

Abstract
Integral membrane proteins perform crucial cellular functions and are the targets for the majority of pharmaceutical agents. However, the hydrophobic nature of their membrane-embedded domains makes them difficult to work with. Here, we describe a shotgun proteomic method for the high-throughput analysis of the membrane-embedded transmembrane domains of integral membrane proteins which extends the depth of coverage of the membrane proteome.
Introduction
Integral membrane proteins (IMPs) carry out crucial cellular functions and, as such, are one of the most important classes of proteins on which to direct proteomic investigations. To enrich for this class of proteins, most proteomic workflows begin with a purification step for membranes (for a comprehensive review on membrane proteomics, see Speers and Wu,1). This approach effectively enriches for membrane proteins, both IMPs and peripheral membrane proteins (lipid-anchored and membrane associated proteins), as well as attached cytoskeletal components. To further enrich for IMPs, some workflows also incorporate a high salt or high pH extraction step, to remove loosely bound membrane-associated proteins from enriched membranes.1 However, IMP enrichments provided by these extractions are modest and distinctions from membrane-anchored proteins are not achieved. Despite the incorporation of these membrane enrichment strategies, to date, the highest enrichment of IMPs reported is ∼65%.2
The most distinguishing feature of IMPs is that they contain one or more membrane-embedded transmembrane domains (TMDs) that are amphipathic or hydrophobic in nature, making them experimentally challenging. Most proteomic strategies make use of detergents,3 organic solvents,4 or organic acids5 to solubilize and/or extract proteins from enriched membranes prior to their proteolytic digestion and analysis by mass spectrometry. However, structural information is lost by denaturing the lipid bilayer and IMPs are not distinguished over other membrane proteins.
Previously, we reported a proteomic method for the identification of membrane proteins from complex membrane samples using high pH and proteinase K (the hppK method).6 This method was based on the use of high pH to favor the formation of membrane “sheets” while leaving IMPs embedded in the lipid bilayer and, coupled with the use of the nonse-quence-specific proteolytic enzyme proteinase K (pK), resulted in the production of protease-accessible peptides (PAPs) from both membrane-embedded and soluble proteins alike (Figure 1, left). This strategy offered both increased peptide identifications from membrane proteins and an indirect characterization of membrane protein topology deduced by the analysis of the exposed PAPs(3). The remaining pK “shaved” membrane fraction is specifically enriched for IMPs. However, efforts to analyze this sample were compromised by the abundance of lipids and poor peptide recovery. We present herein a robust proteomic method targeted at the high-throughput analysis of membrane-embedded proteins and peptides (MEPs). The MEPs analysis resulted in identifications that are dramatically enriched for IMPs (on average, they comprise 82% of proteins identified) and, most notably, peptides that overlap with predicted TMDs (in 71% of IMPs identified).
Figure 1.
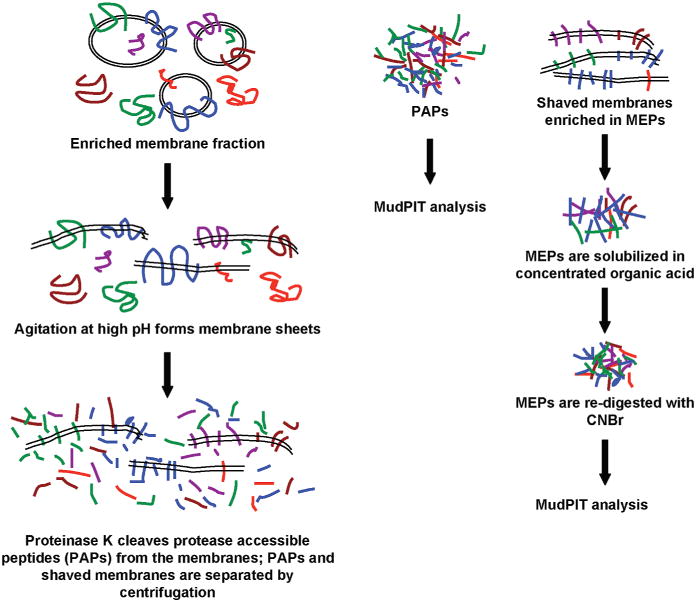
MEPs method. Workflow for the preparation and proteomic analysis of protease-accessible peptides (PAPs) and membrane-embedded peptides (MEPs).
