfiltered spectra but results in a much longer computational processing time. In many cases, in natural product research, the samples are available in limited quantities or the peptide does not fragment well and therefore it is not always possible to produce the best mass spectra. The filters will allow us to work with these spectra, instead of repeating the experiment, which might not be possible in real world drug discovery applications where there is often a limited supply.
Nomenclature used in this paper
For discussion purposes of the results in this paper, we have adapted the nomenclature forwarded by Ngoka and Gross to describe the cyclic peptides in this paper.45 The nomenclature developed by Ngoka and Gross describes the ions with a four-part descriptor with the general formula xnJZ, where ‘x’ is the designation for the type of ion (b, a, etc.) and n is the number of amino acid residues that makes up the ion. J and Z are the one-letter codes for the two amino acid residues connecting the backbone amide bond, J-Z, which is broken to form the linear ion. J is the N-terminal amino acid residue and Z is the C-terminal amino acid residue. To illustrate the nomenclature, we use seglitide, a six-amino acid residues cyclic peptide illustrated in scheme 1 as an example. In Seglitide and tyrocidines the one letter amino acid abbreviation was used to represent each residue, while in other compounds we assigned letters in order of their sequence using the standard alphabet since they contained too many modified residues. For example, in this paper we describe DMMC for which 6 out of 9 are modified or non-standard amino acids, while dudawalamide A has 4 out of 7 that are nonstandard, for mantillamide has 5 out of 9, and cyclomarins have 5 out of 7 (Figure 1). Because the alanine in seglitide has methylation in the nitrogen position, we use A’ to represent this methylated residue. Seglitide using this nomenclature would likely undergo random ring openings following by bn→bn-1 pathway45 resulting in the formation of 6 (n = 6) different series of b ions (Scheme 1).
Scheme 1. Sequences of b ions from the fragmentation of seglitide.
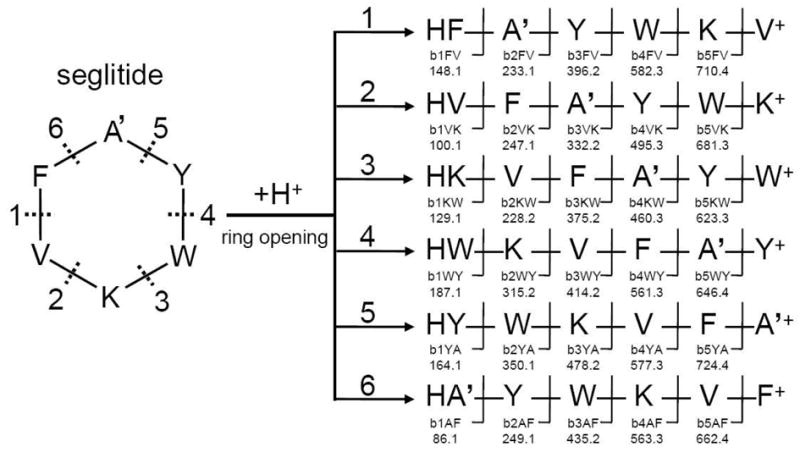
According to the conventional pathway for fragmentation of cyclic peptides, seglitide first undergoes random ring opening at each amide bond, yielding 6 different linear peptides. Sequential C-terminal amino acid cleavage results in six series of ions, for a total 30 b ions.
Cyclic peptide annotation program demonstration-Seglitide
We first illustrate the application and utility of MS-CPA using a simple cyclic peptide, seglitide, a somatostatin receptor antagonist consisting of 6 amino acids and described the results using the nomenclature defined above (Figure 1). Seglitide was analyzed by Fourier-transform ion-cyclotron resonance mass spectrometry (FTICR-MS). A singly protonated ion was observed at 808.4247 Da, which is within 3 ppm of the theoretical mass of seglitide (808.4272 Da). This ion was subjected to CID in a linear ion-trap and the product ions were again analyzed by FTICR-MS (Figure 2). The resulting MS2 spectra were then analyzed by MS-CPA. The spectrum was subject to standard filtering procedures to increase the signal to noise ratio. First, because the raw spectrum was collected in profile mode, only the top peak was retained in a window of +/− 0.05 Daltons. Second, the top 200 most intense peaks were retained. Lastly, isotopic and water-loss peaks were filtered out, yielding 146 final peaks. As shown in Figure 3, the output of MS-CPA includes input residues and the parent mass that is obtained as user input or directly obtained from the input .dta or .mzXML file (A), summary of input filtering parameters and resulting ions counts (B), quantitative statistics of cleavage and total explainable ion intensity (C), a spectrum with color-coded matches (b ions are showed in red; water loss are green; a ions are cyan; NDS’s are blue; unannotated ions are yellow) (D), a plot of mass errors of the annotated ions (E), and a list of matched fragment ions in tabular format (E).
Figure 2. Seglitide MS and MS2 spectrum.
