

Abstract
Current genome-wide surveys of common diseases and complex traits fundamentally aim to detect indirect associations where the single nucleotide polymorphisms (SNPs) carrying the association signals are not biologically active but are in linkage disequilibrium (LD) with some unknown functional polymorphisms. Reproducing any novel discoveries from these genome-wide scans in independent studies is now a prerequisite for the putative findings to be accepted. Significant differences in patterns of LD between populations can affect the portability of phenotypic associations when the replication effort or meta-analyses are attempted in populations that are distinct from the original population in which the genome-wide study is performed. Here, we introduce a novel method for genome-wide analyses of LD variations between populations that allow the identification of candidate regions with different patterns of LD. The evidence of LD variation provided by the introduced method correlated with the degree of differences in the frequencies of the most common haplotype across the populations. Identified regions also resulted in greater variation in the success of replication attempts compared with random regions in the genome. A separate permutation strategy introduced for assessing LD variation in the absence of genome-wide data also correctly identified the expected variation in LD patterns in two well-established regions undergoing strong population-specific evolutionary pressure. Importantly, this method addresses whether a failure to reproduce a disease association in a disparate population is due to underlying differences in LD structure with an unknown functional polymorphism, which is vital in the current climate of replicating and fine-mapping established findings from genome-wide association studies.
The completion of the second phase of the International HapMap Project generated genetic data for over 3 million single nucleotide polymorphisms (SNPs) in samples with African, Asian, and European ancestries, offering maps of common genetic variations found across these populations (The International HapMap Consortium 2007). At a practical level, these maps have aided the design of efficient genotyping arrays for genome-wide studies of common diseases and complex traits, by identifying variants that capture the information from surrounding loci using genetic correlation or linkage disequilibrium (LD) (de Bakker et al. 2006a). Powerful analytical tools have also utilized the comprehensive information from these databases to increase genomic coverage and fine-map association signals through statistical imputation of untyped genetic variants that exist in the databases (Marchini et al. 2007; Servin and Stephens 2007). Central to the selection of tag SNPs and the use of HapMap populations as reference panels in imputation is the assumption that patterns of LD are similar between the target and HapMap populations (Clark and Li 2007; Marchini et al. 2007). Population differences in LD structure between an untyped functional polymorphism and surrounding assayed markers can compromise the effectiveness of pooling genetic data in meta-analyses, as failures to replicate genuine findings may happen when a marker is in substantial LD with the functional polymorphism in one population but not in other populations (Teo et al. 2009).
In order to assess the extent of differences in the patterns of LD between populations on a genome-wide scale, a new method, variation in LD (varLD), was developed. Our method builds upon analytical approaches developed for comparing regional patterns of correlations (Krzanowski 1993) that have been previously implemented for contrasting LD between cases and controls in association mapping (Zaykin et al. 2006). The genome-wide analysis investigates the extent of LD differences in each genomic region relative to the rest of the genome, identifying regions that are found in the right tail of the distribution of varLD scores across the genome when comparing between two populations. This identifies genomic regions where the extent of LD variation between two populations is greater than the rest of the genome. In situations where genome-wide data are unavailable, we introduced a permutation strategy that allows the comparison of a localized region, yielding a statistical significance for testing the null hypothesis of no differences in patterns of LD between two populations.
We apply our method to compare the patterns of LD across all possible population pairs with the three HapMap populations, consisting of: (1) individuals in Utah with European ancestry (CEU); (2) Yoruba people sampled from the Ibadan region in Nigeria (YRI); and (3) a combined group of Han Chinese from Beijing and Japanese from Tokyo (CHB+JPT). We also compare the YRI to another African population, which consists of the Jola people from the Gambia (Jallow et al. 2009), as well as between two populations of European descent given by the CEU and subjects from the British Isles (WTCCC) (The Wellcome Trust Case Control Consortium 2007; see Supplemental material). In addition, we surveyed across 300 random regions identified to be in the top fifth percentile in our comparisons between CEU and CHB+JPT, where for each region we identified the most common haplotype in CEU and compared the frequency of this haplotype in the CEU and CHB+JPT samples. We also assessed the portability of an identified association in each of these regions across the two populations. We observed that the evidence of LD variation for these regions correlated significantly with the degree of difference in haplotype frequencies, and there were greater disparities in the portability of the association signals in these regions. Regions undergoing population-specific positive natural selection are more likely to contain diverse patterns of LD between populations, and we cross-referenced the varLD signals against the established regions carrying signatures of positive selection, showing significant concordance. The use of the permutation strategy to quantify regional LD variations in the absence of genome-wide data also correctly characterized the expected variations at two loci experiencing strong evolutionary pressure, which is expected to introduce haplotypic variations.