Results and Discussion
The hppK method was first applied to an enriched plasma membrane fraction to produce a PAP sample and “shaved” membranes (Figure 1, left). The use of a nonspecific protease at high pH results in the removal of exposed protein domains without sequence constraint on both surfaces of the membrane. The “shaved” membranes were then separated from the PAPs by centrifugation in low levels of organic solvent. We hypothesized that most of the peptides recovered with the shaved membranes should be from TMDs, not digested because they are embedded in intact membranes and inaccessible to pK. Previous studies have reported that high pH does not affect the integrity of the membrane bilayer;7 however, membrane integrity after digestion with pK was of concern. To confirm that lipid bilayers were structurally intact after shaving with pK, we analyzed isolated membranes before (high pH only) and after hppK treatment (pK digestion in 8 M urea at high pH) using electron microscopy and tomography.
Panels A and B in Figure 2 (Panel 1) show representative fields in the high pH treated membrane sample and the shaved membrane sample, respectively. Both samples are homogeneous and enriched in membranes. Protein aggregates (microfilaments and cytoskeletal components adherent to the isolated plasma membrane) are only present in the undigested sample (Figure 2A, Panel 1, arrow). In both samples, many of the membrane structures have free edges. One such membrane structure was selected from each sample (Figure 2A,B, Panel 1, arrowheads) for further 3D analysis. Representative slices from the top, middle, and bottom of each reconstruction show a closed membrane (Figure 2A,B, Panel 2), open with edges (Figure 2A,B, Panel 3), and then closed again (Figure 2A,B, Panel 4) at different points through the volume of the structures. Three-dimensional models of these membranes (Figure 2A,B, Panel 5) show that they are not sheets, but rather, largely intact structures with an opening in one side. When analyzed in 3D (Quicktime Movies 1 and 2 in Supporting Information), most of the membrane structures in both samples show this conformation. This suggests that, even under the harsh conditions of our digestion strategy, the integrity of the membranes is preserved, and they are capable of reforming into relatively native membrane compartments. As shown previously,7 the lipid bilayers of the membranes in the undigested sample are intact (Figure 2A, Panel 6). Importantly, the lipid bilayers in the “shaved” membranes are also intact (Figure 2B, Panel 6) and, without the attached protein aggregates, seem even more defined (Figure 2B, Panel 6). Therefore, we predicted that IMPs resident to these membranes should remain embedded in the lipid bilayer and their TMDs should be protected from protease digestion and be enriched in this “shaved” membrane sample.
Figure 2.
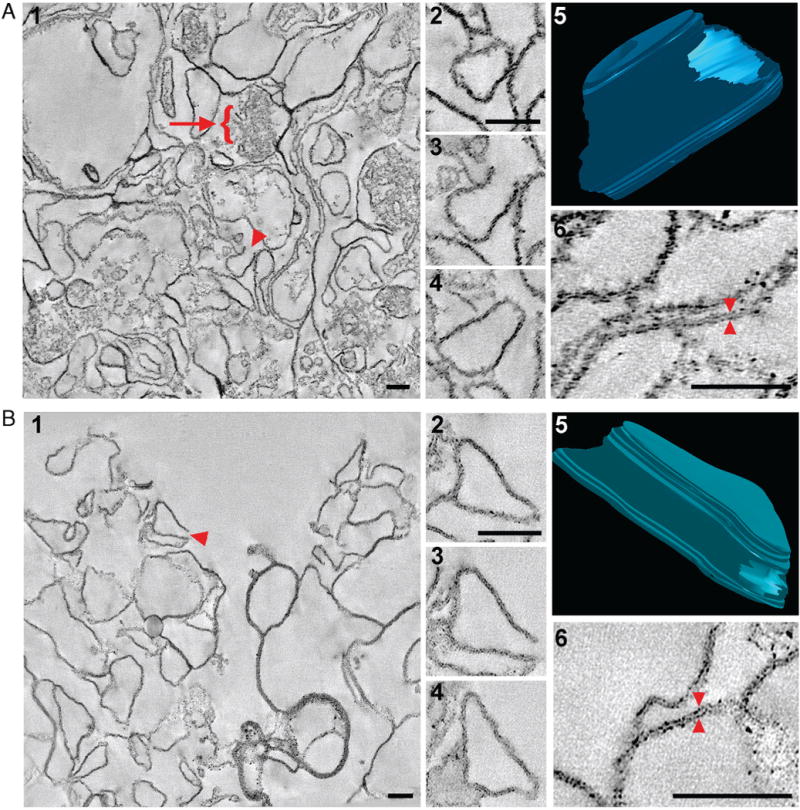
Three-dimensional structural analysis of isolated membranes using EM tomography. (A) Tomographic reconstruction of isolated membranes treated with high pH. A low-magnification overview of the reconstruction. Panel 1 shows that the sample consists primarily of membranes and protein aggregates (arrow). Many of the membrane components appear to be open with free edges (arrowhead). One such membrane structure (Panel 1, arrowhead) was modeled, and representative tomographic slices (7.6 nm thick) taken from different planes through its volume are shown in Panels 2 (closed), 3 (open), and 4 (closed). A 3D model (Panel 5), drawn from relevant slices of the tomogram, illustrates that the free edges are due to a large opening on one end of the membrane compartment. A higher magnification image confirms that lipid bilayers are intact (Panel 6). (B) Tomographic reconstruction of isolated “shaved” membranes. Membranes were agitated in high pH, then treated with 8 M urea and digested with proteinase K. The sample consists primarily of membranes without protein aggregates (Panel 1), and many of the membrane structures are open and have free edges (arrowhead). A selected membrane structure (Panel 1, arrowhead) was modeled, and representative slices are shown in Panels 2 (closed), 3 (open), and 4 (closed). The modeled reconstruction (Panel 5) again reveals that the free edges are due to an opening on one end of the membrane compartment. A higher magnification view of the tomographic slice confirms that lipid bilayers are intact (Panel 6). (Bars = 100 nm.)