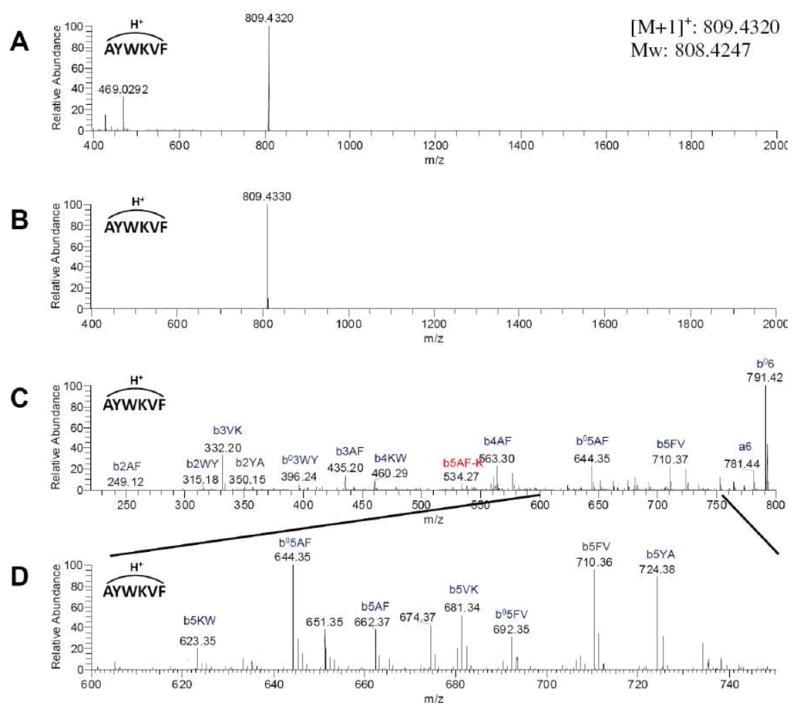
MS and MS2 spectrum were collected by ESI-LTQ-FTICR MS. A: Broadband spectrum. B: Spectra obtained with an isolation window set for seglitide parent ion (M+H)+ C: MS2 spectrum of seglitide. D: Zoom in spectrum of 600~750 m/z region.
Figure 3. MS-CPA output from analysis of seglitide MS2 data.
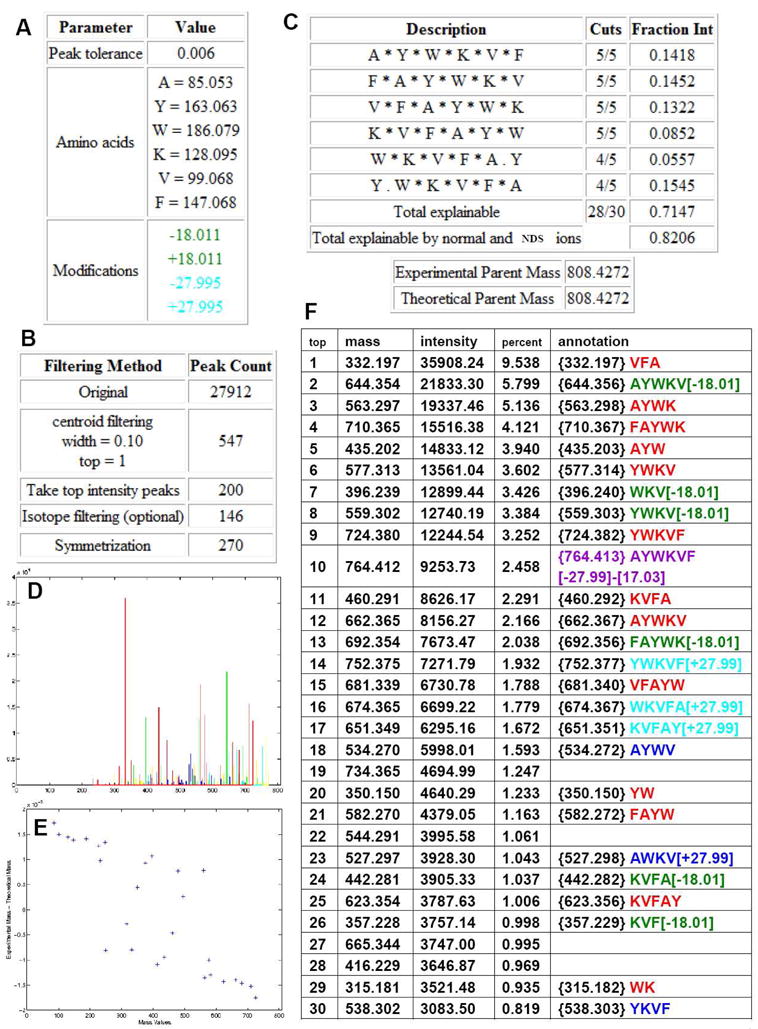
MS-CPA input parameters summary (A, B), number of cleavages and explainable intensity. The * indicates an ion that cleaves here was annotated in the spectrum. (C), annotated spectrum (D), accuracy analysis (E), annotated ions list (unsymmetrized) (to save space, only the top 30 intensity ions were displayed) (The annotation is the recommend arrangement of input sequence computed by program based on specified parameters, users can further manually verify to increase the confidence.) (F). In the output spectrum and annotation list, the b ions are showed in red; H2O loss are green; a ions are cyan; NDS’s are blue; unannotated ions are yellow in the spectrum and unlisted in the table. Symmetric ions are not shown in D or F.
For seglitide, the MS-CPA output indicates that 28 out of the 30 possible b ions were matched to observed masses. The explainable ion intensity of the b ions combined with possible a ions and loss-of-water ions was 71.5% of the total ion intensity. The absolute difference between the calculated and the experimental masses was less than 0.004 Da. Among the annotations, some of the ions with high intensity contained water loss even though there was no serine or threonine in the sequence. In addition, masses corresponding to addition of 28 Da (plus CO) were observed. These ions were not expected thus we subjected these ions to additional rounds of tandem mass spectrometry (MS3 and MS4) to verify if the annotations were real or not. With these additional rounds of fragmentation, the authenticity of MS-CPA annotations was verified and that these ions are indeed correctly annotated (MSn spectra were showed in supporting information). Although the mechanisms behind the formation of these unusual fragments are still elusive, MS-CPA enabled us to discover the existence of these ions.