Methods
Data sets
We used the genotype data for autosomal chromosomes from Phase 2 of the International HapMap Project in our analyses, which consisted of 3,790,590 SNPs for the 60 unrelated parent individuals from the CEU panel, 3,733,291 SNPs for the 60 unrelated parent individuals from the YRI panel, and 3,821,888 SNPs for the 90 unrelated individuals from the CHB and JPT panels. Only SNPs from the HapMap samples with <20% missingness and with minor allele frequencies >5% were used. To avoid the effects of sample size differences in the calculation of LD, 60 control samples were chosen from each of two separate case-control studies carried out in the Gambia by the MalariaGEN Consortium (Jallow et al. 2009) and in Great Britain (The Wellcome Trust Case Control Consortium 2007). The samples from the Gambia consist of 60 individuals who reported their ethnic group as Jola, and for which their ethnic memberships were subsequently genetically verified, out of a total of 1382 control subjects; the samples from Great Britain consisted of 60 randomly chosen subjects from the possible set of 1481 controls for the 1958 British Birth Cohort that formed part of the control samples used in The Wellcome Trust Case Control Consortium (2007). Data from the MalariaGEN and the Wellcome Trust Case-Control Consortium were downloaded from the European Genome-phenome Archive with permission from the respective Data Access Committee. These two sets of samples have been genotyped on the Affymetrix GeneChip 500K set, yielding 490,032 SNPs on the autosomal chromosomes. The genotypes for these data have been called using the CHIAMO algorithm (The Wellcome Trust Case Control Consortium 2007), and only SNPs with <5% missingness across all the control samples in each study and with minor allele frequencies >5% have been included in our analysis.
Quantification of LD
The r2 is a popular measure in population genetics for assessing the strength of the genetic correlation between the alleles of two SNPs. Here, we consider the signed r2 to quantify the extent of LD between two SNPs since this measure additionally reflects the direction of the correlation between the two SNPs, and has thus been shown to be more appropriate in comparing LD (Teo et al. 2009). For two biallelic SNPs with alleles (A, a) and (B, b), respectively, the signed r2 is defined as
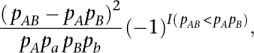 |
where pAB denotes the frequency of haplotype AB; pA, pa, pB, and pb denotes the respective allele frequencies; and I(pAB < pA pB) denotes an indicator function taking a value of one when pAB < pA pB, and zero otherwise.
Eigen-analysis of regional LD between two populations
Let G1 and G2 denote the genotype data for a common set of S SNPs across two populations. For each population, we divide the set of S SNPs into (S − L + 1) overlapping windows of L SNPs, where each consecutive window is obtained by shifting the existing window in the direction of the forward strand by one SNP. For a particular window of L SNPs, let M1 and M2 denote the two L × L symmetric matrices for the two populations, respectively, such that the (i, j) entry in each matrix represents the signed r2 between SNP i and SNP j. Thus, each of these matrices effectively represents a correlation matrix between the L SNPs in the respective population. It has been shown that testing for equality between the elements of two correlation matrices can be achieved by comparing the extent of departures between the ranked eigenvalues of the two matrices (Krzanowski 1993; Zaykin et al. 2006). We perform an eigen-decomposition on each of the two LD matrices such that for population k we obtain Mk = Γk Δk ΓkT, with columns of Γk denoting the eigenvectors of Mk, and Δk is a diagonal matrix with entries comprising the sorted eigenvalues of Mk in descending order. We define the raw varLD score as the trace of |Δ1 − Δ2|, and the magnitude of this score provides a measure for the extent of dissimilarity between the correlation matrices M1 and M2 that we subsequently use to quantify the extent of regional LD differences between the two populations. We ran an initial analysis on the effects of varying the window size L in our comparison between the HapMap CEU and the WTCCC 58C populations, with L as 25, 50, 100, and 200. As varying L has been observed to yield consistent results (see Results), we have chosen a window size L of 50 in all subsequent analyses. We did not choose to define the sizes of the windows by the genetic or physical distance spanned, as such definitions will result in windows encapsulating a different number of SNPs, which does not allow for genome-wide comparison across different windows.
Computation of Monte Carlo statistical significance
We can use permutational procedures to obtain a Monte Carlo statistical significance for each window of L SNPs in order to evaluate the strength of the evidence against the null hypothesis of no differences in regional LD structure between two populations (Krzanowski 1993). For each window of L SNPs at any two populations with n1 and n2 samples, respectively, we can calculate the raw varLD score and define this as the empirical test statistic Temp. Under the null hypothesis of no differences in the LD structure in this region of L SNPs, we can merge the data from both populations. By resampling n1 and n2 samples from this combined data with replacement to yield two ”populations” of sample sizes identical as previously observed, we can calculate the corresponding varLD score, which is effectively a random draw from the null distribution of no differences in regional LD. The Monte Carlo significance is thus defined as (M + 1) / (Niter + 1), where M denotes the number of varLD score obtained from the resampling scheme that is larger than the empirical test statistic Temp, and Niter denotes the total number of permutations. Significance values reported in the paper for the LCT and DARC genes are calculated with Niter of 10,000.
