

Abstract
Staphylococcus aureus produces a golden carotenoid virulence factor called staphyloxanthin (STX), and we report here the inhibition of the enzyme, dehydrosqualene synthase (CrtM), responsible for the first committed step in STX biosynthesis. The most active compounds are halogen-substituted phosphonosulfonates, with Ki values as low as 5 nM against the enzyme and IC50 values for STX inhibition in S. aureus as low as 11 nM. There is, however, only a poor correlation (R2 = 0.27) between enzyme and cell pIC50 (= −log10 IC50) values. The ability to predict cell from enzyme data improves considerably (to R2 = 0.72) with addition of two more descriptors. We also investigated the activity of these compounds against human squalene synthase (SQS), as a counterscreen, finding several potent STX biosynthesis inhibitors with essentially no squalene synthase activity. These results open up the way to developing potent and selective inhibitors of an important virulence factor in S. aureus, a major human pathogen.
Introduction
Staphylococcus aureusis a major human pathogen, producing a wide spectrum of clinically significant hospital- and community-acquired infections. Methicillin-resistant strains of S. aureus (MRSA) have now reached epidemic proportions and pose a significant challenge to the public health. A recent CDC study has shown that more people in the U.S. die from invasive MRSA each year than do from HIV/AIDS.1,2 There is, therefore, an urgent need to find new therapies. One unconventional approach to anti-infective therapy involves blocking bacterial virulence factors,3 a potential benefit of this strategy being that without the “life or death” selective pressure exerted by classical antibiotics, bacteria may be less prone to develop drug resistance.
An important virulence factor of S. aureus is the golden carotenoid pigment, staphyloxanthin (STXa), whose numerous double bonds can react with, and thus deactivate, the reactive oxygen species (ROS) generated by neutrophils and macrophages, making S. aureus resistant to innate immune clearance.4,5 STX has been shown to be essential for infectivity: bacteria that lack staphyloxanthin are nonpigmented, are susceptible to neutrophil killing, and fail to produce disease in mouse skin and systemic infection models.4,6 STX biosynthesis is thus a novel target for preventing or treating S. aureus infections. The first committed step in STX biosynthesis is catalyzed by the enzyme dehydrosqualene synthase, also called diapophytoene synthase or CrtM, and involves the head-to-head condensation of two molecules of farnesyl diphosphate (FPP) to produce the C30 species, presqualene diphosphate, which is then converted to dehydrosqualene (Figure 1A).5 Since this condensation is remarkably similar to the first step in mammalian cholesterol biosynthesis (Figure 1B), we reasoned that known squalene synthase inhibitors, developed in the context of cholesterol-lowering therapy, might also inhibit dehydrosqualene synthase. This turns out to be the case, and we recently reported that phosphonosulfonates such as 1 (BPH-652 or rac-BMS-187745), developed by Bristol-Myers Squibb and advanced through phase I/II human clinical trials,7,8 potently inhibit S. aureus CrtM, as well as STX biosynthesis in the bacterium.6 Upon treatment with 1, the resulting nonpigmented S. aureus are much more susceptible to killing by hydrogen peroxide and are less able to survive in freshly isolated human whole blood than are normally pigmented S. aureus. Moreover, in an in vivo systemic S. aureus infection model, the bacterial counts in kidneys of mice treated with 1 were reduced by 98%, compared to those of a control group. These results show that 1 represents a novel lead compound for virulence factor-based therapy of S. aureus infection. Here, we report the synthesis and testing of a library of 38 phosphonosulfonates and related bisphosphonates against CrtM, against STX biosynthesis in S. aureus, and as a counterscreen, against an expressed human squalene synthase. We report both qualitative and quantitative (QSAR, quantitative structure—activity relationships) results for CrtM and STX biosynthesis inhibition, as well as CrtM/SQS selectivity. In addition, we investigate how cell activity can be predicted from enzyme (CrtM) inhibition results, opening the way to the further development of potent and selective inhibitors of S. aureus virulence.
Figure 1.

(A) Pathway for staphyloxanthin biosynthesis (in S. aureus). (B) Pathway for cholesterol biosynthesis. Each biosynthetic pathway is thought to involve initial formation of presqualene diphosphate, catalyzed by CrtM (S. aureus) or squalene synthase. In the bacterium, the NADPH reduction step is absent, resulting in production of dehydrosqualene.
Results and Discussion
Qualitative Structure—Activity Relationships in CrtM Inhibition
The inhibitor class we investigate here originated in early research aimed at developing human squalene synthase inhibitors as cholesterol lowering drugs. Early SQS inhibitors were based on the structure of FPP, the substrate for SQS, with the labile diphosphate group being replaced by a bisphosphonate. However, isoprenyl side chains were metabolized in vivo. This could be overcome by incorporation of a biphenyl isostere (e.g., 2),9 but these compounds potently bound to bone, in addition to causing elevation in liver enzymes. Phosphonosulfonates, on the other hand, bound only weakly to bone, and the diphenyl ether phosphonosulfonates (such as 1) were not broken down and did not cause liver function abnormalities.10 Plus, in our previous work we found that 1 had promising activity against CrtM as well as cellular  and in vivo activity.6 In the present study, we sought to develop more potent CrtM inhibitors with improved activity in S. aureus cells that, at the same time, have poor activity against human SQS, reducing formation of the 1,10-dioic acid FPP metabolite that is formed as a result of SQS inhibition. We first synthesized a small library of five diphenyl ether phosphonosulfonates, five biphenyl phosphonosulfonates, and three biphenyl bisphosphonates, based in part on the types of compound tested previously as SQS inhibitors, and examined them for their activity against CrtM. The structures and IC50 values (in parentheses) in CrtM inhibition of these compounds are shown in Figure 2. Bisphosphonates (2 and 3) are the most potent CrtM inhibitors (IC50 = 0.5 and 0.2 μM; Ki ≈ 1 and 0.5 nM), being an order of magnitude more active than any of the phosphonosulfonates. However, these compounds were found to have only modest activity in inhibition of STX biosynthesis in S. aureus (see below), due perhaps to poor cell uptake. Compound 4 (IC50 = 5.4 μM), an analogous phosphinophosphonate compound, was ∼10× less active than was 2, showing similar activity in CrtM inhibition as the phosphonosulfonates, which are likely to have the same formal charge. For the phosphonosulfonates with diphenyl ether side chains, 5 and 6 (which contain para-substituted fluoro and n-propyl groups, respectively) were ∼4× as active (IC50 = 2.3 and 2.2 μM; Ki ≈ 5 nM) as 1. Compound 7 (IC50 = 7.2 μM), containing a p-trifluoromethyl substituent, showed activity similar to that found with 1, while 8, which contains an o-benzyl group, was ∼5× less active than 1 (IC50 = 37.2 μM). The phosphonosulfonates containing biphenyl side chains (9−13) had, in general, significantly reduced activities.
and in vivo activity.6 In the present study, we sought to develop more potent CrtM inhibitors with improved activity in S. aureus cells that, at the same time, have poor activity against human SQS, reducing formation of the 1,10-dioic acid FPP metabolite that is formed as a result of SQS inhibition. We first synthesized a small library of five diphenyl ether phosphonosulfonates, five biphenyl phosphonosulfonates, and three biphenyl bisphosphonates, based in part on the types of compound tested previously as SQS inhibitors, and examined them for their activity against CrtM. The structures and IC50 values (in parentheses) in CrtM inhibition of these compounds are shown in Figure 2. Bisphosphonates (2 and 3) are the most potent CrtM inhibitors (IC50 = 0.5 and 0.2 μM; Ki ≈ 1 and 0.5 nM), being an order of magnitude more active than any of the phosphonosulfonates. However, these compounds were found to have only modest activity in inhibition of STX biosynthesis in S. aureus (see below), due perhaps to poor cell uptake. Compound 4 (IC50 = 5.4 μM), an analogous phosphinophosphonate compound, was ∼10× less active than was 2, showing similar activity in CrtM inhibition as the phosphonosulfonates, which are likely to have the same formal charge. For the phosphonosulfonates with diphenyl ether side chains, 5 and 6 (which contain para-substituted fluoro and n-propyl groups, respectively) were ∼4× as active (IC50 = 2.3 and 2.2 μM; Ki ≈ 5 nM) as 1. Compound 7 (IC50 = 7.2 μM), containing a p-trifluoromethyl substituent, showed activity similar to that found with 1, while 8, which contains an o-benzyl group, was ∼5× less active than 1 (IC50 = 37.2 μM). The phosphonosulfonates containing biphenyl side chains (9−13) had, in general, significantly reduced activities.
Figure 2.

Structures of phosphonosulfonate and bisphosphonate compounds and their activities (IC50 values, μM, in parentheses) against dehydrosqualene synthase.
These results, together with the bacterial cell-based results discussed below, are of interest because they show that the biphenyl bisphosphonates are the most potent CrtM inhibitors, with IC50 values of <1 μM. They do, however, have poor cell-based activities, with IC50 > 1 μM, and consequently were not selected for further development. The biphenyl phosphonosulfonates, on the other hand, had generally poor activity against CrtM (average IC50 for the five compounds investigated of ∼50 μM), making them also less attractive candidates for development. However, the diphenyl ether phosphonosulfonates had, on average, an IC50 value of ∼11 μM (or a Ki of ∼30 nM), and these compounds were very potent in cell based assays (i.e., in inhibiting STX biosynthesis by S. aureus), as discussed in detail below, and were thus selected for further development.
To see if major improvements in CrtM activity might be obtained by modifying the phosphonosulfonate side chain, we next synthesized the eight analogues of 1, shown in Figure 3, in which we modified the side chain heteroatom (O → NH, CH2, and a carbazole), the position of the heteroatom (meta → para), and the length of the alkyl side chain (N = 1, 2, and 3 CH2 groups), and we synthesized the (R)- and (S)-enantiomers of 1, since previously the S-form was found to be far more potent than the (R)-form in inhibiting SQS.11 The optically active (S)-1 (IC50 = 1.4 μM) was ∼30× more active than its (R)-enantiomer in CrtM inhibition (Figure 3). Shorter linkers between the aromatic ring and phosphonosulfonate headgroup, as found in 14 and 15, led to essentially no CrtM activity (IC50 > 500 μM). As for 16 (IC50 = 43.7 μM) and 17 (IC50 = 20.4 μM), replacing the bridging —O— atoms with —NH— and —CH2—, respectively, resulted in reduced CrtM inhibition activity. The 4-phenoxyphenyl analogue (18) was slightly more active than its 3-phenoxyphenyl counterpart (1) in CrtM inhibition but was less active in the cell based assay, while compound 19, a fused tricyclic analogue, had very poor activity against CrtM (IC50 = 170 μM). These results, together with those discussed above, clearly indicated that investigating additional diphenyl ether phosphonosulfonates would be of interest.
