

Abstract
Methionine synthase reductase (MSR; gene name MTRR) is responsible for the reductive activation of methionine synthase. Cloning of the MTRR gene had revealed two major transcription start sites which, by alternative splicing, allows for two potential translation products of 698 and 725 amino acids. While the shorter protein was expected to target to the cytosol where methionine synthase is located, the additional sequence in the longer protein was consistent with a role as a mitochondrial leader sequence. The possibility that MSR might target to mitochondria was also suggested by the work of Leal et al. (2004) who showed that it can act as the reducing enzyme in combination with MMAB (ATP:cob(I)alamin adenosyltransferase) to generate adenosylcobalamin from cob(II)alamin in vitro. Here we examined directly whether MSR protein is found in mitochondria. We show that, while two transcripts are produced by alternative splicing, the N-terminal segment of the putative mitochondrial form of MSR fused to GFP does not contain a sufficiently strong mitochondrial leader sequence to direct the fusion protein to the mitochondria of human fibroblasts. Further, antibodies to MSR protein localized MSR to the cytosol but not to the mitochondria of human fibroblasts or the human hepatoma line Huh-1, as determined by Western blot analysis and immunofluorescence of cells in situ. These data confirm that MSR protein is restricted to the cytosol but, based on the Leal study, suggest that a similar protein may interact with MMAB to reduce the mitochondrial cobalamin substrate in the generation of adenosylcobalamin.
Keywords: Methionine synthase reductase, mitochondrial localization, immunofluorescence, cobalamin, Vitamin B12
Introduction
Vitamin B12, also known as cobalamin, is a micronutrient synthesized only by microorganisms, yet is essential to human health. Two distinct, but overlapping, metabolic pathways contribute to the intracellular processing of cobalamin, converting it from the hydroxo (OHCbl) or cyano (CNCbl) forms usually ingested and culminating in the production of methylcobalamin (MeCbl), the cofactor form required by the cytosolic enzyme methionine synthase (MS), and adenosylcobalamin (AdoCbl), the cofactor form usable by the mitochondrial enzyme methylmalonyl-CoA mutase (MUT)[1]. Methionine synthase catalyzes the transfer of a methyl group from methyltetrahydrofolate to homocysteine to form methionine and is a critical constituent of both the folate and methionine cycles. Methylmalonyl-CoA mutase catalyzes the reversible isomerization of methylmalonyl-CoA to succinyl-CoA, an important step in the catabolism of branched chain amino acids, odd-numbered chain fatty acids, cholesterol and other metabolites. In patients, blocks in MeCbl synthesis result in homocystinuria while blocks in AdoCbl synthesis result in methylmalonic aciduria. Patients with an early block affecting both pathways have combined homocystinuria and methylmalonic aciduria.
Genetic disorders linked to Cbl metabolism have been assigned to nine complementation groups – designated cblA-cblH and mut. Three complementation groups - cblF, cblC and cblD -correspond to blocks in steps that are common to the synthesis of both cofactors. The cblF group is predicted to be responsible for transport of Cbl out of the lysosome [2]. The gene responsible for the cblC group, called MMACHC, was identified recently and is predicted to function in a manner similar to TonB, a bacterial protein involved in the energy-coupled transport of cobalamin into gram negative bacteria [3]. The cblD group is predicted to be responsible for the branching of the Cbl pathway to the cytosol or mitochondria, since dysfunction of this protein can affect either or both pathways [4]. Four complementation groups, cblA, cblB, cblH and mut, affect steps occurring in the mitochondrion only and thus affect the production or utilization of AdoCbl. The cblA group is caused by mutations in MMAA, a gene that encodes a protein that belongs to the G3E class of P-loop GTPases, but whose function in the Cbl pathway is unkown [5; 6]. By analogy to bacterial orthologues, it may interact directly with mutase [7; 8]. The cblB group was found to be due to defective MMAB (MMAB gene), which codes for ATP:cobalamin adenosyltransferase, the protein responsible for the transfer of the adenosyl group to cob(I)alamin [9]. Only one patient represents the cblH group [10]. Despite affecting only the mitochondrial pathway, cblH is thought to be allelic with the cblD group [4]. The mut group (MUT gene) was found to be due to mutations in MUT itself. The remaining groups, cblE and cblG, have blocks only in the cytosolic pathway leading to MeCbl synthesis. The gene responsible for cblG was shown to be MTR, which encodes MS [11-13], while cblE (MTRR gene) corresponds to defects in methionine synthase reductase (MSR), the protein responsible for the reduction of Cbl on MS [14] as well as for the reduction of aquacobalamin (cob(III)almin) to cob(II)alamin in the cytosol [15].
Thus far, the enzyme responsible for the mitochondrial reduction of cob(II)alamin to cob(I)alamin, a step required for the synthesis of AdoCbl, has yet to be described. MSR, the protein responsible for this same reaction in the cytosolic pathway, might be a candidate for this role for two reasons. 1) When the MTRR gene was originally cloned, it was found to produce two mRNA splice variants with different 5′ ends [14] (Fig. 1AB). While the first variant produced a protein whose predicted cellular location was consistent with being cytosolic, the second and larger variant had a 58 bp 5′ extension that was predicted to produce an extra 27 N-terminal amino acids, generating a peptide sequence predicted by the mitochondrial prediction program TRANSPEP to constitute a mitochondrial leader sequence [16] (Fig. 1C). These data suggested that a version of MSR might be normally present in the mitochondria. 2) An investigation by Leal et al. demonstrated that, in vitro, MSR and MMAB (ATR in their nomenclature) were able to convert cob(II)alamin to AdoCbl at a level that might be sufficient to meet physiological needs [17]. In this way MSR may participate in the reduction of cobalamin for MMAB in the same way it reduces cobalamin for MS (Fig. 1D). With these studies in mind, we set out to determine if MSR does localize to the mitochondria in vivo and therefore has the possibility of filling the role of the missing AdoCbl synthesizing reductase.
Figure 1. Splice variants of MSR and the predicted mitochondrial leader.