Genome-wide quantification of varLD scores
It can be computationally expensive to calculate Monte Carlo significance when assessing genome-wide data. In such situations, we do not define the statistical significance of the score for each window but instead make use of the relative rank of the score as a surrogate measure of the extent of LD differences in that region relative to the rest of the genome. While an empirical significance may be approximated for each observed score as the proportion of scores larger than itself, this is misleading and does not provide any conventional interpretation of statistical significance, as such a statistic (1) can be affected by the density of the SNPs both within and outside of the considered region, since a densely genotyped region with real LD differences will yield more occurrences of high varLD scores compared with a region that is sparsely assayed, and (2) is not drawn from the true distribution under the null hypothesis of no differences. Instead, we flag a region if the associated score si is greater than or equal to the score at the 95th percentile, and consider this as a candidate region containing LD differences. While this strategy is also affected by SNP density, we avoid any reference to the term “statistical significance” and thus any subsequent interpretations that are associated with it. As the magnitude of the raw varLD scores is affected by the size of the windows L and the populations being compared, we prefer to use the standardized score 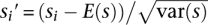 , where E(s) and var(s) denote the empirical mean and variance of the collection of scores across the genome. We reiterate, however, that this serves only to center the distribution of the scores around a mean of zero and a standard deviation of one, and the standardized scores are in no way related to the quantiles of a standard normal distribution (Supplemental Fig. 1).
, where E(s) and var(s) denote the empirical mean and variance of the collection of scores across the genome. We reiterate, however, that this serves only to center the distribution of the scores around a mean of zero and a standard deviation of one, and the standardized scores are in no way related to the quantiles of a standard normal distribution (Supplemental Fig. 1).
Comparison of haplotype frequencies
To assess whether there are different dominant haplotypes in a region identified by our approach between two populations, we randomly selected 300 regions in the top fifth percentile from our comparison between the HapMap CEU and HapMap CHB+JPT samples. In each of these regions, we identified the most common haplotype form seen in the 120 chromosomes for the CEU samples and compared the frequency of this haplotype between the CEU and CHB+JPT chromosomes. As a control, we also selected 300 regions randomly across the genome where each of these regions spans a physical distance that is matched to one of the 300 regions identified by varLD. In order to assess the relationship between the evidence of LD variation and the difference in haplotype frequencies across these 600 regions, we perform a linear regression of the absolute difference in haplotype frequencies with the standardized varLD score and assess the correlation using the Pearson's correlation coefficient.
Simulation of replication across CEU and CHB+JPT
In each of the 300 selected regions identified by our approach in the genome-wide comparison of LD variation between CEU and CHB+JPT, we selected a focal SNP that is typed in both the CEU and CHB+JPT samples and was located nearest to the center of the region. We simulated an effect size corresponding to a multiplicative risk of 1.3 for the minor allele (defined in CEU) at this focal SNP in both CEU and CHB+JPT, assuming a baseline penetrance of 20% for the genotype homozygous for the major allele. We simulated 2000 cases and 2000 controls, where each subject is sampled by drawing two chromosomes randomly from the phased haplotypes from the relevant HapMap population, and the phenotype status assigned as a binomial draw with probability given by the penetrance associated with the genotype at the focal SNP. This method of simulating cases and controls maintains the empirical LD structure observed in the actual HapMap samples. We subsequently mask the focal SNP and identify the SNP in CEU that carries the strongest association signal that was also typed in the CHB+JPT samples. Consequently, we compared the statistical evidence of this “indirect-associated” SNP across the CEU and CHB+JPT samples.
Web resources
Information on established structural variants was obtained from the Database of Genomic Variants (http://projects.tcag.ca/variation/tableview.asp?table=DGV_Content_Summary.txt) maintained by the Center for Applied Genomics, Department of Genetics and Genomic Biology, MaRS Center, Toronto, Canada. The database in Build 35 (hg17) coordinates was used.
Results
We first investigated whether the size of the window used affects the outcome of the genome-wide comparison, by considering four separate genome-wide comparisons between the HapMap CEU and WTCCC 58C samples with the window size L set at 25, 50, 100, and 200 SNPs, respectively. We observed that the size of the window can affect the resolution of the boundaries of the regions, and the sensitivity toward identifying smaller regions. In particular, signals near the 95th quantile that correspond to smaller regions are more sensitive to window sizes. However, the top regions that are identified are consistent across the window sizes (Fig. 1). The robustness of the top signals to the variation in the window sizes is reassuring, and we chose a window size of L = 50 for all subsequent analyses.
Figure 1.

Comparisons across different window sizes L. Comparisons of the standardized scores for regions identified in our analysis of LD differences between HapMap CEU vs. WTCCC 58C with different numbers of SNPs in each window. Four separate analyses were run with L = 25, 50, 100, and 200 SNPs, respectively, where comparisons were made against the regions identified with L = 50. For each of the regions identified for L = 50, we noted the maximum standardized varLD scores in this region in the analyses with L = 25 (A), 100 (B), and 200 (C). Each point in the figures represents a region identified in the original analysis with L = 50. The size and shade of each point indicates the relative size of the region, with larger circles and darker shades of gray indicating larger regions. (Black shading) Regions with sizes >500 kb.