The observation of NonDirect Sequence ions in Seglitide
Because more than 28% of the ion intensity remained unexplained, we explored the nature and significance of the remaining ion intensity. Because these data were acquired with high-resolution, the molecular mass of each ion could be determined. First, we analyzed these for alternate combinations of amino acids that would result from peptide residues rearrangements. We found 58 such ions comprising roughly 10% of the total ion intensity. Each of these scrambled sequence ions had mass errors within 0.004 Da, in agreement with all of the other masses we had annotated. The fact that so many of the ions could be explained by a rearrangement of the amino acid sequence is unlikely be coincidental or due to noise. In fact, some of these scrambled ions are of relatively high abundance. In Seglitide the most abundant NDS ion was up to 16% of the normalized ion intensity when the most intense ion was set to 100%. These kinds of scrambled sequence ions have previously been observed in peptides and described as nondirect sequence ions.28, 30–32, 34–37 Because of their relatively high abundance, they are included into our annotation program MS-CPA. By their inclusion, the accountable signal intensity increases from 71.5% to 82.1%. Notably, some ions are still remain unannotated, these ions are likely resulted in side-chain fragmentations, unknown fragmentations or noise inherently present in mass spectrometry data set.
To confirm the presence of NDS ions from seglitide, the two most intense of these ions, AYWV and YKVF (b5AF-K, b5YA-W), and each b5 ion (i.e., the parent ion minus one amino acid), were isolated and subjected to an additional round of CID. The b5 ions were chosen for comparison and were anticipated to be linear by conventional fragmentation pathways bx->bx−146. Surprisingly, the MS3 spectra indicated that none of these selected ions simply followed the conventional rules for fragmentation which state that cyclic peptides sequentially lose amino acid residues from the C-terminus after the initial ring opening event (Figure 4).26 Instead, we observed a mixed series of b ions (Scheme 1) which suggest that the precursors for the MS3 experiment are still cyclic. For example, if the b5 ion FAYWK was of linear structure, only the bnFK ion series should be present in the associated MS3 spectrum (Figure 4A); however, we observed relatively intense bnYA and b2WY ions. These additional ion fragments most likely originate from cyclic peptide precursors.
Figure 4. MS3 spectra of representative seglitide sequence ions.
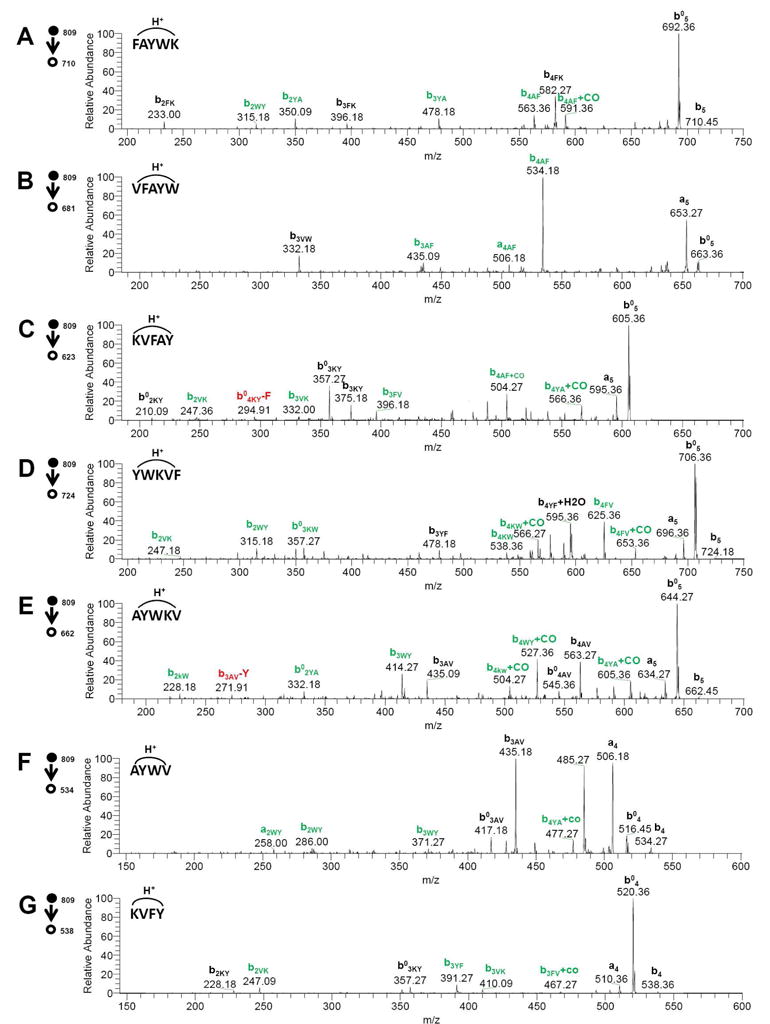
The presence of daughter b-ions from different linearized parent ions suggests that the parent ion is cyclic (as opposed to linear, as initially assumed). MS3 spectra were collected by ESI-LTQ MS. A–E: b5 ions. F–G: top two NDS ions observed. Expected sequence ions of a linear peptide are showed in black color. Expected ions for cyclic peptide are showed in green color combined with black color ones. Red color represents for NDS ions.