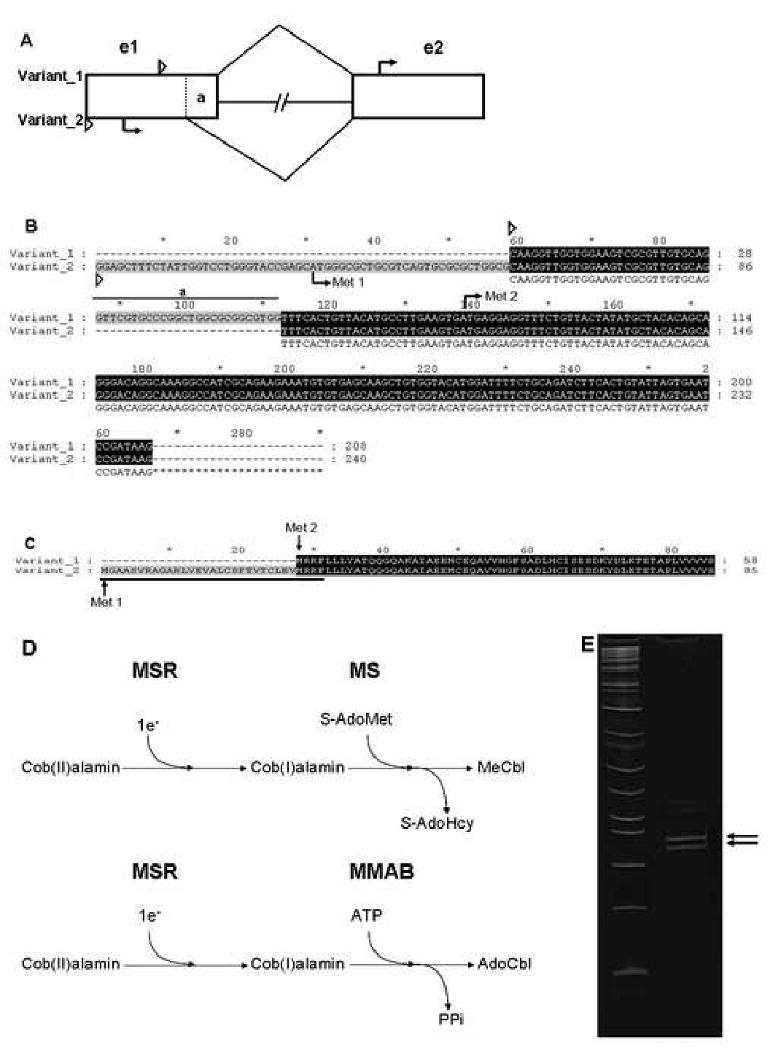
A. A schematic of the two splice variants of MSR mRNA. Represented here are the first two exons (rectangles, e1 and e2), the first intron (lines connecting rectangles), the beginning of the 5′ untranslated region (small triangles) for each splice variant, the start site (arrow) of each, and the 26 bp region at the end of exon 1 that is found as 5′UTR in variant 1, but spliced out in variant 2 (“a”) of MSR. B. Alignment of the mRNA transcribed from the first 2 exons in both splice variant 1 and 2. The red “Met” shows the position of the initiation codon of each variant splice form. The rest of the markings are the same as in A. C. The predicted protein translation of the N-terminus of the two splice variants. Variant 2 is predicted to have an extra N-terminal 27 amino acids, which was originally predicted to make up the bulk of the putative mitochondrial leader sequence (underlined) [16]. D. Schematic of the reduction of cob(II)alamin to cob(I)alamin by MSR for the generation of MeCbl by MS or AdoCbl for MMAB. E. RT-PCR of MSR mRNA from exon 1-2 run on an 8% acrylamide gel demonstrating the two different bands which correspond to the 26 base-pair difference between the two splice variants. Upper arrow: Variant 1. Lower arrow: Variant 2.
Here we report that both isoforms of MSR mRNA are detectable by RT-PCR, as previously reported. We also confirm that MSR and MMAB can indeed function together, in vitro, to produce AdoCbl. However, we show that, unlike other mitochondrial proteins involved in the AdoCbl pathway, variant 2 MSR does not produce a functional mitochondrial leading sequence. We also show that MSR protein is localized to the cytoplasm but not to the mitochondria, in both Western blot and immunofluorescent studies. These data confirm that MSR is restricted to the cytosol and therefore does not function as the missing mitochondrial reductase.
Methods and Materials
RT-PCR
RNA isolation, reverse-transcription and PCR reactions were performed as described [12].
Plasmid Construction
The plasmid pAcGFP1-Mito was purchased from Clontech (Mountain View, CA). This vector contains the mitochondrial targeting sequence from subunit VIII of human cytochrome C oxidase consisting of the first 29 residues of that protein, followed by the green fluorescent protein (GFP) variant AcGFP1. Flanking the mitochondrial leader sequence are the unique restriction enzyme sites NheI (5′) and BamHI (3′). For construction of the GFP plasmid without the mitochondrial leader sequence, we cut pAcGFP1-Mito with NheI and BamHI, filled in the ends with Klenow enzyme and blunt-end ligated the vector to itself using T4 DNA Ligase (all from New England Biolabs; Mississauga, ON). For construction of mmabLeader-GFP, mutLeader-GFP and msrLeader-GFP we used the PCR product from the amplification of the N-terminal regions of each cDNA as specified by the primers shown in Table 1, and ligated them into pAcGFP1 using NheI and BamHI.
Table 1. Primers used to make GFP mitochondrial leader constructs.
| Gene | Primers | Sequence | Product Length | Amino acids covered |
|---|---|---|---|---|
| MMAA | cblA 5′ | GGGCTAGCATGCCCATGCTGCTACCACAT | 180 bp | 60 aa |
| cblA 3′ | GGGATCCCCATCTGACAGCAGCATCCAC | |||
| MMAB | cblB 5′ | GGGCTAGCATGGCTGTGTGCGGCCTGG | 261 bp | 87 aa |
| cblB 3′ | GGGATCCCCCACGGCTTCAAACACTT | |||
| MUT | MUT 5′ | GGGCTAGCATGTTAAGAGCTAAGAA | 183 bp | 61 aa |
| MUT 3′ | GGGATCCCATATTAGGTCTTCTGGGT | |||
| GCTAGCGGAGCTTTCTATTGGTCCTGGGTACC | ||||
| MSR | MSR 5′ | GAGCATGGGCGCTGCGTCAGTGC | 173 bp | 47 aa |
| MSR 3′ | GGGATCCTCTTCTGCGATGGCCTTT |
Antibody Generation
Antibodies against MSR were produced at the Sheldon Biotechnology Centre (Montreal, Canada) by immunizing rabbits with either i) a synthetic peptide encompassing amino acids 37-56 of MSR (MSR-FMN-domain) (Sheldon Biotechnology Centre; Montreal, Canada) or ii) a protein fragment of amino acids 155-306 of MSR plus a 6× His-tag expressed from the plasmid pQE-30 (Qiagen; Mississauga, ON) (MSR-linker-domain). To ensure antigenic specificity before use in Western blot and immunofluorescence, both antibodies were affinity purified to expressed and purified full-length MSR protein using HiTrap NHS-activated HP columns, following manufacturer's protocols (Amersham/GE Healthcare; Baie d'Urfe, QC). Antibodies against MUT were generated at the SACRI Hybridoma Facility (University of Calgary) by immunizing mice against a protein fragment encompassing amino acids 1-371 of MUT with a C-terminal 6× His-tag expressed from pET28a (Novagen; Madison, WI). The primary antibody, anti-p38MapK, was purchased from BD Biosciences (Mississauga, ON). The secondary antibodies used for Western blotting: Goat anti-Rabbit IgG HRP and Goat anti-Mouse IgG HRP were purchased from Biorad (Mississauga, ON) and the secondary antibodies used for immunofluorescence: Cy3-conjugated donkey anti-rabbit IgG and Cy3-conjugated donkey anti-mouse IgG were purchased from Jackson Immunoresearch (West Grove, PA).