Our analysis identifies regions where the extent of variation in LD between two populations is greater than in the rest of the genome. Owing to the use of consecutive windows in our approach for comparing regional LD, we observed that the sharpness of the varLD signals depends on the SNP density in each region. Comparisons between the HapMap populations, which genotyped in excess of 3 million polymorphisms, therefore yielded sharper and narrower regions of differences in comparison to analysis involving the Gambian and WTCCC samples, which assayed only half a million polymorphisms.
Haplotype frequency and varLD
One of the consequences for the presence of different dominant haplotypes between populations is the possibility that patterns of LD will differ between these populations. Here, we define a dominant haplotype in a population as the most common haplotype that is seen in this population. We undertook a survey of the differences in haplotype frequencies in 300 randomly selected regions that have been identified to be in the top fifth percentile in our comparison between the HapMap CEU and HapMap CHB+JPT samples. For each of these regions, we also randomly selected a region across the genome that spans the same physical distance and evaluated the haplotype frequencies observed in CEU and CHB+JPT. The dominant haplotype in CEU in each of these regions is identified, and the absolute difference in the frequencies of this haplotype in CEU and CHB+JPT is calculated. Comparing against the evidence from varLD in each region, we observed a significant correlation between the standardized varLD scores and the absolute difference in haplotype frequencies (Pearson's correlation coefficient = 0.25, P = 5.1 × 10−10), where every unit increase in the standardized varLD score results in an expected increase of 0.018 (95% CI: 0.013 − 0.024) in the difference of the haplotype frequencies. This indicates that, for the most common haplotype seen in CEU, there is a greater disparity in the frequency of this haplotype between CEU and CHB+JPT in regions with stronger evidence of LD variation.
An example where identified regions of LD variation corresponded with established literature of haplotypic differences is in the region encapsulating the NRG1 gene. This region was identified in comparisons between the HapMap populations, but not in comparisons between the Gambian and YRI, or between CEU and WTCCC 58C (Fig. 2; Supplemental Fig. 2). A specific haplotype in the NRG1 gene was implicated in schizophrenia in haplotype-based and fine-mapping studies conducted in Icelandic and Scottish populations (Stefansson et al. 2002, 2003). This association, however, was not replicated in the Han Chinese (Zhao et al. 2004), and separate studies with both microsatellite markers and SNPs in the NRG1 gene identified different haplotypes to be associated instead (Li et al. 2004; Zhao et al. 2004). A detailed survey of the genetic variation in this gene across 39 populations revealed allelic and haplotypic frequency differences that correlate with geographical regions, particularly in SNPs located in an intron of the gene (Gardner et al. 2006). A survey of the haplotype diversity across this region with the data from the Human Genome Diversity Panel similarly indicated significant heterogeneity in the distribution of haplotypes in this region, particularly between the East Asian and European samples (Supplemental Fig. 3; Pickrell et al. 2009). This concurs with our observation of significant LD variation in this region between the broad continental areas but not between populations with similar ancestries.
Figure 2.

LD variation at the NRG1 gene on chromosome 8. (Upper panel) Standardized varLD scores across the region encapsulating the NRG1 gene. (Red points) LD comparisons between HapMap Europeans (CEU) and HapMap Asians (CHB and JPT); (purple points) LD comparisons between HapMap Europeans (CEU) and HapMap Africans (YRI); (cyan points) LD comparisons between HapMap Africans (YRI) and HapMap Asians (CHB and JPT). (Dotted lines) Values of the corresponding thresholds. (Middle panel) Fine-scale recombination rates in the region from the combined HapMap samples. Positions of genes in the region shown in the bottom panel were obtained from Ensembl. All coordinates shown are in NCBI Build 35 (dbSNP build 125).
Simulating the portability of association signals
The most important application of a method for assessing LD variation between populations is in addressing the portability of any signals of phenotypic association across these populations. In the above simulations with the HapMap CEU- and CHB+JPT-phased haplotypes, we modified the procedure to artificially introduce a disease effect at a focal SNP that we subsequently masked from the association analysis (see Methods). We can thus additionally investigate whether the neighboring SNP that carries the strongest association signal in the CEU samples will similarly exhibit a significant association signal in the CHB+JPT samples. This simulation exercise is akin to the popular approach where the phenotype-associated SNPs that emerged from a genome-wide scan performed in a European population are typed in other populations around the world to assess the replicability of the initial findings. Replicating an association across diverse populations fundamentally relies on the existence of similar patterns of LD between the unknown causal variants and the genotyped SNPs, and we expect greater inconsistencies in replication success in genomic regions where patterns of LD are more dissimilar across populations. In our assessment of the same 300 regions identified by varLD in the comparison between CEU and CHB+JPT, we noticed that there was considerably greater variability in the statistical evidence observed at the same SNP across the two populations (Fig. 3A), compared with the 300 matched regions randomly selected across the genome where there was less variation in the association signal at the same SNP in the two populations (Fig. 3B).