To explore these NDS ions behavior, we first compared the total ion intensities explained by assuming a linear precursor with those explained by assuming a circular precursor. For example, CID on the ion b5KW (KVFAY) would yield K, KV, KVF, KVFA, Y, AY, FAY and VFAY fragments if the b5KW ion was linear. However, if this ion was circular, we would observe 20 possible fragments. In the case of b5KW (Figure 4C), the ions annotated as AYKV, FAYK, and YKVF show high intensity and are easily explained if the precursor ion is considered to be circular. In fact, 88% of total ion intensity can be explained by assuming a circular precursor, while only 42% can be explained by assuming a linear precursor. Table 1 summarizes the analysis of the seven MS2 ions that were subjected to additional CID and annotated as either linear or circular. Among these seven MS2 ions, the only one that gave poor fragmentation is b5VK (VFAYW), with 12 cleavages out of 20. However, this ion produced a very intense peak (b4AF) that corresponds to loss of phenylalanine. This peak would not have been the most intense ion in the MS3 spectrum if the initial cyclic peptide had first undergone linearization and then eliminated the C-terminal residue (i.e., tryptophan) as predicted by conventional fragmentation rules.26, 45 While all of the foregoing results strongly support the cyclic nature of the MS2 ions resulting from CID of seglitide, it is likely that a mixture of cyclic and linear forms ultimately contribute to the MS3 spectrum.
Table 1.
MS-CPA analysis[b] of the two most intense NDS ions and b5 ions of seglitide.
| Linear annotation | Circular annotation | Number of critical fragments[a] | |||
|---|---|---|---|---|---|
| cuts | explained intensity | cuts | explained intensity | ||
| b5FV | 6/8 | 64.75% | 13/20 | 89.24% | 3 |
| b5VK | 5/8 | 20.46% | 12/20 | 88.82% | 4 |
| b5KW | 4/8 | 42.39% | 11/20 | 88.36% | 4 |
| b5YA | 6/8 | 46.15% | 14/20 | 92.54% | 6 |
| b5AF | 5/8 | 55.19% | 8/20 | 82.01% | 1 |
| b5AF-K | 4/6 | 73.16% | 7/12 | 85.38% | 2 |
| b5YA-W | 4/6 | 50.47% | 8/12 | 76.81% | 3 |
Fragments that cover the linear breakpoint.
Results were analyzed by isotope removal, water removal, NH3 removal and window filtering with width 10, top 10, unsymmetric.
Although the formation of these NDS ions have been recognized since 2003,30 the actually mechanisms behind are still a hot research topic. Several groups argued the importance of understanding this phenomenon in the development of de novo sequence programs. Therefore, a few mechanisms have been proposed to account NDS ions.28, 30–32, 34 The general consensus involves a cyclic intermediate occurred by recyclization. The presence of recyclized intermediates have been verified by Riba-Garcia and co-workers using ion-mobility MS.36–37 The tendency of generating NDS ions was also studied under N-acetylation modification or various activation energy.35 Recently, just after this current manuscript was submitted, a more thoroughly mechanism and pathway was published by Bleiholder et al., in which a sequence-scrambling fragmentation pathway was proposed describing the mechanism of NDS ions based on experimental and energetic calculations in agreement with the cyclic NDS ions we observed.34 Therefore, our program, MS-CPA provides solid evidence showing the existence and abundance of these NDS ions with non-ribosomally derived cyclic peptides.
The dependence of the intensity of NDS ions on activation time and activation q
Because we anticipated that changing the activation time and energy would provide control of the intensities of NDS ions, we analyzed the effects of the different CID parameters on NDS ion abundance (Figure S-3). Surprisingly, and somewhat unsatisfying, the amount of these NDS ions did not demonstrate a significantly change with increased activation time and energy (q). This phenomenon can be attributed to the fact that when the activation q increases, the m/z range of a frequency sweep decreases (Figure S-4) because low m/z product ions starting to lose stable trajectories as activation energy q rising.47,48 This is a well documented flaw with linear ion traps. Due to the large activation q the fragment ions and NDS ions no longer fall within the acquired scan. Therefore, the cleavage coverage and NDS abundance decrease drastically (Figure S-3B). In addition, there does not appear to be significantly added benefit from changing the activation time and activation q in the increased coverage when the spectra are merged. Unfortunately we do not have PQD (Plused-Q Dissociation) on our instrument that could partially overcome this limitation found with ion traps.49,50 Although not tested in this work as the authors do not have such instrumentation in their laboratories, it is anticipated that Q-tofs, triple quads do not have this limitation.
Capability of MS-CPA in analyzing antibiotic mixture
In addition to seglitide, we investigated the antibiotic mixture tyrothricin, which contains more than 28 different compounds and is readily available commercially due to its clinical utility as a typical antibiotic. Some of these compounds, individually called tyrocidines, are known to be cyclic peptides.51 We used MS-CPA to analyze several ions from this mixture (Figure 5, Table S-2). In the case of tyrocidine A, the program successfully annotated 74 b ions out of 90 possible. In contrast, only 17 b ions were identified through manual annotation of tandem mass spectra from tyrocidine A, despite this being one of the most thorough studies of cyclic peptides available to date in the literature demonstrating a significant advantage of spectra using our approach.52
Figure 5. Tyrocidines MS and MS2 spectra.