Difficulties in analyzing this “shaved” membrane fraction derive from the combined length and hydrophobicity of the membrane-embedded protein segments. TMDs are on average 20–25 amino acids long; however, they can sometimes exceed 30 amino acids.8 These domains frequently lack arginines and lysines,,9 and therefore, digestion of membrane proteins by trypsin results mostly in intact large, hydrophobic peptides. Although methionines occur relatively infrequently in proteins, they are found in approximately 50% of Homo sapiens protein TMDs.10 Therefore, incorporating a membrane solubilization step using concentrated formic acid combined with redigestion using cyanogen bromide (CNBr)5 results in solubilization of the MEPs and chemical cleavage of the long transmembrane segments at methionines to produce smaller, less hydrophobic peptides (Figure 1, right).
The solubilized shaved membrane sample is composed of mainly lipids and hydrophobic MEPs and, therefore, is highly viscous. Dilution of the acidified sample with buffer containing low levels of organic solvent resulted in the precipitation of most of the membrane lipids and facilitated their separation by centrifugation. Mass spectrometric (MS) analysis of the precipitated lipid pellet identified phosphatidylcholine, phos-phatidylethanolamine, and phosphatidylserine as the major lipid species (data not shown). MEPs remained in the supernatant and were analyzed by Multidimensional Protein Identification Technology (MudPIT). Conventional nanoflow separation conditions routinely used in shotgun proteomic applications resulted in limited peptide recovery. However, heated chromatography has been observed to improve peak resolution and recovery of hydrophobic proteins;11,12 therefore, chromatography was conducted at 60 °C, resulting in a 4-fold increase in protein identifications and hydrophobic peptide recovery when compared to room temperature.13
A protein/peptide list of the results from eight replicate 12-step MudPIT runs of the MEPs is available in Supplementary Table 1. When all eight replicates were combined, a total of 670 unique proteins and 4471 peptides were identified (Supplementary Table 3). Of the 670 protein identifications, 479 (or 72%) proteins were predicted by TMHMM14 to be IMPs, of which 67% were identified with TMD coverage. Of the 4471 peptide identifications, 55% were calculated to be hydrophobic using the GRAVY method (grand average of hydropathicity15) with GRAVY scores >0 and 43% overlapped with a predicted TMD. When all eight replicates were averaged, a single analysis identified 316 nonredundant proteins and 1368 unique peptides. A majority of the nonredundant proteins identified (82%) were predicted by TMHMM14 to be IMPs, and of these, 70% were predicted to have >2 TMDs. In fact, when identified IMPs were categorized according to the number of predicted TMDs (Supplementary Figure 1), the distribution mirrored that reported for in silico genome-wide analyses,16 suggesting that there is little to no bias against any particular class of IMPs. A majority of peptides identified (59%) were calculated to be hydrophobic using GRAVY (Supplementary Figure 2); 49% of all unique peptides identified in this sample overlapped at least partially with a predicted TMD, and 7% overlapped completely with a predicted TMD (Supplementary Figure 3). Furthermore, IMPs were identified with greater sequence coverage and average spectral count when compared with corresponding numbers for identified soluble proteins (Supplementary Figures 4A,B), indicating that soluble proteins were significantly de-enriched.