Figure 3.

Structures of compounds and their activities (in parentheses) against dehydrosqualene synthase.
We therefore next synthesized 17 more substituted phosphonosulfonates whose structures and activities in CrtM inhibition are shown in Table 1, together with the previously described results for 1 and 5−7. The following structure—activity relationship features can be seen from these results: (1) Diphenyl ether phosphonosulfonates substituted with F or CF3 had diminished activity when the substituent was in the 2′-position relative to the corresponding 3′- or 4′-substituted analogues. (2) Halogen-containing groups, including F, CF3, and Cl, generally enhance activity. (3) Alkyl groups with various shapes and sizes located at the 4′-position result in modest activity changes (when compared to 1), while oxygen-containing groups at the same position significantly decrease activity. (4) Dihalogen substituted compounds are more active than the parent compound 1. (5) Substitution at the 6-position with F has little effect on activity, but incorporation of an OMe group in this location abrogates most activity. Clearly, the above represents a complex set of empirical observations that require a more quantitative analysis.
Table 1.
IC50 Values of Diphenyl Ether Phosphonosulfonates in CrtM Inhibition
 | ||||
|---|---|---|---|---|
| Entry | Compound | R1 | R2 | CrtM IC50 values (μM) |
| 1 | 1 | H | H | 7.9 |
| 2 | 5 | 4′-F | H | 2.3 |
| 3 | 20 | 3′-F | H | 2.2 |
| 4 | 21 | 2′-F | H | 6.9 |
| 5 | 7 | 4′-CF3 | H | 7.2 |
| 6 | 22 | 3′-CF3 | H | 9.8 |
| 7 | 23 | 2′-CF3 | H | 53.7 |
| 8 | 24 | 4′-Cl | H | 2.5 |
| 9 | 6 | 4′-n-propyl | H | 2.2 |
| 10 | 25 | 4′-tert-butyl | H | 10.5 |
| 11 | 26 | 4′-CH2Ph | H | 13.5 |
| 12 | 27 | 4′-OH | H | 28.2 |
| 13 | 28 | 4′-OPh | H | 169.8 |
| 14 | 29 | 4′-(furan-2-yl) | H | 26.3 |
| 15 | 30 | 3′, 4′-2F | H | 1.7 |
| 16 | 31 | 3′, 4′-2Cl | H | 2.2 |
| 17 | 32 | 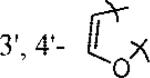 |
H | 2.1 |
| 18 | 33 | 3′, 5′-2F | H | 4.6 |
| 19 | 34 | 3′, 5′-2Cl | H | 6.3 |
| 20 | 35 | 4′-F | 6-F | 2.1 |
| 21 | 36 | 4′-F | 6-OCH3 | 81.3 |
Quantitative Structure—Activity Relationships in CrtM Inhibition
To investigate the structure—activity relationships outlined above, we carried out a comparative molecular similarity indices analysis (CoMSIA)12 of the CrtM inhibition activities of these phosphonosulfonate compounds. All molecules were constructed and minimized using the MMFF94x force field in the program MOE.13 Compounds were then aligned using the flexible alignment module14 in MOE, which perceives common features within the molecules (e.g., similar partial charge, H-bond acceptor, aromaticity, hydrophobicity, etc.). We used fully deprotonated phosphonosulfonate headgroups, based on the observation that 1 binds two Mg2+ in the CrtM active site6 and our previous NMR and quantum chemical studies on bisphosphonates, which indicate deprotonation when bisphosphonates bind to Mg2+.15 All compounds were constructed with the (S)-configuration, based on the observation that (S)-1 is far more active than is its (R)-enantiomer in CrtM inhibition (as also found in SQS inhibition10). The aligned compounds were exported into Sybyl.16 Then, we used a partial least-squares (PLS) method to regress the CrtM inhibitory activity and CoMSIA field data. The 3D QSAR model yielded r2 = 0.98, q2 (number of components) = 0.72 (7), Ftest = 245.8, and a pIC50 error of 0.12, as shown in Table 2. To further validate the model, we performed five leave-five-out training/test sets, obtaining on average a factor of 1.8× error between predicted and experimental IC50 values (Table 2). The compound alignment and the CoMSIA fields are shown in Figure 4. There is a relatively large hydrophobic contribution (46.6%) to the CoMSIA model, followed by steric (25.2%), electrostatic (19.9%), and H-bond donor (8.3%) interactions. The CoMSIA results are in good agreement with the more qualitative experimental observations: steric and hydrophobic-favorable areas (in green and magenta, respectively, in Figure 4B,C) at the 3′- and 4′-positions correspond to generally increased activities of 1 analogues with para- and meta-substituents on the distal phenyl ring. Two large steric penalty areas (in red in Figure 4B) at the 4- and 6- positions of the proximal phenyl ring account for the decreased activity of the biphenyl phosphonosulfonates (9−13 in Figure 2) and 36 (entry 21 in Table 1). In addition, a positive-charge-favored region (in blue, shown in Figure 4D) within the distal phenyl ring helps to account for the enhanced activity of the (electron-withdrawing) halogen containing phosphonosulfonates (e.g., 5, 24, and 30). The negative-charge favored area (in yellow, Figure 4D) obviously correlates with the activity of the bisphosphonates (2 and 3) in CrtM inhibition, since the sulfonate group is replaced by the more negatively charged phosphonate group.
Table 2.
CoMSIA Results for CrtM Inhibition
| CoMSIA pIC50 predictions |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| CrtM enzyme experimental activity |
test setsb |
||||||||
| compda | IC50 (μM) | pIC50 (M) | training set | residual | 1 | 2 | 3 | 4 | 5 |
| 3 | 0.18 | 6.74 | 6.61 | 0.13 | 6.54 | 6.53 | 6.53 | 6.49 | 6.58 |
| 2 | 0.48 | 6.32 | 6.55 | −0.23 | 6.35 | 6.41 | 6.19 | 6.29 | 6.49 |
| 30 | 1.7 | 5.76 | 5.62 | 0.14 | 5.61 | 5.60 | 5.56 | 5.55 | 5.70 |
| 35 | 2.1 | 5.68 | 5.69 | −0.01 | 5.69 | 5.41 | 5.51 | 5.58 | 5.76 |
| 32 | 2.1 | 5.67 | 5.69 | −0.02 | 5.65 | 5.46 | 5.44 | 5.54 | 5.49 |
| 6 | 2.2 | 5.66 | 5.64 | 0.02 | 5.82 | 5.12 | 5.60 | 5.63 | 5.61 |
| 20 | 2.2 | 5.66 | 5.34 | 0.32 | 5.33 | 5.32 | 5.29 | 5.25 | 5.41 |
| 31 | 2.2 | 5.65 | 5.62 | 0.03 | 5.77 | 5.90 | 5.97 | 5.88 | 5.69 |
| 5 | 2.3 | 5.63 | 5.44 | 0.19 | 5.45 | 5.34 | 5.32 | 5.37 | 5.48 |
| 24 | 2.5 | 5.6 | 5.46 | 0.14 | 5.61 | 5.49 | 5.54 | 5.52 | 5.51 |
| 33 | 4.6 | 5.34 | 5.37 | −0.03 | 5.35 | 5.38 | 5.36 | 5.28 | 5.44 |
| 4 | 5.4 | 5.27 | 5.21 | 0.06 | 5.09 | 5.13 | 4.87 | 5.06 | 5.23 |
| 18 | 5.8 | 5.24 | 5.34 | −0.10 | 5.28 | 5.10 | 5.20 | 5.22 | 5.32 |
| 34 | 6.3 | 5.2 | 5.21 | −0.01 | 5.24 | 5.47 | 5.54 | 5.40 | 5.23 |
| 21 | 6.9 | 5.16 | 5.18 | −0.02 | 5.16 | 5.01 | 5.03 | 5.04 | 5.25 |
| 7 | 7.2 | 5.14 | 5.30 | −0.16 | 5.59 | 5.33 | 5.51 | 5.47 | 5.32 |
| 1 | 7.9 | 5.1 | 5.15 | −0.05 | 5.15 | 5.05 | 5.03 | 5.05 | 5.18 |
| 10 | 9.1 | 5.04 | 4.92 | 0.12 | 4.94 | 4.99 | 5.04 | 4.94 | 4.88 |
| 22 | 9.8 | 5.01 | 5.05 | −0.04 | 5.23 | 5.28 | 5.32 | 5.43 | 5.05 |
| 25 | 10.5 | 4.98 | 5.10 | −0.12 | 5.37 | 4.74 | 5.18 | 5.15 | 4.95 |
| 26 | 13.5 | 4.87 | 4.91 | −0.04 | 4.86 | 5.03 | 4.67 | 4.69 | 4.83 |
| 17 | 20.4 | 4.69 | 4.72 | −0.02 | 4.89 | 4.81 | 4.82 | 4.81 | 4.67 |
| 9 | 21.9 | 4.66 | 4.73 | −0.07 | 4.79 | 4.79 | 4.85 | 4.77 | 4.68 |
| 11 | 24.5 | 4.61 | 4.75 | −0.14 | 4.89 | 4.80 | 5.03 | 4.82 | 4.65 |
| 29 | 26.3 | 4.58 | 4.71 | −0.13 | 4.81 | 4.44 | 4.66 | 4.57 | 4.61 |
| 27 | 28.2 | 4.55 | 4.55 | 0.00 | 4.55 | 4.55 | 4.55 | 4.55 | 4.55 |
| 8 | 37.2 | 4.43 | 4.43 | 0.00 | 4.35 | 4.31 | 4.27 | 4.34 | 4.68 |
| 16 | 43.7 | 4.36 | 4.36 | 0.00 | 4.36 | 4.36 | 4.80 | 4.36 | 4.36 |
| 23 | 53.7 | 4.27 | 4.23 | 0.04 | 4.28 | 4.37 | 4.32 | 4.33 | 4.24 |
| 12 | 72.4 | 4.14 | 4.15 | −0.01 | 4.10 | 3.72 | 4.06 | 3.89 | 4.08 |
| 36 | 81.3 | 4.09 | 4.16 | −0.07 | 4.28 | 4.48 | 4.46 | 4.50 | 4.15 |
| 13 | 123 | 3.91 | 3.86 | 0.05 | 3.91 | 4.16 | 4.08 | 4.12 | 3.84 |
| 19 | 170 | 3.77 | 3.79 | −0.02 | 3.91 | 4.09 | 3.92 | 4.07 | 3.70 |
| 28 | 170 | 3.77 | 3.80 | −0.03 | 3.90 | 3.85 | 3.96 | 4.20 | 3.82 |
| 15 | 500 | 3.3 | 3.33 | −0.03 | 3.24 | 3.25 | 3.12 | 3.20 | 3.40 |
| 14 | 1700 | 2.77 | 2.65 | 0.12 | 2.53 | 2.65 | 2.50 | 2.56 | 2.71 |
| r2 | 0.98 | 0.97 | 0.94 | 0.95 | 0.95 | 0.99 | |||
| q2 | 0.72 | 0.69 | 0.71 | 0.72 | 0.67 | 0.70 | |||
| Ftest | 245.8 | 151.6 | 82.4 | 113.9 | 97.5 | 280.1 | |||
| N | 7 | 6 | 5 | 4 | 5 | 7 | |||
| n | 36 | 31 | 31 | 31 | 31 | 31 | |||
Figure 4.