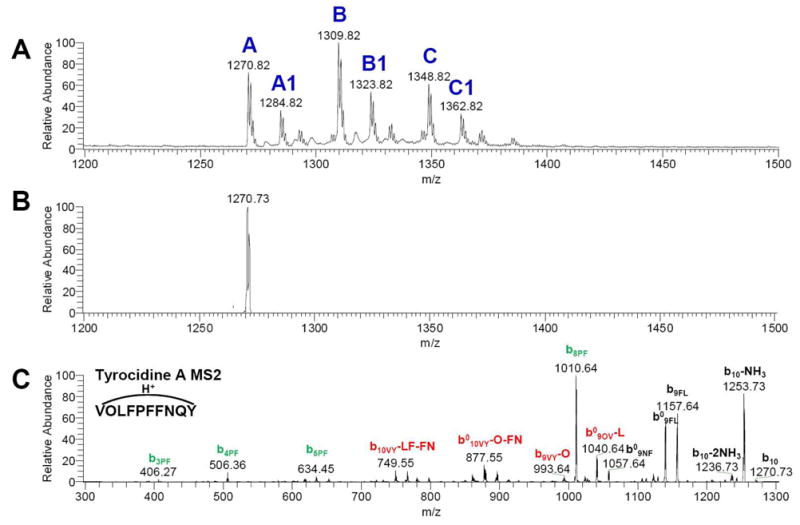
MS and MS2 spectra were collected by ESI-LTQ MS. A: Broadband spectrum showed different species of tyrocidines in tyrothricin antibiotic mixture. B: Isolation of tyrocidine A (protonated form) C: MS2 spectrum of tyrocidine A.
Using MS-CPA to annotate cyclic peptides containing non-standard subunits
Seglitide and the tyrocidines have a uniform peptidic backbone with standard amino acids. However, many non-ribosomal cyclic peptides are cyclized via lactone formation and include nonstandard amino acids.53 Theoretical calculations suggested that cyclic peptides favor lactone bond as the initial ring opening site and also the fragmentation pathway of cyclic peptides differs when lactone bond(s) were involved.29 It is therefore important to establish how these other structural features impact the fragmentation data and the results analyzed by the MS-CPA program. Thus, we analyzed several non-ribosomal cyclic peptide natural products containing lactone linkages and non-standard amino acids by tandem MS followed by MS-CAP (Table 2 and Figures S-5, 6, 7, 8). These included three marine cyanobacterial depsipeptides: desmethoxymajusculamide C (DMMC), mantillamide, and dudawalamide A, all three of which were isolated because of their biological activity to cancer cells or malaria parasites (Figure 1).54–56 Analysis of DMMC by MS-CPA uncovered 36 of the 72 b ions are expected from the standard fragmentation. Including NDS ions, the proportion of explained total ion intensity increased from 71.1% to 78.3%. Similar results were obtained for mantillamide. These data indicate that non-standard residues and ester linkages do not diminish the program’s ability to insightfully annotate a tandem mass spectrum.
Table 2.
Summary of MS-CPA analysis of various cyclic peptide natural products discussed in text.
| explainable ion intensity |
|||
|---|---|---|---|
| name | cuts | w/o NDS ions | with NDS ions |
| DMMC | 36/72 | 71.10% | 78.30% |
| Dudawalamide A | 18/42 | 96.00% | 97.30% |
| Mantillamide | 34/72 | 65.21% | 81.99% |
| Cyclomarin A | 12/42 | 50.34% | 72.48% |
| Cyclomarin C | 16/42 | 36.98% | 47.20% |
| Des[a]-cyclomarin C | 8/42 | 46.74% | 55.48% |
| Dehy[b] Cyclomarin A | 16/42 | 73.79% | 87.88% |
| Dehy[b] Cyclomarin C | 12/42 | 76.35% | 91.83% |
| Dehy[b] Des[a]-cyclomarin C | 12/42 | 72.16% | 79.09% |
Des: Desprenyl.
Dehy: dehydrated.
For the cyclomarins, manual annotations for ions reflecting loss of MeOH were included in the calculations of explainable ion intensity.
Dudawalamide A was isolated from the marine cyanobacterium Lyngbya majuscula and its structure was determined by NMR methods. A full report on the structure and bioactivity of dudawalamide will be published elsewhere.55 A high-resolution MS2 spectrum of this compound was submitted to MS-CPA for annotation. The program was also provided with the masses of the dudawalamide subunits determined by NMR (Figure S-7). The fragmentation behavior of dudawalamide, also a lactone, was found to be very different from the fragmentation behavior of mantillamide and DMMC. Although 96.0% of the total ion intensity was explained by b ions with absolute mass errors smaller than 0.008 Da, only 18 of the predicted 42 b ions were identified by the program. Thus, a high proportion of total ion intensity was accounted by a small fraction of the expected b ions. This phenomenon can be explained by the presence of labile connections between residues within dudawalamide. Such weak connections are represented in normal peptides by amides N-terminal to prolines, amides C-terminal to Asp and Glu, or amides involving tertiary amines.28 Three such linkages are present in dudawalamide; one at the N-terminus of proline and the other two at the N-termini of the N-methylated phenylalanine and the N-methylated isoleucine. Because of these three labile connections, the fragmentation of dudawalamide produced only a few ions, which were consistent with the known structure of dudawalamide but provided little sequence coverage.
Lastly, we used MS-CPA to investigate the structures of cyclomarin A, cyclomarin C, and desprenylcyclomarin C. The natural products cyclomarin A and C were originally isolated, based on their strong anti-inflammatory activity, from the marine bacterium Streptomyces sp. CNB-982.57 Subsequently, desprenylcyclomarin C was isolated from a prenyltransferase mutant of Salinispora arenicola CNS-205, but could not be produced in amounts sufficient to enable structural characterization by NMR.1 We therefore subjected all three cyclomarins to mass spectrometry and acquired MS2 spectra of each analogue. The broadband mass spectra of each of these cyclomarins showed a protonated ion species and a even much more stronger species corresponding to dehydrated forms (Figure S-8 A, D, G), providing evidence that these natural products are prone to water loss. The MS2 spectra of both protonated form and dehydrated form showed on each cyclomarin analog were collected and subjected to MS-CPA.