Cell Culture and Protein Preparation
Huh-1 (human hepatoma) [18], MCH65 (normal human fibroblasts) and WG1296 (cblE patient fibroblasts) cells were cultured in DMEM (Invitrogen; Burlington, ON) supplemented with 10% serum (5% fetal bovine serum, 5% bovine calf serum) and antibiotics-antimycotics. MCH65 and WG1296 were from the Cell Repository, Montreal Children's Hospital. Confluent cells were harvested by trypsinization, washed in PBS, resuspended in 2× cell pellet volume of NETN buffer (150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, pH8.0, 0.01% NP-40) plus protease inhibitors and disrupted using an Ultrasonic Processor XL (Mandel Scientific; Guelph, ON). The cell lysate was then centrifuged at 10,000 × g for 10 minutes at 4°C, and the supernatant containing total soluble cell protein was recovered and protein concentration determined using the Bradford Assay (BioRad).
Cellular fractionation
Cells were fractionated according to methods described by Isaya et al. [19] with a few exceptions. Briefly, cells were grown in a monolayer on a 10 cm plate, washed with PBS, then incubated with 1.5 ml of digitonin release buffer (250 mM sucrose; 17 mM MOPS, pH 7.5; 2.5 mM EDTA; 0.8 mg of digitonin per ml) for exactly 2 minutes. Treatment was stopped by diluting with 3 ml of buffer without digitonin. The solution was aspirated and transferred to two 2 ml microcentrifuge tubes and centrifuged at 7,500 × g at 4°C for 5 minutes. The supernatant (containing the cytosolic fraction) was moved to a new tube and held at 4°C. The “perforated” cell monolayer remaining on the plate was scraped from the plate after addition of 0.5 ml release buffer (without digitonin) and homogenized by 20 passages of a tight-fitting Teflon pestle in a 1 ml tapered glass tissue grinder (Wilmad-Labglass; Buena, NJ) at 4°C. One volume of release buffer without digitonin was added to the homogenate, followed by centrifugation at 1500 × g for 15 minutes at 4°C, resulting in a low speed pellet (unbroken cells, cell debris and nuclei) and a low speed supernatant. The supernatant was centrifuged at 15,000 × g for 15 minutes at 4°C, resulting in a pellet (the mitochondrial enriched fraction) and a high-speed supernatant. The cytosolic and mitochondrial enriched fractions were then used for subsequent experiments. Purity of the fractions was tested by the detection of anti-MUT as a mitochondrial marker and by the detection of anti-p38 as a cytoplasmic/nuclear marker for each fraction.
Western Blots
Cell lysates or immunoprecipitations were resolved by SDS-PAGE and transferred to nitrocellulose membranes by capillary action. The membranes were blocked with 5% skim milk in PBS for 1 hour at room temperature with shaking and then incubated with primary antibody (anti-MSR-FMN-domain at 1:100; anti-MSR-linker-domain at 1:100; anti-MUT at 1:2000; anti-p38 at 1:2000) overnight at 4°C. Membranes were then washed 3 × with TBST and incubated with secondary antibody (1:5000 Goat anti-Rabbit IgG HRP; 1:5000 Goat anti-Mouse HRP IgG) for 45 minutes at room temperature. Membranes were washed 3 × with TBST and immune complexes were detected using ECL Reagent (Amersham/GE Healthcare) according to manufacturer's recommendations and exposed to Hyperfilm (Amersham/GE Healthcare).
Nucleofection and Immunofluorescence
Plasmids noLeader-GFP, mitoLeader-GFP (pAcGFP1-mito), msrLeader-GFP, mutLeader-GFP and mmabLeader-GFP were introduced into cells by nucleofection as per manufacturer's protocol (Amaxa Biosystems; Gaithersburg, MD). Immunofluorescence was performed according to Narang et al. [20] with a few exceptions. Cells were grown on coverslips and fixed with fresh 4% paraformaldehyde in PBS for 20 minutes at 37°C. Cells transfected with GFP constructs were stained with DAPI (Sigma-Aldrich; Oakville, ON) (1:5000 in PBS) for 30 seconds and visualized. Cells to be probed with antibody were washed 1 × with PBS, and permeabolized by 0.1% Triton X-100 (Sigma-Aldrich) in PBS for 15 minutes at room temperature. Cells were washed 3 × PBS and blocked in 10% BSA in PBS with slow rocking for 1 hour at room temperature, washed 3 × PBS and incubated with anti-MSR-linker-domain or anti-MUT (1:250) for 1 hour at 37°C, washed 3 × PBS, and incubated with secondary antisera (Cy3-conjugated donkey anti-rabbit, 1:250 or donkey anti-mouse IgG, 1:250) for 1 hour at 37°C. Cells were then counterstained with DAPI (1:5000 in PBS) for 30 seconds. Cells were mounted and visualized using a Leica DMR microscope with Metamorph 6.1 (Universal Imaging). Images involving cellular localization of GFP were not processed further; however, images involving the colocalization of MSR or MUT and mitochondria were deconvolved using a nearest-neighbors algorithm to reduce background haze and improve image contrast with Autodeblur 7.0 (AutoQuant) software.