Compound alignment and CoMSIA fields for CrtM inhibition: (A) flexible alignment of compounds, with only heavy atoms displayed for clarity; (B) steric fields (green, favorable; red, disfavored); (C) hydrophobic fields (magenta, favorable; white, disfavored); (D) electrostatic and H-bond donor fields (blue, positive-charge favorable; yellow, negative-charge favorable; cyan, H-bond donor disfavored).
Staphyloxanthin Biosynthesis Inhibition
The results described above are of interest since they indicate that dehydrosqualene (CrtM) can be potently inhibited by phosphonosulfonates, and by bisphosphonates. The question next arises: how potent are these compounds in inhibiting STX biosynthesis in S. aureus? We thus treated S. aureus bacteria with serially diluted compounds at 37 °C for 3 days, after which the STX pigment was extracted with methanol. Optical densities were measured at 450 nm, and IC50 values for inhibition of pigment formation for each compound were calculated using a standard dose—response curve. The rank ordered IC50 values of the 38 compounds investigated are shown in Table 3, and dose—response curves of representative compounds are shown in the Supporting Information (Figure S1). Surprisingly, we found that STX biosynthesis inhibition in S. aureus was poorly correlated with CrtM (enzyme) inhibition, with R2 = 0.27 for the CrtM/STX inhibition pIC50 values, as shown in Figure 5A.
Table 3.
Staphyloxanthin Inhibition in S. aureus
| compda | staphyloxanthin IC50 (μM) | compda | staphyloxanthin IC50 (μM) |
|---|---|---|---|
| 31 | 0.011 | 11 | 0.18 |
| 33 | 0.015 | 10 | 0.18 |
| 32 | 0.015 | 18 | 0.19 |
| 8 | 0.019 | 20 | 0.22 |
| 25 | 0.021 | (R)-1 | 0.31 |
| 24 | 0.039 | 9 | 0.35 |
| 7 | 0.040 | 23 | 0.46 |
| 26 | 0.049 | 13 | 0.49 |
| (S)-1 | 0.050 | 16 | 0.50 |
| 34 | 0.051 | 21 | 0.51 |
| 5 | 0.052 | 3 | 1.1 |
| 35 | 0.059 | 29 | 1.1 |
| 22 | 0.074 | 4 | 1.4 |
| 6 | 0.093 | 2 | 2.2 |
| 28 | 0.10 | 27 | 6.8 |
| 1 | 0.11 | 36 | 13.8 |
| 30 | 0.11 | 15 | 20.4 |
| 12 | 0.16 | 14 | 33.9 |
| 17 | 0.17 | 19 | 263 |
Figure 5.

(A) Correlation between staphyloxanthin (cell) activity and CrtM (enzyme) activity, showing R2 = 0.27. (B) Improved correlation between cell activity and enzyme activity with incorporation of a molecular descriptor, SlogP, with R2 = 0.60. (C) Best cell activity prediction obtained by using CrtM inhibition plus two additional descriptors; R2 = 0.72 and Q2 = 0.62. (D) correlation between CrtM activity and human SQS activity, showing R2 = 0.50.
Presumably, this poor correlation is due to the fact that no consideration of cell permeability or drug uptake is involved in the in vitro enzyme inhibition data. To try to take this into account, we therefore chose to use SlogP (the logarithm of the octanol/water partition coefficient17) to describe this effect by using the following equation:
| (1) |
where a, b, and c are regression coefficients from a linear regression analysis. This yielded R2 = 0.60 for the experimental-versus-predicted pIC50 values (Figure 5B) or R2 = 0.53 for a leave-two-out (L2O) prediction test set, to be compared with R2 = 0.16 for a L2O test set of predictions using solely the enzyme pIC50 results. Of course, there is no a priori reason to use SlogP as the extra descriptor or to limit the method to use of a single extra descriptor. So we next used a three descriptor model:
| (2) |
where B and C are all possible descriptor pairs available in MOE that have non-Boolean values (i.e., the properties do not contain 0's or 1's). The top 10 “enzyme plus two descriptor” search results are shown in the Supporting Information (Table S1), rank-ordered by R2 value. Each of the top 10 results contains pIC50 (CrtM) as the major descriptor, and the R2 value obtained for the top solution, R2 = 0.72, is clearly an improvement over that obtained using CrtM, or CrtM and SlogP results (Figure 5C). We then used a leave-two-out method to produce a test set result in which we recomputed all training sets minus the two compounds of interest, then used the coefficients and descriptors to predict the two omitted compounds. In this way, the activity of each compound was predicted 37 times. The R2 in this leave-two-out test set was 0.62, a major improvement over the R2 = 0.16 using solely enzyme inhibition data (and the same leave-two-out test set approach). To verify that this predictivity did not occur by chance, we repeated the leave-two-out process, using scrambled cell activity data. This process was repeated 10 times, with the average R2 values (leave two out, scrambled) being 0.10 (Supporting Information Table S1). Clearly, then, phosphonosulfonates are potent inhibitors of the CrtM enzyme, and their activity can be relatively well predicted by using the combinatorial descriptor search method, even when the cell/enzyme data are very poorly correlated. Moreover, as might be expected on the basis of the enzyme inhibition results (Figure 3), (S)-1 is far more active than is (R)-1 in cells (Table 3).
Selectivity of CrtM Inhibition
Since phosphonosulfonates, such as 1, were originally developed as inhibitors of squalene synthase in the context of cholesterol-lowering therapy,7,8,10 we were also interested to see how they inhibit human SQS, since SQS inhibition results in formation of a 1,10-dioic acid metabolite (from unused FPP). We thus screened each of the compounds described above for their activity against an expressed human SQS enzyme. Results are shown in Table 4. We find that CrtM inhibition is moderately correlated with human SQS inhibition, R2 = 0.50, as shown in Figure 5D, consistent with the modest sequence homology of S. aureus CrtM and human SQS (30% identity, 36% similarity).6 Of course, this modest correlation reflects something that is potentially beneficial: that some good CrtM inhibitors are poor hSQS inhibitors. Consider, for example, the 4′-n-propyl species 6 (Table 1). This compound has a 2.2 μM IC50 versus CrtM and a 1.9 μM IC50 versus hSQS, Table 4, and is therefore a much poorer hSQS inhibitor than is the parent compound 1. For compound 1 (used previously in vivo6), the CrtM IC50 is 7.9 μM but 1 is a very potent hSQS inhibitor with an IC50 of 23 nM, Table 4. The results shown in Table 4 are rank-ordered in terms of selectivity for CrtM over hSQS inhibition or IC50(hSQS)/IC50(CrtM) such that a larger number means more CrtM selectivity, and are also given in terms of selectivity relative to 1, which is (selectivity of compound)/(selectivity of 1). The compound with the highest relative selectivity is thus 6, which is (1.9/2.2) ÷ (0.023/7.9) or ∼300× more selective a CrtM inhibitor than is 1. Remarkably, both 1 and 6 have, however, very similar IC50 values for STX biosynthesis (Table 3): 110 nM for 1 and 93 nM for 6. These results strongly suggest that it is possible to make potent inhibitors of STX biosynthesis (such as 6) that have much better relative selectivity in inhibiting CrtM than does 1.
Table 4.