Previously, other groups have utilized trypsin to remove exposed hydrophilic domains and membrane-associated proteins from membranes to enrich for TMDs.1 Indeed, a recent proteomic study conducted on membrane samples after trypsin treatment and methanol extraction reported only 22% IMPs, of which only 13% were identified with a peptide overlapping a TMD (numbers extracted from supplementary tables).17 These modest numbers most likely resulted from the use of a sequence-specific protease to digest intact membrane vesicles, restricting cleavage to accessible proteins domains containing Arg or Lys on the external surface of the vesicle. Consequently, upon methanol extraction, all remaining proteins within the sample were analyzed, resulting in minimal enrichments for IMPs.
In addition to facilitating the identification of TMDs, the MEP analysis is complementary with the PAP analysis and facilitates the identification of different categories of IMPs (see Supplementary Table 3 and Supplementary Figure 5 for comparison). From the combined MEPs analysis (Supplementary Tables 1 and 3), 57% of the proteins identified were unique to the MEP sample and not identified in the corresponding PAPs sample. (Eight MudPIT analyses of the PAP sample identified 1772 proteins with 30% predicted IMPs; Supplementary Tables 2 and 3.) The addition of MEP analysis increased the total nonredundant protein identifications for this enriched plasma membrane sample to 2156 and the percent IMPs to 38%. When the two analyses are combined, both the number and sequence coverage of identified IMPs are increased, providing a more comprehensive and thorough characterization of the membrane proteome.
To illustrate the unique nature of the TMD coverage acquired from analysis of the MEPs, topological maps of predicted TMDs (depicted using TOPO2: http://www.sacs.ucsf.edu/TOPO2/) are displayed in Figure 3 for 10 representative proteins from the MEP MudPIT analyses. The majority of the amino acid sequence of most of these proteins is predicted to be embedded in the lipid bilayer (between the horizontal lines). Residues colored in black designate peptides identified in the MEP analysis. Targeting a shotgun proteomics analysis toward a sample enriched in TM segments optimizes the identification and sequence coverage of these IMPs. In fact, seven of the 10 proteins shown in Figure 3 were only found in the MEP analysis. Of the three proteins that were also identified in the complementary PAP analysis, all three had less sequence coverage when compared with the MEP analysis. Interestingly, even though the interferon-induced protein (IPI00008922) was identified in both the MEP and PAP analyses, because isoforms 2 (IFITM2) and 3 (IFITM3) are 90% identical with most of the sequence variability occurring in the hydrophobic C-terminus,18 the peptides identified from the membrane-embedded C-terminus in the MEP analysis facilitated the unambiguous identification of IFITM2.
Figure 3.
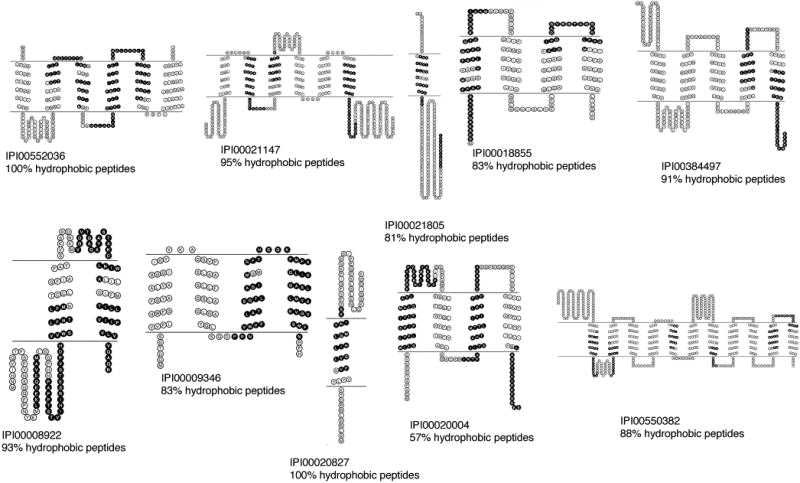
Topological maps of representative identified proteins. Ten IMPs identified in the MEP MudPIT analyses with sequence coverage shown in black. The diagrams show the predicted protein topology, with predicted TMDs located between the black lines for each protein. The percent of hydrophobic peptides (with GRAVY score >0) for each protein is listed. IPI numbers from IPI database downloaded April 2007.