Protein expression and purification
A human MMAB clone in a pET3a vector (Novagen) was kindly provided by H.L. Schubert and C.P. Hill, University of Utah. MMAB protein was expressed and purified as previously described [21]. A human MSR clone was generated by RT-PCR of mRNA from MCH65 cells using the oligonucleotides 5′GGATCCATGAGGAGGTTTCTGTTACTA-3′ and 5′AAGCTTTTATGACCAAATATCCTGAAG-3′, where underlined base-pairs represent addition of restriction endonuclease sites. After restriction with BamHI and HindIII, the MSR product was cloned into the pQE-30 vector (Qiagen) in the 5′-BamHI and 3′-HindIII restriction sites. The recombinant MSR was overexpressed in BL21 (DE3) cells in LB with induction by 1 mM IPTG. Cell pellets were lysed by passing through a French Pressure Cell (SLM Instruments Inc) × 2 at 12,000 psi in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM Imidazole; pH 8.0) and centrifuged at 15,000 rpm for 30 minutes. The soluble fraction was incubated with a 5 mL slurry of Ni-NTA beads (Qiagen), rinsed in lysis buffer and allowed to mix by end-over-end rotation overnight at 4°C. Once washed in wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM Imidazole; pH 8.0), the purified protein was eluted in elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM Imidazole; pH 8.0). Protein was further purified using an FPLC Superose 6 HR 10/30 prepacked column (Pharmacia/GE Healthcare). The protein solution was applied to the column which had been pre-equilibrated with 100 mM potassium phosphate, pH 7.4. Elution was then carried out at 8°C with 100 mM potassium phosphate, pH 7.4, at a flow rate of 0.5 ml/min with protein detection at 280 nm. Protein purity was monitored by SDS-PAGE.
ATP:Cob(I)alamin Adenosyltransferase Assays
Adenosyltransferase assays were performed inside an anaerobic chamber (Coy Laboratories; Ann Arbor, MI) as previously described [17] with a few exceptions. Reaction mixtures contained a final concentration of 200 mM Tris (pH 8.0), 1.6 mM KH2PO4, 2.8 mM MgCl2, 100 mM KCl, 0.1 mM HOCbl, 0.4 mM ATP, 1 mM DTT, 1 mM NADPH and 10 uM MMAB. Reactions were initiated by the addition of purified recombinant MSR and AdoCbl formation was measured by following the increase in absorbance at 525 nm (Δε525 = 4.8 cm-1 mM-1).
Results
Alternative splicing of MSR
We first re-examined the splice isoforms generated by alternative splicing in exon 1. These two forms contain the distinct translation initiation codons corresponding to putative cytosolic (variant 1) and mitochondrial (variant 2) coding sequences. We performed RT-PCR using the primers AB191: 5′ CAAGTTGGTGGAAGTCGCGTTG and 1803E: 5′ AACCCATACCGCAGGTGAGCAAA, as originally described [14; 22]. Under similar PCR conditions, we were able to identify the same two bands, corresponding to the two different isoforms of MSR mRNA (Fig 1E), as seen previously [22]. The identity of these bands was confirmed by sequencing (data not shown). Therefore, MSR has 2 distinct splice isoforms, variant 2 of which Leclerc et al. [16], using the cellular localization prediction program TRANSPEP, predicted to have mitochondrial localization signal.
Interaction of MSR with MMAB
The other rationale for predicting mitochondrial expression is the finding by Leal et al. [17] that MSR can provide the reducing activity for the MMAB-catalyzed production of AdoCbl from cob(II)alamin. We set out to confirm this result by incubating MMAB with MSR and assessing the production of AdoCbl. We found no significant production of AdoCbl when MMAB was incubated in OHCbl, ATP, MgCl2, DTT and NADPH (Fig. 2). However, upon addition of MSR there was immediate production of AdoCbl, and the rate of product formation increased with increasing concentration of MSR (Fig. 2). Thus, MSR and MMAB can combine to produce AdoCbl from cob(II)alamin in vitro. If MSR is targeted to mitochondria along with MMAB in vivo, it may act as the elusive mitochondrial reductase.
Figure 2. MSR acts with MMAB to produce AdoCbl.
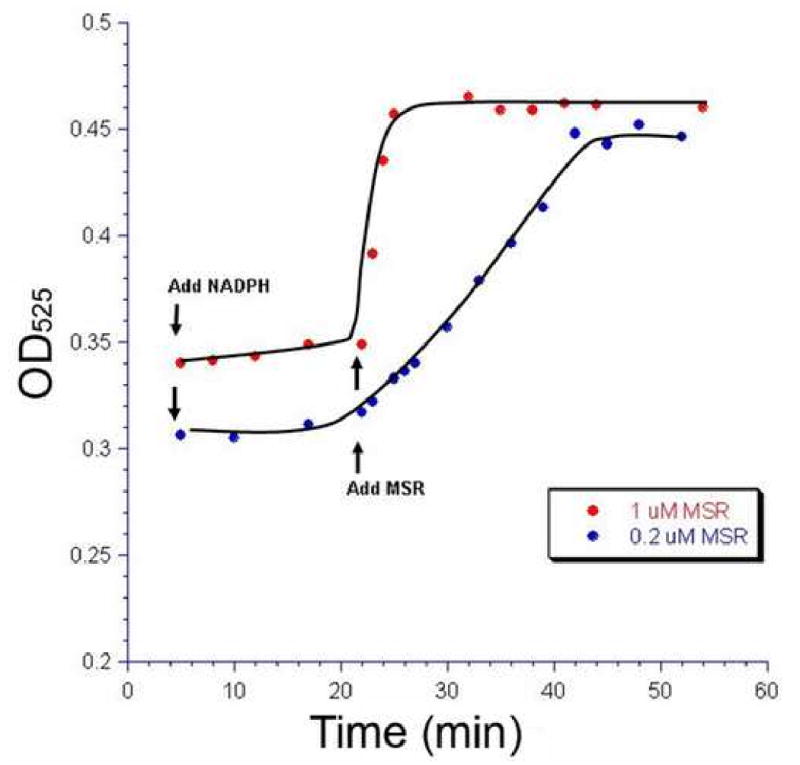
MMAB protein (10 µM) was incubated in the presence of DTT, MgCl2, ATP, OHCbl and NADPH with no significant production of AdoCbl as monitored by absorption at 525 nm. Upon addition of MSR (0.2 µM or 1 µM), AdoCbl was produced. Adding an increased concentration of MSR produced a corresponding increase in the rate of AdoCbl production (Red vs. Blue). Therefore, as was shown previously by Leal et al. [17], MSR can act with MMAB to make AdoCbl.