CoMSIA Results for CrtM/hSQS Selectivity
| CoMSIA predictionsd |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| experimental data |
test sets |
||||||||||
| compda | hSQS IC50 (μM) |
CrtM/hSQS selectivityb |
relative CrtM/hSQS selectivityc |
pIC50(CrtM) – pIC50(hSQS) (M) |
training set |
residual | 1 | 2 | 3 | 4 | 5 |
| 6 | 1.9 | 0.85 | 295 | −0.07 | −0.16 | 0.09 | −0.17 | −0.18 | −0.18 | −0.16 | −0.17 |
| 25 | 4.0 | 0.38 | 132 | −0.42 | −0.38 | −0.04 | −0.28 | −0.26 | −0.31 | −0.43 | −0.27 |
| 19 | 11.7 | 0.068 | 24.0 | −1.16 | −0.99 | −0.17 | −1.13 | −1.15 | −1.17 | −0.97 | −1.13 |
| 26 | 0.72 | 0.054 | 18.6 | −1.27 | −1.29 | 0.02 | −1.41 | −1.40 | −1.43 | −1.29 | −1.42 |
| 7 | 0.17 | 0.024 | 8.3 | −1.62 | −1.52 | −0.10 | −1.51 | −1.49 | −1.53 | −1.52 | −1.51 |
| 10 | 0.22 | 0.024 | 8.3 | −1.62 | −1.52 | −0.10 | −1.60 | −1.45 | −1.60 | −1.51 | −1.89 |
| 29 | 0.54 | 0.021 | 7.1 | −1.69 | −1.70 | 0.01 | −1.75 | −1.73 | −1.77 | −1.71 | −1.71 |
| 30 | 0.033 | 0.019 | 6.6 | −1.72 | −2.12 | 0.40 | −2.22 | −2.21 | −2.38 | −2.09 | −2.22 |
| 27 | 0.47 | 0.016 | 5.8 | −1.78 | −2.34 | 0.56 | −2.37 | −2.39 | −2.44 | −2.34 | −2.40 |
| 9 | 0.35 | 0.016 | 5.6 | −1.79 | −1.98 | 0.19 | −2.01 | −1.81 | −1.90 | −2.00 | −2.02 |
| 13 | 1.8 | 0.015 | 5.1 | −1.83 | −1.93 | 0.10 | −2.04 | −2.01 | −2.00 | −1.90 | −2.02 |
| 3 | 0.003 | 0.014 | 4.8 | −1.86 | −2.00 | 0.14 | −1.84 | −1.80 | −1.82 | −1.99 | −1.88 |
| 4 | 0.072 | 0.014 | 4.7 | −1.87 | −1.93 | 0.06 | −1.74 | −1.71 | −1.75 | −1.97 | −1.70 |
| 11 | 0.29 | 0.012 | 4.1 | −1.93 | −1.64 | −0.29 | −1.62 | −1.39 | −1.50 | −1.63 | −1.64 |
| 36 | 0.79 | 0.0098 | 3.4 | −2.01 | −1.88 | −0.13 | −1.97 | −1.99 | −2.00 | −1.86 | −1.95 |
| 24 | 0.020 | 0.0079 | 2.8 | −2.10 | −2.15 | 0.05 | −2.12 | −2.12 | −2.18 | −2.09 | −2.13 |
| 18 | 0.042 | 0.0072 | 2.5 | −2.14 | −2.23 | 0.09 | −2.30 | −2.32 | −2.30 | −2.27 | −2.38 |
| 5 | 0.017 | 0.0071 | 2.5 | −2.15 | −2.29 | 0.14 | −2.32 | −2.33 | −2.42 | −2.29 | −2.34 |
| 33 | 0.032 | 0.0071 | 2.5 | −2.15 | −2.20 | 0.05 | −2.33 | −2.28 | −2.34 | −2.19 | −2.30 |
| 22 | 0.069 | 0.0071 | 2.5 | −2.15 | −2.31 | 0.16 | −2.31 | −2.31 | −2.29 | −2.47 | −2.30 |
| 28 | 1.1 | 0.0065 | 2.2 | −2.19 | −2.12 | −0.07 | −2.15 | −2.16 | −2.16 | −2.12 | −2.14 |
| 31 | 0.014 | 0.0062 | 2.1 | −2.21 | −2.08 | −0.13 | −2.08 | −2.06 | −2.16 | −1.99 | −2.05 |
| 20 | 0.008 | 0.0036 | 1.3 | −2.44 | −2.31 | −0.13 | −2.38 | −2.35 | −2.45 | −2.31 | −2.34 |
| 17 | 0.069 | 0.0034 | 1.2 | −2.47 | −2.63 | 0.16 | −2.54 | −2.47 | −2.47 | −2.55 | −2.56 |
| 23 | 0.18 | 0.0033 | 1.1 | −2.48 | −2.48 | 0.00 | −2.47 | −2.49 | −2.47 | −2.53 | −2.87 |
| 34 | 0.020 | 0.0032 | 1.1 | −2.49 | −2.43 | −0.06 | −2.47 | −2.40 | −2.34 | −2.41 | −2.40 |
| 1 | 0.023 | 0.0029 | 1.0 | −2.54 | −2.50 | −0.04 | −2.50 | −2.49 | −2.50 | −2.52 | −2.48 |
| 14 | 3.7 | 0.0022 | 0.8 | −2.66 | −2.50 | −0.16 | −2.56 | −2.58 | −2.63 | −2.51 | −2.52 |
| 2 | 0.001 | 0.0019 | 0.6 | −2.73 | −2.84 | 0.11 | −2.66 | −2.62 | −2.62 | −2.90 | −2.60 |
| 35 | 0.004 | 0.0018 | 0.6 | −2.74 | −2.65 | −0.09 | −2.62 | −2.62 | −2.67 | −2.66 | −2.61 |
| 16 | 0.079 | 0.0018 | 0.6 | −2.74 | −2.68 | −0.06 | −2.78 | −2.83 | −2.77 | −2.72 | −2.82 |
| 12 | 0.13 | 0.0018 | 0.6 | −2.74 | −2.70 | −0.04 | −2.74 | −2.72 | −2.76 | −2.68 | −2.74 |
| 32 | 0.002 | 0.0011 | 0.4 | −2.96 | −2.60 | −0.36 | −2.51 | −2.53 | −2.55 | −2.66 | −2.55 |
| 15 | 0.55 | 0.0011 | 0.4 | −2.96 | −2.92 | −0.04 | −2.91 | −2.96 | −2.96 | −2.93 | −2.89 |
| 21 | 0.005 | 0.00073 | 0.3 | −3.14 | −2.78 | −0.36 | −2.75 | −2.74 | −2.77 | −2.81 | −2.74 |
| 8 | 0.003 | 7.1 × 10−5 | 0.02 | −4.15 | −4.16 | 0.01 | −4.17 | −4.16 | −4.16 | −4.18 | −4.18 |
| r2 | 0.94 | 0.93 | 0.93 | 0.94 | 0.95 | 0.92 | |||||
| q2 | 0.54 | 0.50 | 0.52 | 0.51 | 0.55 | 0.51 | |||||
| Ftest | 76.5 | 71.6 | 75.6 | 82.5 | 75.0 | 64.5 | |||||
| N | 6 | 5 | 5 | 5 | 6 | 5 | |||||
| n | 36 | 33 | 33 | 33 | 33 | 33 | |||||
To put these observations on a more structural basis, we carried out a QSAR/CoMSIA analysis, using the relative selectivity results. Using the same compound alignment as shown in Figure 4A, we obtained r2 = 0.94, q2 = 0.54, Ftest = 76.5, and a pIC50 error of 0.20, as shown in Table 4. We also validated this model by using five leave-three-out training/test sets, finding on average a factor of 1.8× error between predicted and experimental IC50 values (Table 4). There is a large hydrophobic contribution (54.8%) to the CoMSIA field results, followed by steric (29.5%) and electrostatic (15.7%) fields. The CoMSIA fields are shown in Figure 6, and it is clear that overlapping steric (green, Figure 6A) and hydrophobic (magenta, Figure 6B) field features near the 4′-position are favored for CrtM selectivity. Hydrophobic (white, Figure 6B) and steric (red, Figure 6A) disfavored regions are located near the 2′- and 3′-positions, respectively, and are responsible for the poor selectivity of compounds such as 8 and 21.
Figure 6.

CoMSIA fields for CrtM/SQS selectivity: (A) steric fields (green, favorable; red, disfavored); (B) hydrophobic fields (magenta, favorable; white, disfavored); (C) electrostatic fields (blue, positive-charge favorable).
Conclusions
The results described above are of interest for a number of reasons. First, we made 38 phosphonosulfonate and bisphosphonate compounds and investigated their activity in inhibiting S. aureus dehydrosqualene synthase (CrtM), the enzyme involved in the first committed step in the biosynthesis of the virulence factor, STX, in S. aureus. The most active compounds were bisphosphonates, but they had poor activity in cells, while many phosphonosulfonates were active in both enzyme and cell assays. Second, we developed a QSAR model for CrtM inhibition that had good predictivity. Third, we determined the activity of each compound against STX biosynthesis, in S. aureus. The pIC50 results were poorly correlated (R2 = 0.27, training; R2 = 0.16, test set) with the enzyme inhibition pIC50 values. However, using a combinatorial descriptor search, we obtained significant improvements in cell activity predictions with R2 = 0.72 (training) and R2 = 0.62 (test set) results. Fourth, we investigated the inhibition of human squalene synthase by these compounds and used a QSAR method to enable good predictions of CrtM/SQS selectivity, relative to that exhibited by 1. Several CrtM inhibitors (e.g., 6 and 25) also showed very potent activity in bacterial cell-based assays. Overall, these results are of general interest, since they demonstrate that diphenyl ether phosphonosulfonates are potent inhibitors of dehydrosqualene synthase and of the STX biosynthesis in S. aureus, and as such, they have considerable potential for further development in a new approach to combating S. aureus infections.
Chemistry: General Aspects
A general synthetic route to the diphenylether phosphonosulfonate compounds is shown in Scheme 1. If not commercially available, a 3-phenoxybenzaldehyde 37 can be prepared with a copper(I) iodide mediated coupling reaction18 from a substituted halobenzene and a substituted hydroxybenzaldehyde, in a yield of 70−90%. The aldehyde 37 was reacted with sodium triethylphosphonoacetate in THF to give an α, β-unsaturated carboxylate, which was hydrogenated, reduced to the alcohol by treatment with LiAlH4, mesylated, then treated with NaI to afford the iodide 38. This was then reacted with the sodium salt of cyclohexyl diethylphosphonomethylsulfonate to give the triester 39,10 typically in an overall yield of 40% from the aldehyde 37. The triester 39 was deprotected by successive treatments with ammonia in methanol, then bromotrimethylsilane, followed by alkaline hydrolysis, affording the phosphonosulfonates as a tripotassium salt in ∼70% yield.
Scheme 1.

General Synthesis of Diphenyl Ether Phosphonosulfonatesa
Biphenyl bisphosphonates and phosphonosulfonates were made similarly, as shown in Scheme 2. Iodide 41 was made from a biphenylaldehyde 40, which is either commercially available or prepared using a Suzuki coupling reaction from 4-bromobenzaldehyde and a substituted phenylboronic acid. Compound 41 was reacted with the sodium salt of tetraethyl methylenediphosphonate or triethyl methylphosphinomethylphosphonate,19 followed by treatment with bromotrimethylsilane, to give bisphosphonates (2 and 3) or phosphinophosphonate (4), respectively. Following steps vi—viii in Scheme 1, biphenyl phosphonosulfonates (9−13) can be obtained from the iodide 41.