Figure 3.

Differences in statistical evidence at the associated SNP in CEU and CHB+JPT. Comparison of the −log10 P-value from a test of association between 2000 simulated cases and 2000 simulated controls at an associated SNP in each of the HapMap CEU and CHB+JPT populations. For each SNP, the larger −log10 P-value is set as the baseline and is mapped to zero, and we only plot the difference of the −log10 P-values. The regions are then ranked from left to right by increasing the degree of the difference in statistical evidence between CEU and CHB+JPT. (A) Three hundred randomly selected regions that have been identified by varLD to be in the top fifth percentile of the genome-wide distribution. (B) Three hundred regions that have been randomly selected across the genome, where each region spans an identical physical distance to one of the 300 varLD-identified regions from A. (Green circles) Differential statistical evidence observed in the CEU; (red circles) differential statistical evidence observed in the CHB+JPT.
An example where patterns of LD differ between the causal variant and the surrounding markers across populations can be found at the hemoglobin beta (HBB) gene region, which encapsulates the locus (rs334) that causes sickle cell anemia and confers protection against malaria. This region forms a useful case study for investigating the effects of interpopulation LD variation on the portability of association signals since the underlying causal variant is actually known. In a recent genome-wide survey on the genetic etiology of severe malaria in the Gambia, the SNP on the Affymetrix 500K array carrying the strongest association signal in the HBB region is most correlated with rs334 with an LD of r2 = 0.32. This SNP, however, has negligible LD in the HapMap YRI samples (r2 = 0.009) (Jallow et al. 2009), and any attempts to reproduce the association in Yoruba samples by typing this identified SNP will not result in a successful replication experiment. Conversely, the SNP with the strongest LD with rs334 in the Yoruba samples (r2 = 0.35) failed to exhibit any indication of malaria association, since the LD with the causal variant in the Gambian samples was only r2 = 0.005. This illustrates the consequence that variation in patterns of LD between populations can have on replication studies that are executed across different populations. In our comparison of LD between samples from the Gambia and the HapMap African data, the HBB region surrounding rs334 was identified to contain significant LD differences in the top fifth percentile of the genome-wide distribution, indicating a greater degree of variation compared with the rest of the genome. In particular, the LD in this region was also identified to be considerably different when comparing the HapMap Yoruba samples to the HapMap East Asian and European populations (see Fig. 5, below).
Figure 5.

Standardized varLD scores across different population pairs in established regions undergoing positive natural selection or containing high haplotype diversity. The standardized varLD signals for each population pair are shown, and only scores above their respective 95th quantiles are illustrated in a nongray color. (Red points) LD comparisons between HapMap Europeans (CEU) and HapMap Asians (CHB and JPT); (purple points) LD comparisons between HapMap Europeans (CEU) and HapMap Africans (YRI); (cyan points) LD comparisons between HapMap Africans (YRI) and HapMap Asians (CHB and JPT); (green points) LD comparisons between two European populations (HapMap CEU vs. WTCCC 58C); (blue points) LD comparisons between two African populations (HapMap YRI vs. the Gambian Jola). The four regions considered contain the LCT gene in chromosome 2 undergoing selection in European populations (A), the SLC24A5 gene in chromosome 15 reported for association with skin pigmentation in Europeans (B), the HBB gene in chromosome 11 with well-documented haplotypic differences between the two populations considered (C), and the highly polymorphic MHC region in chromosome 6 (D). (Dotted lines) Approximate start and end positions of the gene/region in each panel.
Top signals of LD variation
Genome-wide exploration across multiple population pairs identified 88 regions where signals of significant LD variations were observed in all five pairs of comparisons, of which the majority (75 out of 88) corresponded to regions that encapsulate reported copy number variants. In particular, 16 of the top 20 candidate regions with the strongest signals of LD variation contain copy number differences in multiple populations across Europe, East Asia, and African Yoruba (Table 1; Supplemental Fig. 4). Perhaps not surprisingly, one of these 20 regions contains the highly polymorphic HLA gene cluster in class II of the major histocompatibility complex (MHC), while a region that does not overlap with known structural variants contained an olfactory receptor gene cluster (OR10Z1, OR6K2, OR6K3, OR10K2, OR10T2, OR10X1) on chromosome 1. We observed that one of the signals spans the region on chromosome 12 reported to be associated with Type 1 diabetes (The Wellcome Trust Case Control Consortium 2007) containing the candidate gene PTPN11 implicated in myeloid leukemia (Tartaglia et al. 2003; Bentires-Alj et al. 2004), and insulin and immune signaling (Mustelin et al. 2005).
Table 1.