MSR does not have a functional leader sequence
To assess the functionality of the putative MSR mitochondrial leader sequence, we created different plasmids that have known or candidate mitochondrial leader sequences attached to the N-terminus of GFP from the plasmid pAcGFP1-mito (Fig. 3A) and assessed, by immunofluorescence, whether they direct GFP into the mitochondria. When we transiently transfected the plasmid pAcGFP1-mito, with the mitochondrial targeting sequence of subunit VIII of cytochrome c oxidase, into the human fibroblast cell line MCH65, the expressed GFP was distributed in the typical punctate or worm-like pattern of mitochondrial proteins (Fig. 3B, Panel 1). However, when we excised the mitochondrial leader sequence of pAcGFP1-mito, expressed GFP was found to be distributed throughout the cytoplasm (Fig. 3B, Panel 2). When we replaced the cytochrome c oxidase targeting sequence with the coding sequence of the first 47 amino acids of MSR variant 2, we found that the GFP was again distributed diffusely throughout the cytoplasm, mimicking the pattern of the control plasmid with its mitochondrial targeting sequence removed (Fig. 3B, Panel 3). It is important to note that this protein could only be expressed if we included the 30 bp 5′ UTR in front of the MSR coding sequence. In contrast, when the coding sequences of the N-terminal regions of MUT and MMAB were spliced upstream of the GFP sequence, GFP was again directed to the mitochondria (Fig. 3B, Panels 4 & 5, respectively). These results demonstrate that, unlike MMAB and MUT, two cobalamin metabolism proteins known to target to mitochondria, MSR does not possess a functional mitochondrial leader sequence, at least as evaluated by attachment to GFP.
Figure 3. Assay of functional N-terminal leader sequences.
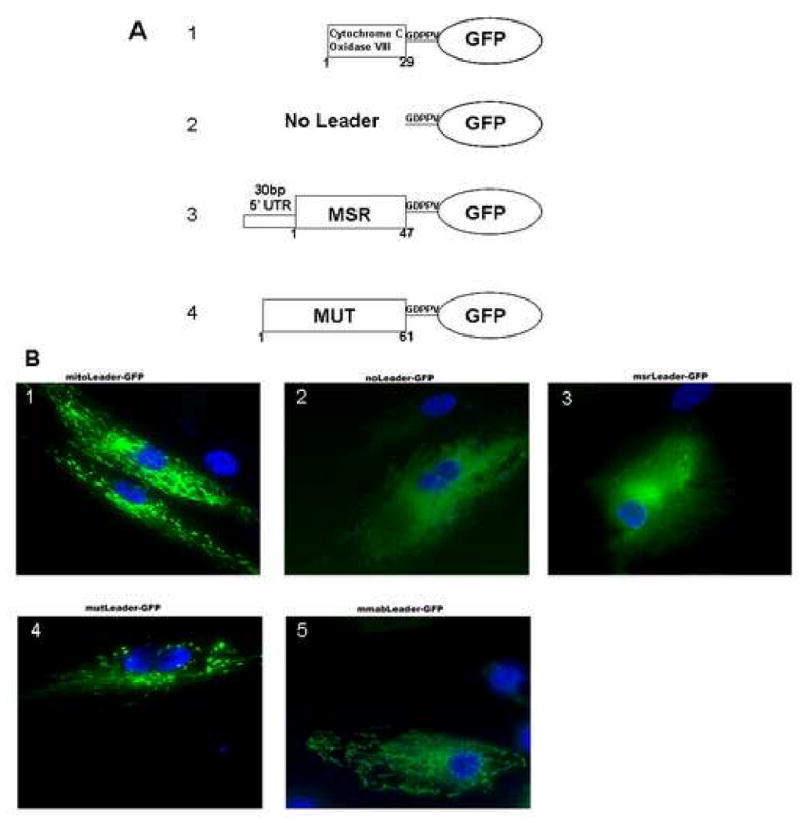
A. Schematic of the different mitochondrial leader constructs made from the pAcGFP1-mito vector. 1. Original pAcGFP1-vector with cytochrome C oxidase subunit VIII leader sequence (positive control). 2. Cytochrome C oxidase subunit VIII leader sequence is cut out, leaving the vector with expression of GFP but no mitochondrial leader (negative control). 3. pAcGFP with the 30bp 5′UTR and first 173 base pairs (47 amino acids) of MSR variant 2 attached to the N-terminus of GFP. 4. The first coding 183 base pairs (61 amino acids) of MUT attached to the N-terminus of GFP. 5. The first coding 261 base pairs (87 amino acids) of MMAB attached to the N-terminus of GFP. B. Brightfield fluorescent images of the constructs described and numbered as above, after transfection into MCH65 fibroblasts. Note the punctate pattern of GFP expression in panels 2, 4 and 5 and diffuse expression in panels 1 and 3.
MSR protein is found in the cytoplasm but not in the mitochondria
To inspect MSR localization directly, we examined the distribution of MSR protein by Western blot of cytosol and mitochondrial extracts and by immunofluorescence analysis of Huh-1 cells and human fibroblasts. The Western blot was accomplished using antibodies independently generated against two different sites in MSR, the FMN-domain at the N-terminus and the central linker-domain. In both cell lines and for both antibodies, MSR was detected in the cytosol but not the mitochondrial extracts (Fig. 4A,B). Note that only one band corresponding to MSR was detected, likely due to the small size difference between the two predicted isofroms, 2.7 kDa, so that if there were two bands they would appear in the same location in this experiment. Antibody specificity was demonstrated by the absence of the MSR band in the MSR deficient cell line WG1296. Successful separation of mitochondria and cytosol was demonstrated by the detection of MUT in the mitochondrial fraction but not cytosolic fractions, and by the presence of the cytoplasmic/nuclear marker p38 in the cytosol and near absence in the mitochondrial fraction (Fig. 4C).
Figure 4. Subcellular localization of MSR. Panels A & B.