Scheme 2a.

Compounds (S)- and (R)-1 were synthesized according to a published method,11 using trans-(1R, 2R)-N,N′-bismethylcyclohexanediamine as a chiral auxiliary. The syntheses of other compounds are illustrated in Scheme 3.
Scheme 3a.

Compounds 14 and 15 were readily prepared from 3-phenoxybenzyl chloride and 3-phenoxyphenylacetic acid, respectively, following the general method in Scheme 1. Compound 16 was prepared from aldehyde 42, which was obtained by the coupling of nitrosobenzene and 3-formylphenylboronic acid, in the presence of copper(I) chloride.20 Suzuki coupling of benzyl bromide and 3-formylphenylboronic acid afforded 3-phenylaminobenzaldehyde 43. Compound 17 was then made from aldehyde 43, following steps ii—viii in Scheme 1. Similarly, compounds 18 and 19 were made following steps ii—viii in Scheme 1, starting from available 4-phenoxybenzaldehyde and 9-ethyl-3-carbazolecarboxaldehyde, respectively.
Experimental Section
All reagents used were purchased from Aldrich (Milwaukee, WI). The purities of all compounds were routinely monitored by using 1H and 31P NMR spectroscopy at 400 or 500 MHz on Varian (Palo Alto, CA) Unity spectrometers. Elemental analysis results are provided in the Supporting Information (Table S2).
General Method A (Steps ii—viii in Scheme 1)
Step ii
Triethyl phosphonoacetate (3.3 mmol) was added dropwise to NaH (145 mg, 60% in oil, 3.6 mmol) suspended in dry THF (7 mL) at 0 °C. To the resulting clear solution was added a benzaldehyde (3 mmol), and after being stirred at room temperature for 0.5 h, the reaction mixture was partitioned between diethyl ether (50 mL) and water (50 mL). The organic layer was dried and evaporated.
Step iii
The residue oil was then hydrogenated in MeOH (15 mL), in the presence of 5% Pd/C (50 mg) or Raney Ni (500 mg) when the aldehyde 37 was prepared using the CuI mediated reaction (Scheme 1). The catalyst was filtered, and the filtrate was concentrated and dried in vacuo.
Step iv
The resulting oil was dissolved in anhydrous THF (8 mL), and LiAlH4 (114 mg) was slowly added to the solution at 0 °C. After 1 h, the reaction was carefully quenched by adding a few drops of water, and the reaction mixture was filtered.
Step v
The filtrate was evaporated to dryness and the alcohol thus obtained redissolved in CH2Cl2 (10 mL) containing NEt3 (0.5 mL, 3.6 mmol). Methanesulfonyl chloride (230 μL, 3 mmol) was added slowly at 0 °C. After 1 h of stirring at room temperature, diethyl ether (50 mL) and water (50 mL) were added and the organic layer was collected, washed with 1 N HCl and saturated NaHCO3, dried, and evaporated to dryness. The oily residue was treated with NaI (1.35 g, 9 mmol) in acetone (7 mL) at 60 °C for 1 h. The reaction mixture was then partitioned between diethyl ether (50 mL) and water (50 mL) and the organic layer washed with 5% Na2S2O3, dried, and evaporated to dryness to give an iodide, such as 38. The iodide thus obtained was quite pure, according to 1H and 13C NMR spectra, and may be used in the next step without purification.
Step vi
Cyclohexyl diethylphosphonomethylsulfonate (470 mg, 1.5 mmol) was added to NaH (60 mg, 60% in oil, 1.5 mmol) suspended in dry DMF (2 mL) at 0 °C. To the resulting clear solution was added an iodide (1 mmol), and after being stirred at room temperature for 3 h, the reaction mixture was partitioned between diethyl ether (50 mL) and water (50 mL). The organic layer was dried and evaporated and the residue subjected to a column chromatography (silica gel, ethyl acetate/hexane, 1:1) to give a phosphonosulfonate triester, such as 39, as a colorless oil.
Step vii
The triester was treated with ammonium hydroxide (12 M, 1 mL) in MeOH (6 mL) at 60 °C for 3 h. The solvents were evaporated, and the residue was subjected to ion exchange chromatography (DOWEX 50WX8−200, H+ form, 3 mL) using MeOH as eluent.
Step viii
The eluent was evaporated to dryness and the resulting diethylphosphonosulfonic acid dissolved in anhydrous CH3CN (3 mL) and treated with Me3SiBr (400 μL, 3 mmol) at 40 °C overnight. The solution was evaporated to dryness and MeOH (5 mL) added to the residue. The solvent was removed in vacuo again, and the residue was redissolved in MeOH (5 mL). Neutralization with 2 M KOH to pH 8 gave a tripotassium salt of a phosphonosulfonate as a white powder.
General Method B (Step i in Scheme 1)18
A mixture containing a halobenzene (3 mmol), a 3-hydroxybenzaldehyde (4.5 mmol), CuI (58 mg, 0.3 mmol), N,N-dimethylglycine (93 mg, 0.9 mmol), and Cs2CO3 (2 g, 6 mmol) in 1,4-dioxane (8 mL) was vigorously stirred at 90 °C for 18 h. The solvent was evaporated and the residue partitioned between diethyl ether (50 mL) and water (50 mL). The organic layer was successively washed with 5% NaOH (2 × 20 mL), water (20 mL), and saturated NaCl (20 mL). It was then dried and evaporated to give aldehyde 37 as a pale-yellow oil, which is quite pure and may be used in the next step directly. It may also be purified via a column chromatography.
1-Phosphono-4-(3-phenoxyphenyl)butylsulfonic Acid Tripotassium Salt (1)
Compound 1 was prepared from 3-phenoxybenzaldehyde (3 mmol), following general method A as a white powder (680 mg, 36% overall yield). Anal. (C16H16K3O7PS·KBr·0.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.50−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.70−2.80 (m, 1H, CHSO3K), 6.70−7.30 (m, 9H, aromatic). 31P NMR (D2O): δ 12.4.
4-(4-Biphenyl)butyldiphosphonic Acid Tetrapotassium Salt (2)
Compound 2 was prepared from 4-phenylbenzaldehyde (3 mmol), following steps ii—vi and then step viii of the general method A, as a white powder (721 mg, 46% overall yield). Purity was determined to be 87.3% by quantitative NMR spectroscopy. 1H NMR (400 MHz, D2O): δ 1.60−1.80 (m, 5H, —CH2CH2— and CH), 2.56 (t, J = 5.6 Hz, 2H, PhCH2), 7.20−7.60 (m, 9H, aromatic). 31P NMR (D2O): δ 21.2.
4-[4-(4-Trifluoromethylphenyl)phenyl)]butyldiphosphonic Acid Dipotassium Salt (3)
Compound 3was prepared from 4-trifluoromethylphenylbenzaldehyde (3 mmol), following steps ii—vi and then step viii of general method A, as a white powder (671 mg, 42% overall yield). Anal. (C17H17F3K2O6P2) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 5H, —CH2CH2— and CH), 2.58 (t, J = 5.6 Hz, 2H, PhCH2), 7.20−7.60 (m, 9H, aromatic). 31P NMR (D2O): δ 21.5.
4-(4-Biphenyl)butyldiphosphonic Acid Dipotassium Salt (4)
Compound 4 was prepared from 4-phenylbenzaldehyde (3 mmol), following steps ii—vi and then step viii of general method A, as a white powder (470 mg, 21% overall yield). Anal. (C17H20K2O5P2) C, H. 1H NMR (400 MHz, D2O): δ 1.19 (d, J = 11.2 Hz, 3H, Me), 1.60−1.80 (m, 5H, —CH2CH2— and CH), 2.56 (t, J = 5.6 Hz, 2H, PhCH2), 7.20−7.60 (m, 9H, aromatic). 31P NMR (D2O): δ 18.3 (s, 1P), 49.6 (s, 1P).
1-Phosphono-4-[3-(4-fluorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (5)
Compound 5 was prepared from 4-fluoroiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (425 mg, 26% overall yield). Anal. (C16H15FK3O7PS·1.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.60−7.10 (m, 8H, aromatic). 31P NMR (D2O): δ 13.6.
1-Phosphono-4-[3-(4-propylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (6)
Compound 6 was prepared from 4-propylbromobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (670 mg, 27% overall yield). Anal. (C19H22K3O7PS·2.4 KBr) C, H. 1H NMR (400 MHz, D2O): δ 0.73 (t, J = 7.2 Hz, 3H, CH3), 1.40−1.50 (m, 2H, CH3CH2), 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 4H, PhCH2 and PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.70−7.20 (m, 8H, aromatic). 31P NMR (D2O): δ 14.4.
1-Phosphono-4-[3-(4-trifluoromethylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (7)
Compound 7 was prepared from 4-trifluoromethyl-iodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (570 mg, 32% overall yield). Anal. (C17H15F3K3O7PS·1.5 H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.80−2.00 (m, 4H, —CH2CH2—), 2.40−2.60 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.80−7.40 (m, 8H, aromatic). 31P NMR (D2O): δ 13.9.
1-Phosphono-4-[3-(2-benzylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (8)
Compound 8 was prepared from 2-benzyliodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and then A, as a white powder (570 mg, 29% overall yield). Anal. (C23H22K3O7PS·4 H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 3.80 (s, 2H, Ph CH2Ph), 6.50−7.30 (m, 13H, aromatic). 31P NMR (D2O): δ 12.7.
1-Phosphono-4-(4-biphenyl)butylsulfonic Acid Tripotassium Salt (9)
Compound 9 was prepared from 4-phenylbenzaldehyde (3 mmol), following general method A, as a white powder (730 mg, 31% overall yield). Anal. (C16H16K3O6PS·2.5KBr) C, H. 1H NMR (400 MHz, D2O): δ 1.70−1.90 (m, 4H, —CH2CH2—), 2.50−2.60 (m, 2H, PhCH2), 2.90−3.00 (m, 1H, CHSO3K), 7.20−7.60 (m, 9H, aromatic). 31P NMR (D2O): δ 14.4.