Top 20 candidate regions with overlapping signals of LD differences in all five sets of comparison madea

aCEU–CHB+JPT; CEU–YRI; CHB+JPT–YRI; CEU–WTCCC 58C; Gambian Jola–YRI.
bCopy number differences refer to the occurrence of both insertions and deletions.
cCEPH/Chinese/Japanese/Yoruba samples: Kidd et al. (2008); German: Pinto et al. (2007); HapMap samples: Conrad et al. (2006), McCarroll et al. (2006), Redon et al. (2006), Pinto et al. (2007); French: de Smith et al. (2007); Canadian Ontario controls: Zogopoulos et al. (2007); HapMap-CEU: Wang et al. (2008); 36 diverse human samples: Mills et al. (2006).
CNV, copy number variation.
In the analyses between the HapMap populations, we observed a consistent trend that regions containing structural variations in the top 20 signals for each comparison carry copy number differences in at least one of the two targeted populations (Supplemental Tables 1–3). Regions identified between CEU and the HapMap East Asian (CHB+JPT) samples contain the progesterone receptor (PGR), the G-protein-coupled receptor gene (GPR74), and the region containing the solute carrier family 24 member 5 (SLC24A5) (Supplemental Table 1). The top signals between CEU and YRI not containing structural variants include the breast carcinoma amplified sequence 3 (BCAS3) gene containing a functional estrogen response element, the X-prolyl aminopeptidase (XPNPEP1) gene important in the digestion of resistant dietary protein, and a member of the calcium-binding protein superfamily (NCALD) that has been suggested to be vital in the regulation of the process of neuronal signal transduction (Supplemental Table 2). For the signals between YRI and the JPT+CHB not involving structural variants, we identified the mitochondrial ribosomal protein S18C (MRPS18C) that contains an element of the mitochrondrial ribosome in the nuclear genome, the BRCA-associated ring domain 1 (BARD1) associated with tumor suppression activities, and the ATP-binding cassette subfamily A member 12 (ABCA12) gene implicated in epidermal conditions (Supplemental Table 3).
Four gene clusters were observed in the top 20 signals comparing between the Gambian Jola samples and the HapMap Yoruba individuals (Supplemental Table 4). These include the late cornified envelope (LCE) gene cluster in the epidermal differentiation complex on chromosome 1, the histone 1 gene family on chromosome 6, the HLA cluster, and the olfactory receptor 4 (OR4) superfamily cluster on chromosome 11. The strongest signal in this comparison was observed in the methylthioadenosine phosphorylase (MTAP) gene that has a major function in the metabolism of polyamine and is implicated in lymphoblastic leukemia. Eleven of these 20 regions have been reported to carry copy number variants in at least the Yoruba samples, consistent with the earlier observations that population-specific copy number variations concur with signals of interpopulation LD differences. Similarly, 12 of the top 20 signals in the comparison between the two sets of samples with European ancestries (HapMap CEU and WTCCC 58C) are found in regions containing copy number differences that have been reported in European populations (Supplemental Table 5). The signals that do not overlap with known copy number variants include regions that encompass the olfactory receptor cluster on chromosome 1 discussed above; the solute carrier family 3 member 1 (SLC3A1), whose protein products have been shown to be involved in cystine, dibasic, and neutral amino acid transport; and the glutamate receptor metabotropic 3 (GRM3) gene involved in presynaptic inhibition of glutamate release.
LD variation and positive natural selection
Genomic regions experiencing strong forces of positive selection can yield haplotypic backgrounds that are substantially different across diverse populations (Sabeti et al. 2007), as the haplotype on which the selected allele sits on is expected to dominate. One possible implication of such haplotypic differences between populations is the existence of significant variations in the patterns of LD in these regions. We thus expect regions where selection pressure exists in one particular population but not the others to present strong evidence of LD differences. Two well-known regions that have undergone strong evolutionary pressure in specific populations are the LCT and DARC genes, which serve as useful examples to illustrate where the conventional reliance on visual tools like heatmaps (The International HapMap Consortium 2007; The Wellcome Trust Case Control Consortium 2007) for qualifying the extent of variation between LD patterns between two populations can be subjective (Fig. 4). Our analysis with the described permutation strategy (see Methods) correctly indicated the regional LD of SNPs in the LCT gene to be significantly different between Europeans and Asians (P < 0.0001), and between Europeans and Africans (P < 0.0001), but less so between Asians and Africans (P = 0.145). This concurred with the evidence that the LCT gene underwent positive natural selection in European populations where dairy products form an integral component of the diet, conferring the ability to metabolize lactose to persist into adulthood (Bersaglieri et al. 2004; Sabeti et al. 2006). Appropriately, at the DARC gene containing the Duffy antigen that swept to fixation in the African continent (Hamblin and Di Rienzo 2000; Hamblin et al. 2002), significant LD differences were observed when comparing the Yoruba samples against either the Europeans (P < 0.0001) or the Asians (P < 0.0001), but not between the Europeans and Asians (P = 0.528).
Figure 4.