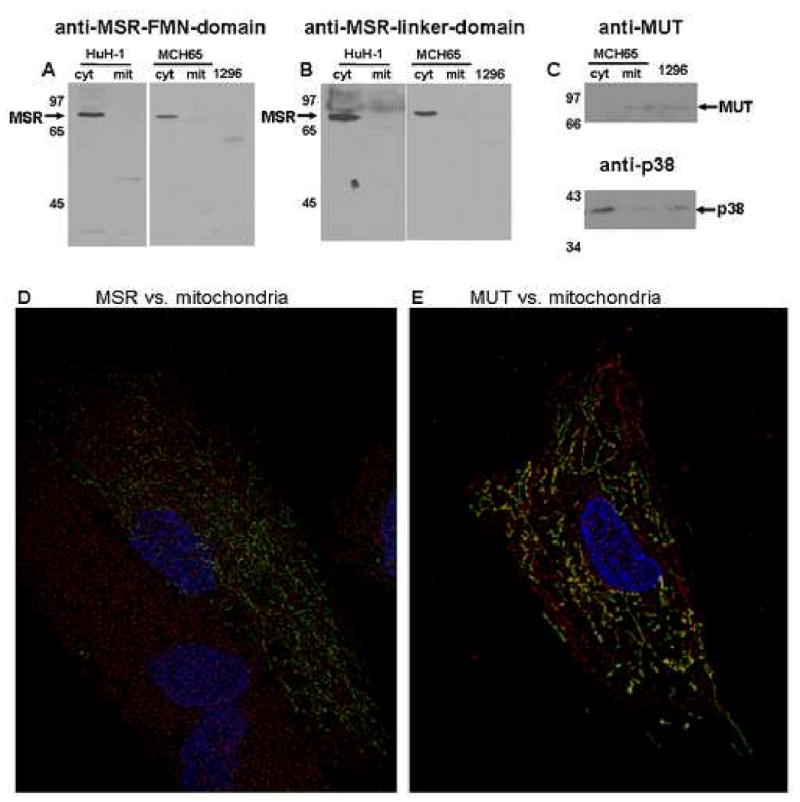
Western blot of cytosolic (cyt) and mitochondrial (mit) fractions of Huh-1 and MCH65 cells and total cell protein of WG1296 (1296) probed with anti-MSR-FMN-domain (panel A) or anti-MSR-linker-domain (panel B). Panel C. Control to test purity of mitochondrial and cytoplasmic fractions. Upper panel shows anti-MUT (mitochondrial protein); lower panel shows anti-p38 (cytosolic and nuclear protein). There appears to be a small amount of cytosolic contamination of the mitochondrial fraction (band in ‘mit’ of p38), but almost no mitochondrial contamination of cytoslic fraction (band ‘c’ in MUT). Lanes are the same as indicated for Panel A and B. Panel D & E. Immunofluorescent localization of MSR. MCH65 cells were transfected with pAcGFP1-mito (mitochondrial label, Green) and were immunostained for either MSR (anti-MSR-linker-domain) Panel D or methylmalonyl-CoA mutase (anti-MUT) Panel E (Red). Nuclei are stained with DAPI (Blue). After deconvolution, MUT was found to co-localize with mitochondria (yellow) but MSR was not.
To evaluate the subcellular distribution of MSR in cells in situ, we probed MCH65 fibroblasts for MSR and compared it with the distribution of GFP (AcGFP1) containing the N-terminal mitochondrial targeting sequence from subunit VIII of human cytochrome C oxidase expressed following nucleofection with the pAcGFP1 plasmid. MSR, detected by immunofluorescence using the MSR linker-domain antibody, was observed to be distributed throughout the cytoplasm and did not colocalize with the GFP mitochondrial marker (Fig. 4D). In contrast, the mitochondrial protein MUT did show colocalization with mitochondrial GFP (Fig. 4E). These experiments demonstrate that MSR protein is not found in the mitochondria of cells and that it is instead restricted to the cytosol.
Discussion
The role of MSR in the reductive activation of MS, and, recently, as an aquacobalamin reductase, has been well documented [15; 23; 24]. In contrast, the reductase responsible for the reduction of cob(II)alamin to cob(I)alamin prior to adenosylation by MMAB in the mitochondria, has yet to be found. Given the ability of MSR to reduce cobalamin for the production of adenosylcobalmin by MMAB in vitro [17] and the presence of a putative mitochondrial leader sequence in one splice form at its N-terminus, it was tempting to evaluate MSR as the missing mitochondrial cobalamin reductase. However, our results show that MSR does not traverse the mitochondrial membrane. These studies showed MSR to be restricted to the cytoplasm in human Huh-1 hepatoma cells and in human fibroblasts. The results indicate that the two splice forms, which differ in utilizing different translation initiation codons separated by 27 amino acids, do not confer any special properties with respect to organelle targeting, leaving it unclear as to why this difference in N-terminal sequence exists. It could affect protein stability and even, potentially, net enzyme activity; however, such alternatives have not been considered in the past. Interestingly, the additional sequence borders the FMN binding domain although it does not show sequence homology with the FMN domain of other proteins. In this regard, it seems unlikely that the additional sequence has a functional role.
One clue to the origin of the additional variant sequence comes from the examination of the N-terminal sequence of MSR from different mammalian species. An alignment of MSR protein shows that the N-terminal extension found in human variant 2 is partially conserved in primates (chimp, macaque, bushbaby) but very poorly conserved in lower mammals (Fig. 5). Moreover, two lower mammals that have an upstream Met-initiated in-frame sequence before Met2, opossum (5 amino acids) and guinea pig (21 amino acids), have much shorter N-terminal extensions (Fig. 5). These data point to the longer version of MSR being introduced as a late evolutionary addition which, due to the variation in sequence in lower mammals, is unlikely to have a functional role.
Figure 5. Multiple sequence alignment of the N-terminal amino acids of MSR from selected mammalian species.

MSR protein sequences of selected mammals (human, ENST00000264668; chimp, ENSPTRP00000028690; macaque, ENSMMUP00000024676; bushbaby, ENSOGAP00000000920; treeshrew, ENSTBEP00000011472; cat, ENSFCAP00000014011; dog, ENSCAFP00000014869; cow, ENSBTAP00000012372; mouse, ENSMUSP00000039810; rat, ENSRNOP00000024041; opossum, ENSMODP00000002568; guinea pig, ENSCPOP00000005952; hedgehog, ENSEEUP00000001324; squirrel, ENSSTOP00000004416; and rabbit, ENSOCUP00000003139) were aligned using Clustalw [33] and visualized using GeneDoc [34]. For clarity, only the first 100 amino acids are shown.
MSR variant 2 was predicted to have a mitochondrial leader sequence by TRANSPEP in 1999, a program that was then part of the PCGENE package from Intelligenetics. It is interesting to assess whether current mitochondrial prediction programs would give the same subcellular location. Table 2 contains the subcellular localization predictions of MSR variant 2 from several targeting sequence prediction programs. Three of the four programs used predict that the variant 2 sequence “is possibly” or “is” mitochondrial. By comparison, two of the four programs predict mitochondrial localization for MSR variant 1, a protein known to be cytosolic, but all four predict MUT to be in the mitochondria (Table 2). It seems that even with the advanced prediction programs of today, there is still an uncertainty in predicting whether proteins like MSR, with putative, if not strong, mitochondrial signals, are targeted to this organelle.