1-Phosphono-4-[4-(2,4-difluorophenyl)phenyl]butylsulfonic Acid Tripotassium Salt (10)
4-(2,4-Difluorophenyl)benzaldehyde was made by a Suzuki coupling reaction from 2,4-difluorophenylboronic acid (3.6 mmol) and 4-bromobenzaldehyde (3 mmol), as described previously.21 Compound 10 was prepared from the aldehyde thus obtained, following general method A, as a white powder (470 mg, 28% overall yield). Anal. (C16H14F2K3O6PS) C, H. 1H NMR (400 MHz, D2O): δ 1.65−2.00 (m, 4H, —CH2CH2—), 2.45−2.60 (m, 2H, PhCH2), 2.80−2.85 (m, 1H, CHSO3K), 6.80−7.40 (m, 7H, aromatic). 31P NMR (D2O): δ 12.5.
1-Phosphono-4-[4-(4-methylphenyl)phenyl]butylsulfonic Acid Tripotassium Salt (11)
Compound 11 was prepared from 4-(4-methylphenyl)benzaldehyde (3 mmol), following general method A, as a white powder (770 mg, 28% overall yield). Anal. (C17H18K3O6PS·3.5KBr) C, H. 1H NMR (400 MHz, D2O): δ 1.70−1.90 (m, 4H, —CH2CH2—), 2.21 (s, 3H, Me), 2.40−2.50 (m, 2H, PhCH2), 2.90−3.00 (m, 1H, CHSO3K), 7.20−7.50 (m, 8H, aromatic). 31P NMR (D2O): δ 14.5.
1-Phosphono-4-[4-(4-butylphenyl)phenyl]butylsulfonic Acid Tripotassium Salt (12)
4-(4-Butylphenyl)benzaldehyde was made by a Suzuki coupling reaction from 4-butylphenylboronic acid (3.6 mmol) and 4-bromobenzaldehyde (3 mmol).21 Compound 12 was prepared from the aldehyde thus obtained, following general method A, as a white powder (650 mg, 25% overall yield). Anal. (C20H24K3O6PS·2.8KBr) C, H. 1H NMR (400 MHz, D2O): δ 0.74 (t, J = 7.2 Hz, 3H, CH3), 1.10−1.20 (m, 2H, CH3CH2), 1.40−1.50 (m, 2H, CH3CH2CH2), 1.650−2.00 (m, 4H, —CH2CH2—), 2.45−2.60 (m, 4H, PhCH2 and PhCH2), 2.70−2.80 (m, 1H, CHSO3K), 7.20−7.50 (m, 8H, aromatic). 31P NMR (D2O): δ 12.4.
1-Phosphono-4-[4-(3-propoxyphenyl)phenyl]butylsulfonic Acid Tripotassium Salt (13)
4-(3-Propoxyphenyl)benzaldehyde was made by a Suzuki coupling reaction from 3-propoxyphenylboronic acid (3.6 mmol) and 4-bromobenzaldehyde (3 mmol).21 Compound 13 was prepared from the aldehyde thus obtained, following general method A, as a white powder (539 mg, 30% overall yield). Anal. (C19H22K3O6PS) C, H. 1H NMR (400 MHz, D2O): δ 0.74 (t, J = 7.2 Hz, 3H, CH3), 1.40−1.50 (m, 2H, CH3CH2), 1.65−2.00 (m, 4H, —CH2CH2—), 2.45−2.60 (m, 2H, PhCH2), 2.80−2.85 (m, 1H, CHSO3K), 3.75 (t, J = 7.2 Hz, 2H, OCH2), 6.80−7.40 (m, 7H, aromatic). 31P NMR (D2O): δ 12.8.
(1S)-1-Phosphono-4-(3-phenoxyphenyl)butylsulfonic Acid Tripotassium Salt [(S)-1]
Compound (S)-1 was prepared from methylphosphonic dichloride (3 mmol) and trans-(1R,2R)-N,N′-bismethylcyclohexanediamine (3 mmol), following a published method,11 as a white powder (640 mg, 29% overall yield). Anal. (C16H16K3O7PS·1.2K2SO4·1.5H2O) C, H. Identical NMR spectra as for 1.
(1R)-1-Phosphono-4-(3-phenoxyphenyl)butylsulfonic Acid Tripotassium Salt [(R)-1]
Compound (R)-1 was obtained as a minor product during the synthesis of (S)-1, as described immediately above (405 mg, 16% overall yield). Anal. (C16H16K3O7PS·2K2SO4) C, H. Identical NMR spectra as for 1.
1-Phosphono-2-(3-phenoxyphenyl)ethylsulfonic Acid Tripotassium Salt (14)
Compound 14 was prepared from 3-phenoxybenzyl chloride (1 mmol), following steps vi—viii of general method A, as a white powder (285 mg, 55% overall yield). Anal. (C14H12K3O7PS·C2H5OH) C, H. 1H NMR (400 MHz, D2O): δ 2.95−3.05 (m, 1H, CH2). 3.10−3.30 (m, 2H, CH and CH2), 6.70−7.30 (m, 9H, aromatic). 31P NMR (D2O): δ 13.8.
1-Phosphono-3-(3-phenoxyphenyl)propylsulfonic Acid Tripotassium Salt (15)
Compound 15 was prepared from 3-phenoxyphenylacetic acid (3 mmol), following steps iv—viii of general method A, as a white powder (380 mg, 25% overall yield). Anal. (C15H14K3O7PS·0.25C2H5OH·0.5H2O) C, H. 1H NMR (400 MHz, D2O): δ δ 2.00−2.10 (m, 2H, CH2), 2.65−2.80 (m, 2H, PhCH2), 2.90−3.00 (m, 1H, CHSO3K), 6.70−7.30 (m, 9H, aromatic). 31P NMR (D2O): δ 14.1.
1-Phosphono-4-(3-phenylaminophenyl)butylsulfonic Acid Tripotassium Salt (16)
A mixture of nitrosobenzene (3 mmol) and CuCl (3 mmol) in anhydrous DMF (8 mL) was heated to 55 °C for 0.5 h. Then 3-formylphenylboronic acid (3.3 mmol) was added to the reaction mixture, which was then stirred for another 16 h.20 The product was then partitioned between diethyl ether (50 mL) and water (50 mL) and the organic layer washed with saturated NaHCO3, dried, then evaporated to dryness, giving 3- phenylaminobenzaldehyde as a pale-yellow oil, which may be used directly without purification. Compound 16 was prepared from the aldehyde thus obtained, following general method A, as a white powder (425 mg, 22% overall yield). Anal. (C16H17NK3O7PS·KBr·1.5H2O) C, H, N. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.70−7.20 (m, 9H, aromatic). 31P NMR (D2O): δ 14.4.
1-Phosphono-4-(3-benzylphenyl)butylsulfonic Acid Tripotassium Salt (17)
3-Benzylbenzaldehyde was prepared from benzyl bromide (3 mmol) and 3-formylphenylboronic acid (3.3 mmol) by a Suzuki coupling reaction.21 Compound 17 was prepared from the aldehyde thus obtained, following general method A, as a white powder (540 mg, 33% overall yield). Anal. (C17H20K3O7PS·0.25KBr·H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.50−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 3.88 (s, 2H, PhCH2Ph), 6.90−7.20 (m, 9H, aromatic). 31P NMR (D2O): δ 13.8.
1-Phosphono-4-(4-phenoxyphenyl)butylsulfonic Acid Tripotassium Salt (18)
Compound 18 was prepared from 4-phenoxybenzaldehyde (3 mmol), following general method A, as a white powder (680 mg, 36% overall yield). Anal. (C16H16K3O7PS) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.80−7.30 (m, 9H, aromatic). 31P NMR (D2O): δ 13.7.
1-Phosphono-4-(9-ethylcarbazole-3-yl)butylsulfonic Acid Tripotassium Salt (19)
Compound 19 was prepared from 9-ethyl-3-carbazolecarboxaldehyde (3 mmol), following general method A, as a white powder (600 mg, 36% overall yield). Anal. (C18H19NK3O6PS·0.5C2H5OH·0.5H2O) C, H, N. 1H NMR (400 MHz, D2O): δ 1.14 (t, J = 7.2 Hz, 3H, CH3), 1.70−1.90 (m, 4H, —CH2CH2—), 2.60−2.70 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 4.22 (q, J = 7.2 Hz, 3H, NCH2), 7.05−7.40 (m, 5H, aromatic), 7.92 (s, 1H, aromatic), 8.00 (d, J = 8.0 Hz, 1H, aromatic). 31P NMR (D2O): δ 14.1.
1-Phosphono-4-[3-(3-fluorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (20)
Compound 20 was prepared from 3-fluoroiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (570 mg, 28% overall yield). Anal. (C16H15FK3O7PS·KBr·2.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.50−2.70 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.70−7.20 (m, 8H, aromatic). 31P NMR (D2O): δ 13.8.
1-Phosphono-4-[3-(2-fluorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (21)
Compound 21 was prepared from 2-fluoroiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (340 mg, 20% overall yield). Anal. (C16H15FK3O7PS·2.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.70−1.90 (m, 4H, —CH2CH2—), 2.60−2.80 (m, 2H, PhCH2), 2.90−3.00 (m, 1H, CHSO3K), 6.60−7.20 (m, 8H, aromatic). 31P NMR (D2O): δ 13.2.
1-Phosphono-4-[3-(3-trifluoromethylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (22)
Compound 22 was prepared from 3-(3-trifluoromethylphenoxy)benzaldehyde (3 mmol), following general method A, as a white powder (600 mg, 30% overall yield). Anal. (C17H15F3K3O7PS) C, H. 1H NMR (400 MHz, D2O): δ 1.80−2.00 (m, 4H, —CH2CH2—), 2.50−2.70 (m, 2H, PhCH2), 2.85−2.95 (m, 1H, CHSO3K), 6.80−7.40 (m, 8H, aromatic). 31P NMR (D2O): δ 13.3.