Heatmap representations of LD in two genomic regions between pairs of populations in HapMap. The upper left and lower right triangles of each plot correspond to the LD in a region for each of two populations, respectively, as measured by the pairwise r2 metric, with the plots in the first column comparing HapMap Europeans with HapMap Asians, the second column comparing HapMap Europeans with HapMap Africans, and the last column comparing HapMap Africans with HapMap Asians. The plots in the first row depict the same genomic region on chromosome 2 of 136.26 Mb–136.38 Mb spanning the LCT gene, while the plots in the second row depict the genomic region on chromosome 1 of 155.9 Mb–156.0 Mb spanning the DARC gene.
In addition to the analyses above, we also illustrated the use of our genome-wide approach in four well-known regions that contain significant haplotype diversity or are subjected to strong evolutionary pressure. These regions are: (1) the LCT gene introduced above and (2) the SLC24A5 gene that has been selected in the European population for skin pigmentation (Lamason et al. 2005), in both of which, we expect LD variations between Europeans (CEU) and non-Europeans (CHB+JPT and YRI; Fig. 5A,B); (3) the HBB region where evolutionary pressure on the sickle-cell mutation resulted in the rising of different haplotypic backgrounds in the Gambia (the Senegal haplotype) and Nigeria (the Benin haplotype) (Hanchard et al. 2007; Daily and Sabeti 2008); thus, we expect LD variations between the two African populations and between YRI and the non-African HapMap populations (Fig. 5C); and (4) the highly polymorphic MHC region (de Bakker et al. 2006b), where we expect LD variations to be present in all our pairwise population comparisons (Fig. 5D). These regions appropriately exhibit strong evidence of LD variations between the relevant population pairs considered. In addition to the four established regions of positive natural selection, we also cross-referenced the genomic regions identified by our approach with the 20 top candidates for positive selection in the HapMap populations (Sabeti et al. 2007). Of these regions, 17 contained varLD signals in the top fifth percentile, of which eight are located in the top percentile of the distribution (Table 2; Supplemental Fig. 5). Four of these regions were located in the top 0.1 percentile of the distribution, including the region encapsulating SLC24A5. Our findings for some of these regions experiencing selection pressure in two populations appropriately indicated that regional LD was not significantly differentiated in both selected populations but was dissimilar against the third population, suggesting the same selection forces may have acted on both populations (for example, 22.55 Mb–22.85 Mb on chromosome 10). The agreement between fundamentally different approaches to assess genomic diversity is reassuring, as this provides independent evidence that these regions may be functionally significant.
Table 2.
Distribution of varLD scores for the 20 autosomal candidate regions with strongest signals for natural selection in the HapMap populations

aApproximate coordinates and gene information as reported in Sabeti et al. (2007).
Discussion
We have introduced a novel method for performing genome-wide comparisons of LD between populations, and have applied the method to the HapMap populations and two other populations with African and European ancestries, respectively. Using our approach, we observe a significant correlation in the evidence of LD variation with the degree of disparity in the frequencies of the common haplotypes between two populations. Regions identified in our analyses to contain variation in regional patterns of LD also exhibited greater differences in the replication signal when attempting to reproduce the primary association in a different population. In the absence of genome-wide data for comparisons, we have introduced a permutation scheme that quantitatively assesses the degree of LD variation between two populations. This produces a statistical significance for testing the null hypothesis that the LD patterns in the region are identical between the two populations. We have illustrated the use of this permutation scheme in two well-established genomic regions with diverse haplotypic structure between global populations.
To minimize false-positive associations identified in genome-wide disease studies, it is necessary to replicate the findings in an independent cohort within the same population or in other populations (Chanock et al. 2007). A number of possible explanations exist when phenotype–genotype associations identified in one population fail to replicate in another population. Assuming that the initial association was not a false-positive result and the same functional variant exists across populations, the inability to reproduce the finding across other populations is most likely due to: (1) lower frequencies of the functional allele in the different populations; (2) underlying differences in environmental influences underpinning a complex gene–environmental effect; (3) variations in patterns of LD between the functional variants and the assayed polymorphisms. The third explanation can even confound replication candidate gene studies with large sample sizes leading to low statistical power, as the associated polymorphism is simply not in sufficiently strong LD with the functional polymorphism in a different population to present any evidence of replicated association (Teo et al. 2009). Knowledge of the extent of LD differences around the functional polymorphisms can thus be valuable when attempting to replicate disease associations across populations. As varLD utilizes regional patterns of LD to identify variations across populations, it does not require the functional polymorphism to be assayed when calculating varLD scores, as long as the extent of LD with surrounding markers is sufficiently long relative to the density of the genotyped SNPs. Given the popularity of genome-wide strategies in disease studies, we foresee that the assessment of LD variation between disparate populations will become increasingly common when confronted with conflicting evidence of disease association.