Because the PAP and MEP analyses are performed in the context of intact membranes, this method facilitates the high-throughput acquisition of MS experimental data characterizing the approximate location of TMDs in the primary protein sequence. An important component to understanding protein function results from insights provided by interrogating protein structure. Unfortunately, IMPs are significantly underrepresented among structures present in the Protein Data Bank.19 To partially compensate for this, algorithms have been developed to predict protein topology from primary protein sequences. Arguably, the greatest limitation in producing an accurate algorithm is the lack of a suitable experimental data set to represent the TMDs of IMPs in each of the diverse membrane microenvironments of the cell.20 High-throughput data such as PAP and MEP analyses may be used to constrain TMD prediction algorithms and improve the overall quality of the TMD prediction.
Conclusion
In conclusion, we present a shotgun proteomic strategy (the MEPs method) that enriches for IMPs, and importantly, targets the identification of TMDs. The MEPs method provides a high-throughput platform biased toward the analysis of membrane-embedded peptides and proteins, which are still largely underrepresented in published proteomes to date. As such, this method facilitates the comprehensive coverage of the membrane proteome.
Methods
Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Mallinckrodt Baker (Phillipsburg, NJ), and all solvents were purchased from Mallinckrodt Baker unless otherwise stated.
Preparation of Enriched Membrane Fractions
HeLa cells expressing the human dopamine transporter (used for other experiments) were grown to 95% confluency on 500 cm2 plates and harvested in ice-cold Dulbecco's Phosphate Buffered Saline (PBS, Gibco, Carlsbad, CA) using a rubber policeman. Cells were pelleted (2 plates each) and stored at −80 °C. Two cell pellets were thawed on ice and 4 mL of ice-cold 0.5 M sucrose buffer (100 mM K2HPO4/KH2PO4, pH 6.7, 5 mM CaCl2, and 500 mM sucrose) with 1:1000 dilution of protease inhibitor cocktail (Sigma-Aldrich, p8340) was added to resuspend the pellet. The cell suspension was homogenized with a loose fitting Teflon pestle in a glass homogenizer for 25 strokes. The homogenate was centrifuged (1000g, 4 °C, 5 min) to pellet unbroken cells and nuclei. The resulting postnuclear supernatant (PNS) was layered into a 4-step discontinuous sucrose gradient with the following sucrose concentrations (in order from bottom to top) in a thin-walled 12.5 mL centrifugation tube: 3 mL of 1.3 M sucrose, 3.5 mL of 0.86 M sucrose, 5 mL of PNS, 2.5 mL of 0.25 M sucrose. All sucrose solutions were made with 100 mM K2HPO4/KH2PO4, pH 6.7, 5 mM CaCl2, and 1:1000 protease inhibitor cocktail. The sucrose gradients were centrifuged in a swinging bucket rotor (80 000g, 4 °C, 60 min). The SIII interface, located between the 1.3 M sucrose and 0.86 M sucrose layer was collected using a wide-bore transfer pipet. One volume of cold phosphate buffer (100 mM K2HPO4/KH2PO4, pH 6.7) was added to the collected SIII sample. The diluted sampled was vortexed and then centrifuged (20 000g, 4 °C, 30 min). The supernatant was discarded and the membrane pellet was resuspended in an equal volume of 200 mM Na2CO3 (pH 11) and agitated on ice by 5 strokes with an insulin syringe every 15 min for 1 h. The sample was centrifuged (135 000g, 45 min, 4 °C) and the supernatant was discarded. The enriched SIII membrane pellet was resuspended in phosphate buffer for protein assay (DC Assay, Bio-Rad, Hercules, CA), and stored at −20 °C until further analysis.