Analysis by MS-CPA consistently revealed the presence of strong b5GF and b4AG ions the MS2 spectra in all these cyclomarin species (Table 2, Figure S-8), thus confirming that desprenylcyclomarin C is structurally related to cyclomarin A and C. Overall, these analysis of cyclomarins identified from 8 to 16 b ions out of 34 possible b ions. The fraction of explained total ion intensity ranged from 37.0 to 50.3% when NDS ions were excluded, and from 47.2 to 72.5% when NDS ions were included. On the other hand, this fraction was much higher for the dehydrated forms of cyclomarins, ranging from 72.2 to 79.1% without NDS ions, and from 76.4 to 91.8% with NDS ions. In addition, we have successfully localized the dehydration site to the tryptophan-derived residue. Because cyclomarins are so prone to dehydration, it is possible that this is the form that provides its anti-inflammatory activity. The most likely path leading to dehydration is the formation of an imine on the tryptophan residue, yielding a conjugated system upon loss of water (Scheme S-1). These examples highlight the usefulness of MS-CPA to assist in the structural characterization of cyclic non-ribosomally encoded natural products even when limited quantities are available.
CONCLUSION
Because cyclic peptides are important class of therapeutics and toxins we have developed a program, MS-CPA, to facilitate the structural characterization of these types of natural products. Users can easily access the program on the World Wide Web in order to annotate their tandem mass spectra of cyclic peptides. Using this program we solidified the amino acid sequence of several recently discovered bioactive natural products such as dimethoxymajuscalide (DMMC), mantillamide, dudawalamide A, verified the structure of desprenycyclomarin C as well as dehydro-desprenylcyclomarin C that were isolated from a desprenyltransferase knockout S. arenicola CNS-205 strain. This analysis demonstrates the strength of this program when combined with tandem mass spectrometry as well as a candidate structure enables the structural characterization of cyclic peptides produced in such low quantities that normally prohibit the use of other structural methods such as NMR.
Using our annotation program we observed that cyclic non-ribosomal peptides fragment in unusual ways. This kind of sequence-scrambling fragmentations results in spontaneous recyclization event. The observation of NDS ions makes the problem of de novo sequencing of cyclic peptides even more challenging than was previously anticipated. Therefore, the annotation and understanding of the fragmentation patterns will, undoubtly, facilitate and improve de novo sequencing algorithm developments.
In sum, our current developed program provides a rapid annotation platform for tandem MS spectra of cyclic peptides. Also, although not designed for this, it can likely also be used to analyze the cyclization phenomenon of linear peptides. We are currently using this program to annotate peptides that have been isolated from marine organisms that have potent cancer, malarial and antibiotic resistant bacterial inhibitory activities. The approach described in this paper should be useful to the studies of cyclic peptide virulence factors, the chemical ecology of cyclic peptides as well as cyclic peptides in drug screening programs.1,58–61
EXPERIMENTAL SECTION
Sample preparation
Seglitide was purchased from Aldrich and was dissolved to a concentration of 20 μg/ml in 50:50 MeOH:water with 1.0% AcOH. Dudawalamide A and DMMC were isolated from cyanobacteria and prepared in a solution of 50 μg/mL in 50:50 MeOH:water with 1.0% AcOH was infused in the mass spectrometer. Cyclomarins were isolated from a marine actinomycete and desalted with C18 ZipTip pipette tips (Millipore) following the manufacturer’s protocol to a final concentration of 50 μg/ml.
Mass spectrometry
All samples were subjected to electrospray ionization on a Biversa Nanomate (Advion Biosystems, Ithaca, NY) nano-spray source (pressure: 0.3 psi, spray voltage: 1.4–1.8 kV). Seglitide, tyrothricin, and DMMC were analyzed a Finnigan LTQ-FTICR-MS instrument (Thermo-Electron Corporation, San Jose, CA) running Tune Plus software version 1.0 and Xcalibur software version 1.4 SR1. Dudawalamide A was analyzed on a Thermo LTQ-Orbitrap-MS instrument (Thermo) running Tune Plus and Xcalibur software version 2.0. Activation time and q experiments, low resolution spectra of seglitide, tyrothricin and cyclomarins were acquired on a Finnigan LTQ-MS (Thermo-Electron Corporation, San Jose, CA) running Tune Plus software version 1.0. The final spectrum was obtained by averaging MS2 scans with QualBrowser software version 1.4 SR1 (Thermo). Generally, the instrument was first auto-tuned on the m/z value of the ion to be fragmented. Then, the [M + H]+ ion of each compound was isolated in the linear ion trap and fragmented by collision induced dissociation (CID). Sets of consecutive, high-resolution, full MS/MS scans were acquired in centroid or profile mode and averaged using QualBrowser software (Thermo). The Thermo-Finnigan RAW files containing the average spectra were then converted to mzXML file format using the program ReAdW (tools.proteomecenter.org).
Supplementary Material
Acknowledgments
This work was supported by PhRMA foundation, NIH GM086283, NIH NS053398, NIH CA100851, FIC ICBG TW006634, and California Sea Grant program (grant 85-MNP-N).