1-Phosphono-4-[3-(2-trifluoromethylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (23)
Compound 23 was prepared from 2-trifluoromethyliodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (205 mg, 12% overall yield). Purity was determined to be 86.5% by quantitative NMR spectroscopy. 1H NMR (400 MHz, D2O): δ 1.70−1.90 (m, 4H, —CH2CH2—), 2.50−2.65 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.70−7.30 (m, 8H, aromatic). 31P NMR (D2O): δ 13.7.
1-Phosphono-4-[3-(4-chlorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (24)
Compound 24 was prepared from 3-(4-chlorophenoxy)benzaldehyde (3 mmol), following general method A, as a white powder (525 mg, 30% overall yield). Anal. (C16H15ClK3O7PS·C2H5OH) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.45−2.55 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.70−7.10 (m, 8H, aromatic). 31P NMR (D2O): δ 14.0.
1-Phosphono-4-[3-(4-tert-butylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (25)
Compound 25 was prepared from 3-(4-tert-butylphenoxy)benzaldehyde (3 mmol), following general method A, as a white powder (610 mg, 35% overall yield). Anal. (C20H24K3O7PS·1.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.10 (s, 9H, CMe3), 1.60−1.85 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.60−7.40 (m, 8H, aromatic). 31P NMR (D2O): δ 14.3.
1-Phosphono-4-[3-(4-benzylphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (26)
Compound 26 was prepared from 4-benzyliodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (510 mg, 28% overall yield). Anal. (C23H22K3O7PS·H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.75−2.85 (m, 1H, CHSO3K), 3.79 (s, 2H, PhCH2), 6.60−7.20 (m, 13H, aromatic). 31P NMR (D2O): δ 13.9.
1-Phosphono-4-[3-(4-hydroxyphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (27)
Compound 27 was prepared from 4-tert-butoxyiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (690 mg, 22% overall yield). Anal. (C16H16K3O8PS·4 KBr·3H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.90−3.00 (m, 1H, CHSO3K), 6.60−7.20 (m, 8H, aromatic). 31P NMR (D2O): δ 14.5.
1-Phosphono-4-[3-(4-phenoxyphenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (28)
Compound 28 was prepared from 4-phenoxyiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (610 mg, 30% overall yield). Anal. (C22H20K3O8PS·0.5KBr·1.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.60−7.25 (m, 13H, aromatic). 31P NMR (D2O): δ 13.7.
1-Phosphono-4-[3-[4-(furan-2-yl)phenoxy]phenyl]butylsulfonic Acid Tripotassium Salt (29)
Compound 29 was prepared from 4-(furan-2-yl)iodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (690 mg, 22% overall yield). Anal. (C20H18K3O8PS·2 KBr·1.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.70−2.80 (m, 1H, CHSO3K), 6.40−7.60 (m, 11H, aromatic). 31P NMR (D2O): δ 12.4.
1-Phosphono-4-[3-(3,4-difluorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (30)
Compound 30 was prepared from 3,4-difluoroiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (525 mg, 29% overall yield). Anal. (C16H14F2K3O7PS·0.5C2H5OH·2.5H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−2.00 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.50−7.30 (m, 7H, aromatic). 31P NMR (D2O): δ 12.8.
1-Phosphono-4-[3-(3,4-dichlorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (31)
Compound 31 was prepared from 3-(3,4-dichlorophenoxy)benzaldehyde (3 mmol), following general method A, as a white powder (540 mg, 28% overall yield). Anal. (C16H14Cl2K3O7PS·0.5C2H5OH·3H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−2.00 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.80−7.40 (m, 7H, aromatic). 31P NMR (D2O): δ 12.4.
1-Phosphono-4-[3-(benzofuran-5-yloxy)phenyl]butylsulfonic Acid Tripotassium Salt (32)
Compound 32 was prepared from 5-bromobenzofuran (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (380 mg, 22% overall yield). Anal. (C18H16K3O8PS·2H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.60−7.60 (m, 9H, aromatic). 31P NMR (D2O): δ 14.1.
1-Phosphono-4-[3-(3,5-difluorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (33)
Compound 33 was prepared from 3,5-difluoroiodobenzene (3 mmol) and 3-hydroxybenzaldehyde (4.5 mmol), following general methods B and A, as a white powder (415 mg, 25% overall yield). Anal. (C16H14F2K3O7PS·H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.40−2.50 (m, 2H, PhCH2), 2.80−2.90 (m, 1H, CHSO3K), 6.40−7.20 (m, 7H, aromatic). 31P NMR (D2O): δ 14.2.
1-Phosphono-4-[3-(3,5-dichlorophenoxy)phenyl]butylsulfonic Acid Tripotassium Salt (34)
Compound 34 was prepared from 3-(3,5-dichlorophenoxy)benzaldehyde (3 mmol), following general method A, as a white powder (570 mg, 26% overall yield). Anal. (C16H14Cl2K3O7PS·C2H5OH·KBr) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.45−2.55 (m, 2H, PhCH2), 2.85−2.95 (m, 1H, CHSO3K), 6.80−7.40 (m, 7H, aromatic). 31P NMR (D2O): δ 12.8.
1-Phosphono-4-[3-(4-fluorophenoxy)-6-fluoro-phenyl]butylsulfonic Acid Tripotassium Salt (35)
Compound 35 was prepared from 4-fluorophenol (4.5 mmol) and 3-bromo-6-fluorobenzaldehyde (3 mmol), following general methods B and A, as a white powder (525 mg, 30% overall yield). Anal. (C16H14F2K3O7PS·0.25KBr·H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.60−1.90 (m, 4H, —CH2CH2—), 2.50−2.70 (m, 2H, PhCH2), 2.75−2.85 (m, 1H, CHSO3K), 6.60−7.00 (m, 7H, aromatic). 31P NMR (D2O): δ 13.6.
1-Phosphono-4-[3-(4-fluorophenoxy)-6-methoxy-phenyl]butylsulfonic Acid Tripotassium Salt (36)
Compound 36 was prepared from 4-fluorophenol (4.5 mmol) and 3-bromo-6-methoxybenzaldehyde (3 mmol), following general methods B and A, as a white powder (470 mg, 25% overall yield). Anal. (C17H17FK3O8PS·0.4KBr·2H2O) C, H. 1H NMR (400 MHz, D2O): δ 1.50−1.90 (m, 4H, —CH2CH2—), 2.30−2.50 (m, 2H, PhCH2), 2.60−2.70 (m, 1H, CHSO3K), 3.62 (s, 3H, OMe), 6.65−7.00 (m, 7H, aromatic). 31P NMR (D2O): δ 13.6.
Computational Methods
CoMSIA analysis was performed with default settings in Sybyl16 (version 7.3). All compounds were geometrically optimized, using the MMFF94x force field, then aligned in the program MOE,13 utilizing the flexible alignment module.14 The alignment was carried out by performing up to 1000 flexible refinement iterations using a gradient test of 0.01−1.0 with hydrophobe, log P, and partial charge similarity features, as well as the default options (H-bond donor, acceptor, aromaticity, polar hydrogens, and volume). The alignments were exported into the Sybyl program, where atomic charges were determined by using the Gasteiger—Marsili method.22 CoMSIA indices were calculated on a rectangular grid containing each of the sets of aligned molecules using steric, electrostatic, hydrophobic, H-donor, and H-acceptor fields, using default grid spacing and probe atoms. We then used a partial least-squares (PLS) method to correlate the 3D structural features with activity. The optimal number of components was determined with the SAMPLS cross-validation method.23 Each of the QSAR models was then validated by performing five training/test set predictions.
For cell activity predictions, a set of 117 molecular descriptors, including SlogP,17 were computed within MOE and exported to MATLAB24 for a combinatorial descriptor search. The entire descriptor space was searched exhaustively to find the combinations of descriptors that gave the best regression coefficient (highest R2) for the equation
| (3) |
where B and C are MOE descriptors and a—d are coefficients. A leave-two-out cross-validation was performed to test the predictivity of the model, where all combinations of two compounds were excluded from the data set and the descriptor combinations reevaluated for the remaining (training set) compounds. The regression equation obtained from each run was then used to calculate the cell activity of the left out compounds (the test set), and the R2 from all such leave-two-out predictions is reported in the text. Finally, a scrambling analysis was performed in which the cell activity values for all the compounds were scrambled and the leave-two-out cross-validation was performed using the scrambled data set.
CrtM Enzyme Inhibition Assay
The expression and purification of the S. aureus CrtM as well as the inhibition assays were carried out by using our previous methods.6 In brief, CrtM with a histidine tag was overexpressed in E. coli BL21 (DE3) cells. After an overnight growth, an initial 50 mL of culture was transferred into 1 L of LB medium supplemented with 100 μg/mL ampicillin. Induction was carried out with 1 mM IPTG for 4 h, when the cell culture reached an OD of 0.6 at 600 nm. Protein was purified with a Ni-NTA column, using a 100 mL linear gradient of 0−0.5 M imidazole in 50 mM Tris-HCl buffer at pH 7.4. Enzyme inhibition assays were carried out, in duplicate, in 96-well plates, with a total of 200 μL of reaction mixture in each well. The reaction was monitored by using a continuous spectrophotometric assay for phosphate releasing enzymes.25 The reaction buffer contained 50 mM Tris-HCl, 1 mM MgCl2, 450 μM FPP, pH 7.4. The compounds investigated were preincubated with 2 μg of CrtM for 30 min at 20 °C. The IC50 values were obtained by fitting the inhibition data to a normal dose—response curve, using GraphPad PRISM, version 4.0, software for windows (GraphPad Software Inc., San Diego, CA, www.graphpad.com). Ki was calculated on the basis of the IC50 value and the reported Km of CrtM.26
Staphyloxanthin Biosynthesis Inhibition Assay
The S. aureus strain used was the WT clinical isolate (Pig1).4 S. aureus was cultured in THB (1 mL) in the presence of inhibitor compounds for 72 h, in duplicate. Cells were then centrifuged and washed twice with PBS. STX was extracted with MeOH, and the OD was determined at 450 nm using a Perkin-Elmer MBA 2000 (Norwalk, CT) spectrophotometer. The IC50 values were obtained by fitting the OD data to a normal dose—response curve, using GraphPad PRISM.