Preparation of Protease-Accessible Peptides (PAPs) and Membrane-Embedded Peptides (MEPs)
An aliquot of the enriched SIII interface (500 μg protein) was pelleted out of phosphate buffer by centrifugation (20 000g, 4 °C, 30 min), resuspended at 1 mg/mL in 200 mM Na2CO3 (pH 11), and agitated on ice by 5 strokes with an insulin syringe every 15 min for 1 h. Solid urea was added to a final concentration of 8 M. The sample was reduced (5 mM dithiothreitol, 60 °C, 20 min) and alkylated (15 mM iodoacetamide, room temperature, in dark, 20 min). Recombinant Proteinase K (Roche Diagnostics Corp, Indianapolis, IN) was added at an enzyme/substrate ratio of 1:50 (w/w) and the sample was incubated overnight at 37 °C. An equal volume of aqueous–organic buffer (10% acetonitrile, 2% formic acid) was added and the sample was incubated for 30 min on ice. The digested (“shaved”) membranes enriched in membrane-embedded peptides (MEPs) were pelleted by centrifugation (20 00g, 4 °C, 30 min) and either prepared for electron microscopy or μLC-MS/MS analysis. The supernatant (PAP sample) was collected, adjusted to 5% formic acid and analyzed by MudPIT as previously described.6 In a chemical fume hood, the membrane pellet was solubilized in 10 μL of cyanogen bromide/90% formic acid (1:2 w/v) and incubated overnight (20 °C, in dark) in a chemical fume hood. After incubation, 1 vol of methanol and 18 vol of 100 mM Tris-HCl (pH 8) were added for a final concentration of 5% formic acid, 5% methanol, 90% 100 mM Tris-HCl. The sample was centrifuged (20 000g, 4 °C, 15 min) to pellet the lipids. The supernatant (MEP sample) was collected and analyzed by MudPIT.6 [Note: The MEPs method has been found to be generally applicable to membrane fractions of different origins (data not shown).]
Electron Microscopy and Tomography
Membranes (undigested or proteinase K digested) were collected by centrifugation (20 000g, 4 °C, 30 min). Membrane pellets were fixed in 100 μL of 2% glutaraldehyde/100 mM sodium cacodylate, pH 7.4, overnight at 4 °C. The pellet was postfixed in 2% OsO4 buffered with 100 mM sodium cacodylate, pH 7.4, overnight at room temperature. The pellets were then washed with water, dehydrated in an increasing ethanol series (50%, 60%, 70%, 80%, 90%, 100%), and embedded in Spurr's resin (Electron Microscopy Sciences, Hatfield, PA) according to manufacturer's instructions. For tomographic reconstructions, semithick (200 nm) sections were cut from each sample on an UltraCut-UCT microtome (Leica Microsystems, Vienna) using a diamond knife (Diatome US, Port Washington, PA). Sections were collected on Formvar-coated copper/rhodium slot EM grids (Electron Microscopy Sciences, Port Washington, PA) and stained with uranyl acetate and lead citrate. Colloidal gold particles (15 nm diameter) were affixed to both surfaces of the grid to serve as fiducial markers for image alignment. Grids were placed in a double-tilt rotation specimen holder (Model 925; Gatan, Inc. Pleasanton, CA) and viewed in a Tecnai F20 transmission electron microscope (FEI Company, Eindhoven) at 200 keV. Areas selected for tomographic reconstruction were imaged at 29 000×, corresponding to a pixel size of 0.76 nm. Specimens were tilted ±60° and images recorded at 1° intervals. The grid was then rotated 90° and a similar tilt-series recorded about the orthogonal axis. Tilt-series data sets were recorded digitally and acquired automatically using SerialEM software.21 Tomograms were calculated from the tilt-series and analyzed using the IMOD software package22 on Macintosh G4 and G5 computers.
MEP Analysis by Multidimensional Protein Identification Technology (MudPIT)
The MEP samples (volume equivalent to 125 μg of predigested protein) was loaded off-line onto a fused-silica microcapillary column (100 μm i.d., 360 μm o.d., Polymicro Tech., Phoenix, AZ) with a ∼5 μm tip (pulled in-house) and packed with 10 cm of Aqua C18 material (Phenom-enex, Torrance, CA), 3 cm of strong cation exchange material (Whatman, Inc., Florham Park, NJ) and 2 cm of Aqua C18 using a high-pressure bomb (in-house). The column was set into a block column heater (built in-house)13 placed in-line with an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) and interfaced with an Agilent 1100 binary HPLC and autosampler system. The mobile phase buffers were the following: Buffer A (95% H2O, 5% acetonitrile, 0.1% formic acid) and Buffer B (95% acetonitrile, 5% H2O, 0.1% formic acid). Samples were analyzed with a 12-step MudPIT at 60 °C during chromatography.13 The mobile phase gradient for Steps 2-11 was 100% Buffer A, 5 min; 100% Buffer A to 35% Buffer A/65% Buffer B, over 80 min; 35% Buffer A/65% Buffer B to 100% Buffer A, over 10 min; 100% Buffer A, 25 min. The mobile phase gradient for Steps 1 and 12 was 100% Buffer A, 5 min; 100% Buffer A to 35% Buffer A/65% Buffer B, over 70 min; 35% Buffer A/65% Buffer B to 10% Buffer A/90% Buffer B, over 10 min; 10% Buffer A/90% Buffer B, 5 min; 10% Buffer A/90% Buffer B to 100% Buffer A, over 10 min; 100% Buffer A, 20 min. Ammonium acetate salt pulses were injected by the autosampler at the beginning of each subsequent step (Step 2, 20 μL, 100 mM; Step 3, 20 μL, 200 mM; Step 4, 20 μL, 300 mM; Step 5, 20 μL, 400 mM; Step 6, 20 μL. 500 mM; Step 7, 20 μL, 600 mM; Step 8, 20 μL, 700 mM; Step 9, 20 μL, 800 mM; Step 10, 20 μL, 900 mM; Step 11, 20 μL, 5 M; Step 12, 20 μL, 12.5 M). Mass spectra were acquired using data-dependent acquisition with a single full mass scan followed by 5 MS/MS scans.