Footnotes
Supporting information available
References
- 1.Schultz AW, Oh DC, Carney JR, Williamson RT, Udwary DW, Jensen PR, Gould SJ, Fenical W, Moore BS. J Am Chem Soc. 2008;130:4507–4516. doi: 10.1021/ja711188x. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Proc Natl Acad Sci USA. 2005;102:7315–7317. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pomilio AB, Battista ME, Vitale AA. Current Organic Chemistry. 2006;10:2075–2121. [Google Scholar]
- 4.Rinehart KL, Harada K, Namikoshi M, Chen C, Harvis CA. J Am Chem Soc. 1988;110:8557–8558. [Google Scholar]
- 5.Gupta N, Pant SC, Vijayaraghavan R, Lakshmana Rao PV. Toxicology. 2003;188:285–296. doi: 10.1016/s0300-483x(03)00112-4. [DOI] [PubMed] [Google Scholar]
- 6.Holden MTG, Chhabra SR, de Nys R, Stead P, Bainton NJ, Hill PJ, Maneeld M, Kumar N, Labatte M, England D, Rice S, Givskov M, Salmond GPC, Stewart GSAB, Bycroft BW, Kjelleberg S, Williams P. Molecular Microbiology. 1999;33:1254–1266. doi: 10.1046/j.1365-2958.1999.01577.x. [DOI] [PubMed] [Google Scholar]
- 7.Ibrahim M, Guillot A, Wessner F, Algaron F, Besset C, Courtin P, Gardan R, Monnetl V. Journal of Bacteriology. 2007;189:8844–8854. doi: 10.1128/JB.01057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poupel O, Tardieux I. Microbes and Infection. 1999;1:653–662. doi: 10.1016/s1286-4579(99)80066-5. [DOI] [PubMed] [Google Scholar]
- 9.Branda SS, Chu F, Kearns DB, Losick R, Kolter R. Molecular Microbiology. 2006;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- 10.Straight PD, Willey JM, Kolter R. Journal of Bacteriology. 2006;188:4918–4925. doi: 10.1128/JB.00162-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturme MHJ, Nakayama J, Molenaar D, Murakami Y, Kunugi R, Fujii T, Vaughan EE, Kleerebezem M, de Vos WM. Journal of Bacteriology. 2005;187:5224–5235. doi: 10.1128/JB.187.15.5224-5235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jegorov A, Hajduch M, Sulc M, Havlicek V. J Mass Spectrom. 2006;41:563–576. doi: 10.1002/jms.1042. [DOI] [PubMed] [Google Scholar]
- 13.Italia JL, Bhardwaj V, Ravi Kumar MNV. Drug Discovery Today. 2006;11:846–854. doi: 10.1016/j.drudis.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Hannon JP, Nunn C, Stolz B, Bruns C, Weckbecker G, Lewis I, Troxler T, Hurth K, Hoyer D. J Mol Neurosci. 2002;18:15–27. doi: 10.1385/JMN:18:1-2:15. [DOI] [PubMed] [Google Scholar]
- 15.Gerding DN, Muto CA, Owens RC., Jr Clin Infect Dis. 2008;46:S43–S49. doi: 10.1086/521861. [DOI] [PubMed] [Google Scholar]
- 16.Kofoed J, Reymond JL. J Comb Chem. 2007;9:1046–1052. doi: 10.1021/cc7001155. [DOI] [PubMed] [Google Scholar]
- 17.Fluxa VS, Reymond JL. Bioorg Med Chem. 2008 doi: 10.1016/j.bmc.2008.01.045. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 18.Berkovich-Berger D, Lemcoff NG. Chem Commun. 2008;14:1686–1688. doi: 10.1039/b800384j. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Joo SH, Voorhees JL, Brooksa CL, Pei D. Bioorg Med Chem. 2008 doi: 10.1016/j.bmc.2008.01.015. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Zhou S, Wavreille AS, DeWille J, Pei D. J Comb Chem. 2008;10:247–255. doi: 10.1021/cc700185g. [DOI] [PubMed] [Google Scholar]
- 21.Feng Y, Carroll AR, Pass DM, Archbold JK, Avery VM, Quinn RJ. J Nat Prod. 2008;71:8–11. doi: 10.1021/np070094r. [DOI] [PubMed] [Google Scholar]
- 22.Linington RG, Edwards DJ, Shuman CF, McPhail KL, Matainaho T, Gerwick WH. J Nat Prod. 2008;71:22–27. doi: 10.1021/np070280x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimokawa K, Mashima I, Asai A, Ohno T, Yamada K, Kita M, Uemura D. Chem Asian J. 2008;3:438–446. doi: 10.1002/asia.200700243. [DOI] [PubMed] [Google Scholar]
- 24.Shindoh N, Mori M, Terada Y, Oda K, Amino N, Kita A, Taniguchi M, Sohda KY, Nagai K, Sowa Y, Masuoka Y, Orita M, Sasamata M, Matsushime H, Furuichi K, Sakai T. International Journal of Oncology. 2008;32:545–555. [PubMed] [Google Scholar]
- 25.Krishnamurthy T, Szafraniec L, Hunt DF, Shabanowitz J, Yates JR, Hauert CR, Carmichael WW, Skulberg O, Coddii GA, Missler S. Proc Nati Acad Sci USA. 1989;86:770–774. doi: 10.1073/pnas.86.3.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ngoka LCM, Gross ML. J Am Soc Mass Spectrom. 1999;10:732–746. doi: 10.1016/S1044-0305(99)00049-5. [DOI] [PubMed] [Google Scholar]
- 27.Jegorov A, Paizs B, Zabka M, Kuzma M, Havlicek V, Giannakopulos AE, Derrick PJ. Eur J Mass Spectrom. 2003;9:105–116. doi: 10.1255/ejms.531. [DOI] [PubMed] [Google Scholar]