Human SQS Enzyme Expression, Purification, and Inhibition Assay
A DNA sequence encoding a double truncated protein lacking residue 31 at the N-terminus and residue 46 at the C-terminus was amplified using the following primers: 5′-CATATGGACCAGGACTCGCTCAGCAGC and 3′-GGATCCTCAATTCTGCGTCCGGATGGT. The corresponding amplified insert was initially cloned in the vector pGEMT (Promega). Plasmid was digested with the endonucleases NdeI and BamHI, and the resulting fragment was cloned into the bacterial expression vector pET-28a to give pET28a-HsSQS, which was used to transform E. coli BL21 (DE3)RP strain (Novagen) for overexpression. This cloning procedure resulted in the addition of a six-histidine tag to the N-terminus of double truncated HsSQS.
Bacteria expressing the constructs were cultured in Luria—Bertani medium supplemented with kanamycin (30 μg/mL) and chloramphenicol (34 μg/mL) at 37 °C, until the cells reached an OD of 0.4 at 600 nm, and were then induced at 37 °C for 4 h by incubation with 1 mM isopropyl-1-thio-β-d-galactopyranoside. Cells were harvested by centrifugation (10 min, 4000 rpm) and resuspended in 10 mL of lysis/elution buffer (20 mM NaH2PO3, pH 7.4, 10 mM CHAPS, 2 mM MgCl2, 10% glycerol, 10 mM mercaptoethanol, 500 mM NaCl, 10 mM imidazole, and a protease inhibitor cocktail (Roche), disrupted by sonication, and centrifuged at 16 000 rpm for 30 min. The supernatant (40 mL) was then applied to a HiTrap nickel-chelating HP column (Amersham Biosciences). Enzyme purification was performed according to the manufacturer's instructions using a Pharmacia FPLC system. Unbound protein was washed off with 50 mM imidazole. Then the His6-HsSQS was eluted with 1 M imidazole. Purity was confirmed by SDS—PAGE electrophoresis. Fractions containing the pure enzyme were pooled and dialyzed against buffer A (25 mM sodium phosphate, pH 7.4, 20 mM NaCl, 2 mM dithiothreitol, 1 mM EDTA, 10% glycerol, 10% methanol), concentrated, then stored at −80 °C.
SQS activity was based on measuring the conversion of [3H]FPP to [3H]squalene. Final assay concentrations were 50 mM MOPS (pH 7.4), 20 mM MgCl2, 5 mM CHAPS, 1% Tween-80, 10 mM DTT, 0.025 mg/mL BSA, 0.25 mM NADPH, and 7.5 ng of purified recombinant human SQS. The reaction was started with the addition of substrate (3HFPP, 0.1 nmol, 2.22 × 106 dpm), and the final volume of the reaction was 200 μL. After incubation at 37 °C for 5 min, an amount of 40 μL of 10 M NaOH was added to stop the reaction, followed by 10 μL of a (100:1) mixture of 98% EtOH and squalene. The resulting mixtures were mixed vigorously by vortexing. Then 10 μL aliquots were applied to 2.5 × 10 cm channels of a silica gel thin layer chromatogram, and newly formed squalene was separated from unreacted substrate by chromatography in toluene—EtOAc (9:1). The region of the squalene band was scraped and immersed in Hydrofluor liquid scintillation fluid and assayed for radioactivity. IC50 values were calculated from the hyperbolic plot of percent of inhibition versus inhibitor concentration, using GraphPad PRISM.
Supplementary Material
Acknowledgment
This work was supported by the U.S. Public Health Service (Grant HD051796 to V.N., Grants AI074233, GM073216, and GM65307 to E.O., and Grants AR045504, CA113874, and AI057160 to C.T.M.). Y.S. was supported by a Leukemia and Lymphoma Society Special Fellowship.
Footnotes
Abbreviations: CrtM, dehydrosqualene synthase; FPP, farnesyl diphosphate; QSAR, quantitative structure—activity relationship; SQS, squalene synthase; STX, staphyloxanthin.
Supporting Information Available: Details of computational cell activity predictions, activity of selected compounds on human cell lines, elemental analysis results, and dose—responsive curves of representative compounds in STX inhibition. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bancroft EA. Antimicrobial resistance: It's not just for hospitals. J. Am. Med. Assoc. 2007;298:1803–1804. doi: 10.1001/jama.298.15.1803. [DOI] [PubMed] [Google Scholar]
- 2.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillinresistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.National Research Council . Treating Infectious Diseases in a Microbial World: Report of Two Workshops on Novel Antimicrobial Therapeutics. National Academies Press; Washington, DC: 2006. p. 21. [PubMed] [Google Scholar]
- 4.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, Nizet V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005;202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clauditz A, Resch A, Wieland KP, Peschel A, Gotz F. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect. Immun. 2006;74:4950–4953. doi: 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma A, Slugg PH, Hammett JL, Jusko WJ. Clinical pharmacokinetics and pharmacodynamics of a new squalene synthase inhibitor, BMS-188494, in healthy volunteers. J. Clin. Pharmacol. 1998;38:1116–1121. [PubMed] [Google Scholar]
- 8.Sharma A, Slugg PH, Hammett JL, Jusko WJ. Estimation of oral bioavailability of a long half-life drug in healthy subjects. Pharm. Res. 1998;15:1782–1786. doi: 10.1023/a:1011977116543. [DOI] [PubMed] [Google Scholar]
- 9.Ciosek CP, Jr., Magnin DR, Harrity TW, Logan JV, Dickson JK, Jr., Gordon EM, Hamilton KA, Jolibois KG, Kunselman LK, Lawrence RM, et al. Lipophilic 1,1-bisphosphonates are potent squalene synthase inhibitors and orally active cholesterol lowering agents in vivo. J. Biol. Chem. 1993;268:24832–24837. [PubMed] [Google Scholar]
- 10.Magnin DR, Biller SA, Chen Y, Dickson JK, Jr., Fryszman OM, Lawrence RM, Logan JV, Sieber-McMaster ES, Sulsky RB, Traeger SC, Hsieh DC, Lan SJ, Rinehart JK, Harrity TW, Jolibois KG, Kunselman LK, Rich LC, Slusarchyk DA, Ciosek CP., Jr. α-Phosphonosulfonic acids: potent and selective inhibitors of squalene synthase. J. Med. Chem. 1996;39:657–660. doi: 10.1021/jm9507340. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence RM, Biller SA, Dickson JK, Logan JVH, Magnin DR, Sulsky RB, DiMarco JD, Gougoutas JZ, Beyer BD, Taylor SC, Lan SJ, Ciosek CP, Harrity TW, Jolibois KG, Kunselman LK, Slusarchyk DA. Enantioselective synthesis of α-phosphono sulfonate squalene synthase inhibitors: chiral recognition in the interactions of an α-phosphono sulfonate inhibitor with squalene synthase. J. Am. Chem. Soc. 1996;118:11668–11669. [Google Scholar]
- 12.Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 13.Molecular Operating Environment (MOE) Chemical Computing Group, Inc.; Montreal, Quebec: 2006. [Google Scholar]
- 14.Labute P, Williams C, Feher M, Sourial E, Schmidt JM. Flexible alignment of small molecules. J. Med. Chem. 2001;44:1483–1490. doi: 10.1021/jm0002634. [DOI] [PubMed] [Google Scholar]
- 15.Mao J, Mukherjee S, Zhang Y, Cao R, Sanders JM, Song Y, Zhang Y, Meints GA, Gao YG, Mukkamala D, Hudock MP, Oldfield E. Solid-state NMR, crystallographic, and computational investigation of bisphosphonates and farnesyl diphosphate synthase—bisphosphonate complexes. J. Am. Chem. Soc. 2006;128:14485–14497. doi: 10.1021/ja061737c. [DOI] [PubMed] [Google Scholar]
- 16.Sybyl. Tripos; St. Louis, MO: 2007. [Google Scholar]
- 17.Wildman SA, Crippen GM. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999;39:868–873. [Google Scholar]
- 18.Ma D, Cai Q. N,N-Dimethyl glycine-promoted Ullmann coupling reaction of phenols and aryl halides. Org. Lett. 2003;5:3799–3802. doi: 10.1021/ol0350947. [DOI] [PubMed] [Google Scholar]
- 19.Teulade M-P, Savignac P, Aboujaoude EE, Collignon N. Carbanions phosphonates [alpha]-lithium: synthèse, basicité comparée et stabilitéà l'autocondensation. J. Organomet. Chem. 1986;312:283–295. [Google Scholar]
- 20.Yu Y, Srogl J, Liebeskind LS. Cu(I)-mediated reductive amination of boronic acids with nitroso aromatics. Org. Lett. 2004;6:2631–2634. doi: 10.1021/ol048982q. [DOI] [PubMed] [Google Scholar]
- 21.Sanders JM, Song Y, Chan JM, Zhang Y, Jennings S, Kosztowski T, Odeh S, Flessner R, Schwerdtfeger C, Kotsikorou E, Meints GA, Gomez AO, Gonzalez-Pacanowska D, Raker AM, Wang H, van Beek ER, Papapoulos SE, Morita CT, Oldfield E. Pyridinium-1-yl bisphosphonates are potent inhibitors of farnesyl diphosphate synthase and bone resorption. J. Med. Chem. 2005;48:2957–2963. doi: 10.1021/jm040209d. [DOI] [PubMed] [Google Scholar]
- 22.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity, a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 23.Bush BL, Nachbar RB., Jr. Sample-distance partial least squares: PLS optimized for many variables, with application to CoMFA. J. Comput.-Aided Mol. Des. 1993;7:587–619. doi: 10.1007/BF00124364. [DOI] [PubMed] [Google Scholar]
- 24.MATLAB, version 7.4. The Mathworks, Inc.; Natick, MA: 2007. [Google Scholar]
- 25.Rieger CE, Lee J, Turnbull JL. A continuous spectrophotometric assay for aspartate transcarbamylase and ATPases. Anal. Biochem. 1997;246:86–95. doi: 10.1006/abio.1996.9962. [DOI] [PubMed] [Google Scholar]
- 26.Ku B, Jeong JC, Mijts BN, Schmidt-Dannert C, Dordick JS. Preparation, characterization, and optimization of an in vitro C30 carotenoid pathway. Appl. Environ. Microbiol. 2005;71:6578–6583. doi: 10.1128/AEM.71.11.6578-6583.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.