PAP Analysis by MudPIT
The PAP sample (125 μg) was loaded off-line onto a fused-silica column (250 μm i.d., Agilent) packed with 2 cm Aqua C18 material using a high-pressure bomb. With the use of an in-line filter assembly (Upchurch, Oak Harbor, WA), the column was connected to a 2-phase fused silica column (100 μm i.d.) with a ∼5 μm tip and packed with 10 cm of Aqua C18 material and 3 cm of strong-cation exchange material. After desalting with Buffer A, the 2-phase separation column with attached desalting column was placed in-line with an LTQ mass spectrometer and interfaced with an Agilent 1100 binary HPLC and autosampler system. The sample was analyzed using an automated MudPIT method containing 12 2-h steps at 45 °C, with the same mobile phase gradients use for the MEP analysis. Ammonium acetate salt pulses were injected by the autosampler at the beginning of each subsequent step (Step 2, 25 μL, 250 mM; Step 3, 25 μL, 500 mM; Step 4, 25 μL, 750 mM; Step 5, 25 μL, 1000 mM; Step 6, 25 μL, 1250 mM; Step 7, 25 μL, 1500 mM; Step 8, 25 μL, 1750 mM; Step 9, 25 μL, 2000 mM; Step 10, 25 μL, 2250 mM; Step 11:, 25 μL, 5 M; Step 12, 50 μL, 12.5 M).
Data Analysis
MS/MS spectra from each analysis were searched using no enzyme specificity and a static modification of +57 on cysteines on a 96 node G5 Beowulf cluster against the IPI human database (downloaded April 2007) concatenated to a shuffled decoy database23 using a normalized implementation of SEQUEST.24 MS/MS spectra from the MEPs analysis were also searched for a variable modification of +48 on methionines, reflecting the homoserine lactone formation of methionines after reaction with CNBr.25 The resulting peptide identifications were assembled into proteins using DTASelect,26 and thresholds were adjusted to maintain a protein false discovery rate (FDR) < 5%.27
Supplementary Material
Acknowledgments
The authors thank Jessica Krank and Dr. Robert Murphy for expert assistance in the MS analysis of lipids and Dr. Michael MacCoss for critical reading of the manuscript. Financial support for this work was provided by National Institutes of Health Grants K22-AI059076 and R21-DA021744 (C.C.W.) and F31-DA0022825 (A.R.B.).