- 28.Yagüe J, Paradela A, Ramos M, Ogueta S, Marina A, Barahona F, López de Castro JA, Vázquez J. Anal Chem. 2003;75:1524–1535. doi: 10.1021/ac026280d. [DOI] [PubMed] [Google Scholar]
- 29.Jegorov A, Paizs B, Kuzma M, Zabka M, Landa Z, Sulc M, Barrow MP, Havlicek V. J Mass Spectrom. 2004;39:949–960. doi: 10.1002/jms.674. [DOI] [PubMed] [Google Scholar]
- 30.Harrison AG, Young AB, Bleiholder C, Suhai S, Paizs B. J Am Chem Soc. 2006;128:10364–10365. doi: 10.1021/ja062440h. [DOI] [PubMed] [Google Scholar]
- 31.Jia C, Qi W, He Z. J Am Soc Mass Spectrom. 2007;18:663–678. doi: 10.1016/j.jasms.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 32.Qi W, Jia C, He Z, Qiao B. Acta Chimica Sinica. 2007;65:233–238. [Google Scholar]
- 33.Tilvi S, Naik CG. J Mass Spectrom. 2007;42:70–80. doi: 10.1002/jms.1140. [DOI] [PubMed] [Google Scholar]
- 34.Bleiholder C, Osburn S, Williams TD, Suhai S, Van Stipdonk M, Harrison AG, Paizs B. J Am Chem Soc. 2008;130:17774–89. doi: 10.1021/ja805074d. [DOI] [PubMed] [Google Scholar]
- 35.Harrison AG. J Am Soc Mass Spectrom. 2008;19:1776–1780. doi: 10.1016/j.jasms.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 36.Riba-Garcia I, Giles K, Bateman RH, Gaskella SJ. J Am Soc Mass Spectrom. 2008;19:1781–1787. doi: 10.1016/j.jasms.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 37.Riba-Garcia I, Giles K, Bateman RH, Gaskell SJ. J Am Soc Mass Spectrom. 2008;19:609–613. doi: 10.1016/j.jasms.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Eng JK, McCormack AL, Yates JR., III J Am Soc Mass Spectrom. 1994;5:976. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 39.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Tanner S, Shu H, Frank A, Wang LC, Zandi E, Mumby M, Pevzner PA, Bafna V. Anal Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 41.Jagannath S, Sabareesh V. Rapid Commun Mass Spectrom. 2007;21:3033–3038. doi: 10.1002/rcm.3179. [DOI] [PubMed] [Google Scholar]
- 42.Bandeira N, Ng J, Meluzzi D, Linington RG, Dorrestein P, Pevzner PA. Proceedings of the Twelfth Annual International Conference in Research in Computational Molecular. Biology. 2008:181–195. [Google Scholar]
- 43.Lin SM, Zhu L, Winter AQ, Sasinowski M, Kibbe WA. Expert Rev Proteomics. 2005;2:839–45. doi: 10.1586/14789450.2.6.839. [DOI] [PubMed] [Google Scholar]
- 44.Pedrioli PGA, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R. Nat Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 45.Ngoka LCM, Gross ML. J Am Soc Mass Spectrom. 1999;10:360–363. doi: 10.1016/S1044-0305(99)00006-9. [DOI] [PubMed] [Google Scholar]
- 46.Paizs B, Suhai S. Mass Spectrom Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 47.Payne AH, Glish GL. Anal Chem. 2001;73:3542–3548. doi: 10.1021/ac010245+. [DOI] [PubMed] [Google Scholar]
- 48.Racine AH, Payne AH, Remes PM, Glish GL. Anal Chem. 2006;78:4609–4614. doi: 10.1021/ac060082v. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz JC, Syka JEP, Quarmby ST. The 53rd ASMS Conference on Mass Spectrometry and Allied Topics; 2005. [Google Scholar]
- 50.Schlabach T, Zhang T, Miller K, Kiyonami R. The 2006 ABRF Conference; 2006. [Google Scholar]
- 51.Eckart K. Mass Spectrome Rev. 1994;13:23–55. [Google Scholar]
- 52.Pittenauer E, Zehl M, Belgacem O, Raptakis E, Mistrik R, Allmaier G. J Mass Spectrom. 2006;41:421–447. doi: 10.1002/jms.1032. [DOI] [PubMed] [Google Scholar]
- 53.Kopp F, Marahiel MA. Nat Prod Rep. 2007;24:735–749. doi: 10.1039/b613652b. [DOI] [PubMed] [Google Scholar]
- 54.Simmons TL. Thesis. University of California; San Diego (USA): 2008. [Google Scholar]
- 55.Gutiérrez M, Gerwick WH. manuscript in preparation. [Google Scholar]
- 56.Linington RG. manuscript in preparation. [Google Scholar]
- 57.Renner MK, Shen YC, Cheng XC, Jensen PR, Frankmoelle W, Kauffman CA, Fenical W, Lobkovsky E, Clardy J. J Am Chem Soc. 1999;121:11273 –11276. [Google Scholar]
- 58.Selim S, Negrel J, Govaerts C, Gianinazzi S, van Tuinen D. Appl Environ Microbiol. 2005;71:6501–6507. doi: 10.1128/AEM.71.11.6501-6507.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nair SS, Romanuka J, Billeter M, Skjeldal L, Emmett MR, Nilsson CL, Marshall AG. Biochimica et Biophysica Acta. 2006;1764:1568–1576. doi: 10.1016/j.bbapap.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 60.Greve H, Kehraus S, Krick A, Kelter G, Maier A, Fiebig HH, Wright AD, König GM. J Nat Prod. 2008;71:309–312. doi: 10.1021/np070373e. [DOI] [PubMed] [Google Scholar]
- 61.Adams B, Pörzgen P, Pittman E, Yoshida WY, Westenburg HE, Horgen FD. J Nat Prod. 2008;71:750–754. doi: 10.1021/np070346o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.