Table 2. Subcellular localization predictions of MSR variant 2, as determined by various subcellular prediction programs.
| MitoProt II 1.0a4 | Predotar v. 1.03 | iPSORT | TargetP1.1 (mTP score) | |||||
|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | Score | Prediction | Score | Prediction | |
| MSR v2 | 0.5931 | Mitochondria-probable | 0.23 | Mitochondria-possible | >0.083? no | Not Mitochondrial | 0.404 | Mitochondrial |
| MSR v1 | 0.3846 | Mitochondria-probable | 0.36 | Mitochondria-possible | >0.083? no | Not Mitochondrial | 0.268 | Other |
| MUT | 0.5959 | Mitochondria-probable | 0.71 | Mitochondrial | >0.083? yes | Mitochondrial | 0.768 | Mitochondrial |
While MSR does not target to mitochondria, the ability of MSR to take part in the adenosylation sequence suggests that a candidate in this role might be a homologous protein. Thus far, only four mammalian proteins are members of the family of dual flavin reductases: MSR, nitric oxide synthase (NOS), cytochrome P450 reductase (POR) and NADPH dependent diflavin oxidoreductase 1 (NDOR1). NOS has three major isoforms and one potential mitochondrial isoform, all of which have been well characterized as producers of NO and citrulline from l-arginine and oxygen [25]. Cytochrome P450 reductase is an endoplasmic reticulum-associated protein that is important in the metabolism of drugs and in steroid biosynthesis [26]. Interestingly, it has been shown, in combination with soluble cytochrome b5, to catalyze the reductive activation of MS in vitro [27]. Less is known about NDOR1; however, it has been shown to be a cytoplasmic protein that is suggested to play a role in the bioreduction of anti-cancer drugs [28], possibly by a mechanism that is modulated by the histidine triad protein DCS-1 [29]. It also can participate in the reductive methylation of MS in vitro [30]. Given the redundancy of functions displayed by this family of proteins in vitro, the ability of MSR to act as the reducing enzyme in conjunction with the adenyltransferase in vitro may be a further reflection of the biochemical breadth of these proteins. In this light, it is possible that the real mitochondrial reductase may be an as yet undiscovered member of this family.
An early clue that MSR was unlikely to be the mitochondrial cobalamin reductase was the absence of methylmalonic acidemia in cblE patients with defects in MSR [31; 32]. However, the absence of any patients at all with a block in the mitochondrial reduction of cobalamin is unexpected, given the occurrence of metabolic defects throughout this pathway including the several complementation groups associated with mitochondrial metabolism of cobalamin. It is conceivable that this is an essential step, incompatible with embryonic development, resulting in the absence of live-born infants with such a disorder. Alternatively, it is also possible that this vital step can be performed by multiple reductases, so that a defect in one of them would be made up by the others. However, such enzymes would almost certainly have to interact closely with MMAB to prevent re-oxidation of the highly reactive cobalt centre on cobalamin. This is a property of MSR [17]. Whether additional proteins with this capacity occur in human cells remains to be investigated.
Acknowledgments
We are grateful to Linda Ayer, Monica Narang and Roya Fattahie for assistance and Shannon Healy for critical discussion. These studies were supported by grants from the Canadian Institute for Health Research (CIHR) and NIH-National Heart, Lung and Blood Institute (NHLBI). Scholarship support to DSF and JZ was provided by the CIHR Training Grant in in Genetics, Child Health and Development.
Abbreviations
The abbreviations used are:
- MS
methionine synthase
- MSR
methionine synthase reductase
- MUT
methylmalonyl CoA-mutase
- MMAB
methylmalonic aciduria cblB type
- GFP
green fluorescent protein
- OHCbl, CNCbl, MeCbl, AdoCbl
hydroxo-, cyano-, methyl-, adenosylcobalamin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosenblatt DS, Fenton WA. Inherited disorders of folate and cobalamin transport and metabolism. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3897–3933. [Google Scholar]
- 2.Vassiliadis A, Rosenblatt DS, Cooper BA, Bergeron JJ. Lysosomal cobalamin accumulation in fibroblasts from a patient with an inborn error of cobalamin metabolism (cblF complementation group): visualization by electron microscope radioautography. Exp Cell Res. 1991;195:295–302. doi: 10.1016/0014-4827(91)90376-6. [DOI] [PubMed] [Google Scholar]
- 3.Lerner-Ellis JP, Tirone JC, Pawelek PD, Dore C, Atkinson JL, Watkins D, Morel CF, Fujiwara TM, Moras E, Hosack AR, Dunbar GV, Antonicka H, Forgetta V, Dobson CM, Leclerc D, Gravel RA, Shoubridge EA, Coulton JW, Lepage P, Rommens JM, Morgan K, Rosenblatt DS. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet. 2006;38:93–100. doi: 10.1038/ng1683. [DOI] [PubMed] [Google Scholar]
- 4.Suormala T, Baumgartner MR, Coelho D, Zavadakova P, Kozich V, Koch HG, Berghauser M, Wraith JE, Burlina A, Sewell A, Herwig J, Fowler B. The cblD defect causes either isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. J Biol Chem. 2004;279:42742–42749. doi: 10.1074/jbc.M407733200. [DOI] [PubMed] [Google Scholar]
- 5.Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dore C, Hudson T, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci USA. 2002;99:15554–15559. doi: 10.1073/pnas.242614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 7.Korotkova N, Lidstrom ME. MeaB is a component of the methylmalonyl-CoA mutase complex required for protection of the enzyme from inactivation. J Biol Chem. 2004;279:13652–13658. doi: 10.1074/jbc.M312852200. [DOI] [PubMed] [Google Scholar]
- 8.Padovani D, Labunska T, Banerjee R. Energetics of interaction between the G-protein chaperone, MeaB, and B12-dependent methylmalonyl-CoA mutase. J Biol Chem. 2006;281:17838–17844. doi: 10.1074/jbc.M600047200. [DOI] [PubMed] [Google Scholar]