Footnotes
Supporting Information Available: Figures of percent of proteins by number of predicted TMDs for MEPs MudPITs, average percent of peptides vs GRAVY score for MEPs MudPITs, percent of amino acids per peptide overlapping with a predicted TMD for MEPs MudPITs, average spectra per protein for identified membrane and soluble proteins in MEPs Mud-PITs; tables of MEPs, PAPs, and MEP and PAP comparison; movies of three-dimensional models of membranes showing conformation of largely intact structures with an opening in one side. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Speers AE, Wu CC. Proteomics of integral membrane proteins—theory and application. Chem Rev. 2007;107(8):3687–3714. doi: 10.1021/cr068286z. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen PA, Olsen JV, Podtelejnikov AV, Andersen JR, Mann M, Wisniewski JR. Proteomic mapping of brain plasma membrane proteins. Mol Cell Proteomics. 2005;4(4):402–408. doi: 10.1074/mcp.T500002-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Chen EI, Cociorva D, Norris JL, Yates JR., III Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J Proteome Res. 2007;6(7):2529–2538. doi: 10.1021/pr060682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blonder J, Conrads TP, Yu LR, Terunuma S, Janini GM, Issaq HJ, Vogel JC, Veenstra TD. A detergent- and cyanogen bromide-free method for integral membrane proteomics: application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics. 2004;4(1):31–45. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- 5.Washburn MP, Wolters D, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 6.Wu CC, MacCoss MJ, Howell KE, Yates JR., III A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21(5):532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 7.Howell KE, Palade GE. Hepatic Golgi fractions resolved into membrane and content subfractions. J Cell Biol. 1982;92(3):822–832. doi: 10.1083/jcb.92.3.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sprong H, van der Sluijs P, van Meer G. How proteins move lipids and lipids move proteins. Nat Rev Mol Cell Biol. 2001;2(7):504. doi: 10.1038/35080071. [DOI] [PubMed] [Google Scholar]
- 9.Hildebrand PW, Preissner R, Frommel C. Structural features of transmembrane helices. FEBS Lett. 2004;559(1–3):145–151. doi: 10.1016/S0014-5793(04)00061-4. [DOI] [PubMed] [Google Scholar]
- 10.Eichacker LA, Granvogl B, Mirus O, Muller BC, Miess C, Schleiff E. Hiding behind hydrophobicity. Transmembrane segments in mass spectrometry. J Biol Chem. 2004;279(49):50915–50922. doi: 10.1074/jbc.M405875200. [DOI] [PubMed] [Google Scholar]
- 11.Martosella J, Zolotarjova N, Liu H, Moyer SC, Perkins PD, Boyes BE. High recovery HPLC separation of lipid rafts for membrane proteome analysis. J Proteome Res. 2006;5(6):1301–1312. doi: 10.1021/pr060051g. [DOI] [PubMed] [Google Scholar]
- 12.Whitelegge JP, Jewess P, Pickering MG, Gerrish C, Camilleri P, Bowyer JR. Sequence analysis of photoaffinity-labelled peptides derived by proteolysis of photosystem-2 reaction centres from thylakoid membranes treated with [14C]azidoatrazine. Eur J Biochem. 1992;207(3):1077–1084. doi: 10.1111/j.1432-1033.1992.tb17144.x. [DOI] [PubMed] [Google Scholar]
- 13.Speers AE, Blackler AR, Wu CC. Shotgun analysis of integral membrane proteins facilitated by elevated temperature. Anal Chem. 2007;79(12):4613–4620. doi: 10.1021/ac0700225. [DOI] [PubMed] [Google Scholar]
- 14.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 15.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 16.Sadka T, Linial M. Families of membranous proteins can be characterized by the amino acid composition of their transmembrane domains. Bioinformatics. 2005;21(Suppl 1):i378–386. doi: 10.1093/bioinformatics/bti1035. [DOI] [PubMed] [Google Scholar]
- 17.Fischer F, Wolters D, Rogner M, Poetsch A. Toward the complete membrane proteome: high coverage of integral membrane proteins through transmembrane peptide detection. Mol Cell Proteomics. 2006;5(3):444–453. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Lewin AR, Reid LE, McMahon M, Stark GR, Kerr IM. Molecular analysis of a human interferon-inducible gene family. Eur J Biochem. 1991;199(2):417–423. doi: 10.1111/j.1432-1033.1991.tb16139.x. [DOI] [PubMed] [Google Scholar]
- 19.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ott CM, Lingappa VR. Integral membrane protein biosynthesis: why topology is hard to predict. J Cell Sci. 2002;115(Pt 10):2003–2009. doi: 10.1242/jcs.115.10.2003. [DOI] [PubMed] [Google Scholar]
- 21.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152(1):36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 23.Finney G, Merrihew G, Klammer A, Frewen B, MacCoss MJ. Protein false discovery rates from MS/MS experiments: decoy databases and normalized cross-correlation. Proceeding of the 53rd ASMS Conference on Mass Spectrometry; San Antonio, TX. 2005. [Google Scholar]
- 24.MacCoss MJ, Wu CC, Yates JR., III Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal Chem. 2002;74(21):5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 25.Kaiser R, Metzka L. Enhancement of cyanogen bromide cleavage yields for methionyl-serine and methionyl-threonine peptide bonds. Anal Biochem. 1999;266(1):1–8. doi: 10.1006/abio.1998.2945. [DOI] [PubMed] [Google Scholar]
- 26.Tabb DL, McDonald WH, Yates JR., III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1(1):21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blackler AR, Klammer AA, MacCoss MJ, Wu CC. Quantitative comparison of proteomic data quality between a 2D and 3D quadrupole ion trap. Anal Chem. 2006;78(4):1337–1344. doi: 10.1021/ac051486a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.