- 9.Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Genet. 2002;11:3361–3369. doi: 10.1093/hmg/11.26.3361. [DOI] [PubMed] [Google Scholar]
- 10.Cooper BA, Rosenblatt DS, Watkins D. Methylmalonic aciduria due to a new defect in adenosylcobalamin accumulation by cells. Am J Hematol. 1990;34:115–120. doi: 10.1002/ajh.2830340207. [DOI] [PubMed] [Google Scholar]
- 11.Li YN, Gulati S, Baker PJ, Brody LC, Banerjee R, Kruger WD. Cloning, mapping and RNA analysis of the human methionine synthase gene. Hum Mol Genet. 1996;5:1851–1858. doi: 10.1093/hmg/5.12.1851. [DOI] [PubMed] [Google Scholar]
- 12.Leclerc D, Campeau E, Goyette P, Adjalla CE, Christensen B, Ross M, Eydoux P, Rosenblatt DS, Rozen R, Gravel RA. Human methionine synthase: cDNA cloning and identification of mutations in patients of the cblG complementation group of folate/cobalamin disorders. Hum Mol Genet. 1996;5:1867–1874. doi: 10.1093/hmg/5.12.1867. [DOI] [PubMed] [Google Scholar]
- 13.Chen LH, Liu ML, Hwang HY, Chen LS, Korenberg J, Shane B. Human methionine synthase. cDNA cloning, gene localization, and expression. J Biol Chem. 1997;272:3628–3634. [PubMed] [Google Scholar]
- 14.Leclerc D, Wilson A, Dumas R, Gafuik C, Song D, Watkins D, Heng HH, Rommens JM, Scherer SW, Rosenblatt DS, Gravel RA. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc Natl Acad Sci USA. 1998;95:3059–3064. doi: 10.1073/pnas.95.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada K, Gravel RA, Toraya T, Matthews RG. Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc Natl Acad Sci USA. 2006;103:9476–9481. doi: 10.1073/pnas.0603694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leclerc D, Odievre M, Wu Q, Wilson A, Huizenga JJ, Rozen R, Scherer SW, Gravel RA. Molecular cloning, expression and physical mapping of the human methionine synthase reductase gene. Gene. 1999;240:75–88. doi: 10.1016/s0378-1119(99)00431-x. [DOI] [PubMed] [Google Scholar]
- 17.Leal NA, Olteanu H, Banerjee R, Bobik TA. Human ATP:Cob(I)alamin adenosyltransferase and its interaction with methionine synthase reductase. J Biol Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 18.Huh N, Utakoji T. Production of HBs-antigen by two new human hepatoma cell lines and its enhancement by dexamethasone. Gann. 1981;72:178–179. [PubMed] [Google Scholar]
- 19.Isaya G, Fenton WA, Hendrick JP, Furtak K, Kalousek F, Rosenberg LE. Mitochondrial import and processing of mutant human ornithine transcarbamylase precursors in cultured cells. Mol Cell Biol. 1988;8:5150–5158. doi: 10.1128/mcb.8.12.5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narang MA, Dumas R, Ayer LM, Gravel RA. Reduced histone biotinylation in multiple carboxylase deficiency patients: a nuclear role for holocarboxylase synthetase. Hum Mol Genet. 2004;13:15–23. doi: 10.1093/hmg/ddh006. [DOI] [PubMed] [Google Scholar]
- 21.Schubert HL, Hill CP. Structure of ATP-Bound Human ATP:Cobalamin Adenosyltransferase. Biochemistry. 2006;45:15188–15196. doi: 10.1021/bi061396f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson A, Leclerc D, Rosenblatt DS, Gravel RA. Molecular basis for methionine synthase reductase deficiency in patients belonging to the cblE complementation group of disorders in folate/cobalamin metabolism. Hum Mol Genet. 1999;8:2009–2016. doi: 10.1093/hmg/8.11.2009. [DOI] [PubMed] [Google Scholar]
- 23.Olteanu H, Banerjee R. Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J Biol Chem. 2001;276:35558–35563. doi: 10.1074/jbc.M103707200. [DOI] [PubMed] [Google Scholar]
- 24.Wolthers KR, Scrutton NS. Protein interactions in the human methionine synthase-methionine synthase reductase complex and implications for the mechanism of enzyme reactivation. Biochemistry. 2007;46:6696–6709. doi: 10.1021/bi700339v. [DOI] [PubMed] [Google Scholar]
- 25.Knowles RG, Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298(Pt 2):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller WL, Huang N, Fluck CE, Pandey AV. P450 oxidoreductase deficiency. Lancet. 2004;364:1663. doi: 10.1016/S0140-6736(04)17344-3. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Banerjee R. Purification of soluble cytochrome b5 as a component of the reductive activation of porcine methionine synthase. J Biol Chem. 1998;273:26248–26255. doi: 10.1074/jbc.273.40.26248. [DOI] [PubMed] [Google Scholar]
- 28.Paine MJ, Garner AP, Powell D, Sibbald J, Sales M, Pratt N, Smith T, Tew DG, Wolf CR. Cloning and characterization of a novel human dual flavin reductase. J Biol Chem. 2000;275:1471–1478. doi: 10.1074/jbc.275.2.1471. [DOI] [PubMed] [Google Scholar]
- 29.Kwasnicka-Crawford DA, Vincent SR. Role of a novel dual flavin reductase (NR1) and an associated histidine triad protein (DCS-1) in menadione-induced cytotoxicity. Biochem Biophys Res Commun. 2005;336:565–571. doi: 10.1016/j.bbrc.2005.08.129. [DOI] [PubMed] [Google Scholar]
- 30.Olteanu H, Banerjee R. Redundancy in the pathway for redox regulation of mammalian methionine synthase: reductive activation by the dual flavoprotein, novel reductase 1. J Biol Chem. 2003;278:38310–38314. doi: 10.1074/jbc.M306282200. [DOI] [PubMed] [Google Scholar]
- 31.Harding CO, Arnold G, Barness LA, Wolff JA, Rosenblatt DS. Functional methionine synthase deficiency due to cblG disorder: a report of two patients and a review. Am J Med Genet. 1997;71:384–390. doi: 10.1002/(sici)1096-8628(19970905)71:4<384::aid-ajmg3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 32.Watkins D, Rosenblatt DS. Functional methionine synthase deficiency (cblE and cblG): clinical and biochemical heterogeneity. Am J Med Genet. 1989;34:427–434. doi: 10.1002/ajmg.1320340320. [DOI] [PubMed] [Google Scholar]
- 33.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholas KB, Nicholas HBJ, Deerfield DW., II GeneDoc: analysis and visualization of genetic variation. EMBNEW NEWS. 1997:14. [Google Scholar]