One important consequence of disparities in patterns of LD between two populations is the effect on imputation strategies that use the haplotypic framework from a reference population to infer probabilistically the genotypes of the unobserved SNPs in a target population (Marchini et al. 2007; Servin and Stephens 2007). While these strategies can yield improved power in disease association studies, they generally rely on the assumption that the structure of genetic correlation and recombination is similar across the two populations. For regions where the LD structure in the reference panel does not reflect that in the target population, imputation may not produce a set of confident genotypes. We explored the relationship between the evidence from varLD and the diagnostics generated from a well-calibrated imputation algorithm IMPUTE (Marchini et al. 2007) by using the HapMap African data (YRI) as a reference panel to impute the data from the Gambia (see Supplemental material). We observed that, across the genome, regions identified with substantial LD differences relative to the genome concurred with greater imputation uncertainty as implied by lower imputation accuracy, information, and posterior probabilities in assigning genotype calls (Fig. 6). This reinforces the understanding that the diagnostics of IMPUTE are well-calibrated and appropriately downweighs the confidence of the genotype inference in regions with substantial disparities in the patterns of LD between the reference and target populations. In the absence of an established strategy for comparing LD, imputation diagnostics serve as useful surrogates for validating the efficacy of varLD, just as candidate regions undergoing population-specific positive natural selection provide empirical regions with putatively different LD patterns across populations for assessing concordance with the varLD signals.
Figure 6.

Imputation diagnostics and standardized varLD scores. Comparison of the standardized varLD score against imputation diagnostics generated by IMPUTE when the HapMap YRI is used as a reference panel against Gambian Jola data. The imputation algorithm calculates a measure of information and a confidence score based on the average maximum posterior probability, which we used as surrogates of imputation accuracy. A composite measure of imputation accuracy as measured by the product of call rate and genotype concordance is calculated for the 10 deciles of varLD scores found in the top 20th percentile of the genome-wide distribution of varLD scores. As concordance is measured as the proportion of agreement between the imputed and observed genotypes for the Gambian Jola samples, we only consider autosomal SNPs on the Affymetrix array that are found in the regions identified by varLD.
The inability to accurately impute and fine-map disease association signals using a reference panel that has dissimilar regional haplotype structure to the target population was also clearly demonstrated in the HBB gene region: The use of HapMap YRI as the reference to impute the sickle-cell anemia mutation (rs334) in a case-control study of malaria in the Gambia did not yield any association, whereas the use of sequence data from the Gambia as a reference panel correctly imputed and fine-mapped the association signal to rs334 directly (Jallow et al. 2009). Although this is attributed to the well-known fact that different haplotypic structure exists in the HBB region between different African populations (Hanchard et al. 2007), it is unclear how prevalent such interpopulation differences will be across the genome. As imputation strategies are increasingly being proposed as a tool for fine-mapping, particularly with the availability of whole genome sequence data from the 1000 Genomes Project (http://www.1000genomes.org), it is important to acknowledge that LD differences between the reference panel and the target population can potentially confuse the process of localizing the causal variant. While it was recently established that incorporating reference samples from a mixture of two or more HapMap populations can improve imputation performance in non-African populations (Huang et al. 2009), it was also observed that for African populations there was no significant boost in performance beyond that accorded by the use of just the HapMap YRI as reference. This is not unexpected given that (1) the HapMap YRI samples consist of 30 trios sampled from a considerably homogeneous group in Nigeria, which is unlikely to be representative of the genetic diversity in Africa, and (2) LD is observed to span a shorter distance in YRI compared with European and Asian populations. The ability to quantify the extent of LD variations between populations will undoubtedly be useful in the context of genome-wide association studies conducted in African populations, especially since these studies have been postulated to provide greater success in localizing the functional variants.
As preliminary data from targeted-sequencing studies become available, it is increasingly evident that the presence of long-range and high LD that was advantageous for genome-wide association studies in European populations is instead an impediment for the localization of the causal variants. The approach of sequencing a specific region carrying a veritable association signal aims to fine-map the functional variant by assaying every genetic position in that region, and subsequently testing each polymorphic position for correlation with the phenotype. Ideally, the functional variant will present the strongest association signal, conspicuously overshadowing the evidence from neighboring markers. However, in a population with long-range LD in the implicated region, multiple nearby markers may be in perfect LD with the causal variant, which results in a plateau of signals of similar magnitude that does not allow the functional polymorphism to be easily distinguishable. Knowledge of which populations contain significantly diverse patterns of LD in this region becomes useful, since the integration of haplotype data and signals from genome-wide scans across these populations can refine the boundaries within which the causal variant is expected to lie, and potentially elucidate the causal SNP from surrogate markers in high LD should the haplotype structure across different populations be sufficiently diverse (Y Teo and E Tai, in prep.). This approach relies on identifying populations with diverse haplotypes and containing significant variations in patterns of LD, which is conceptually distinct but complementary to the use of imputation strategies for fine-mapping the causal variant.
The concurrence between top signals of LD differences and population-specific copy number variants suggests that conventional quantification of LD may be biased and potentially erroneous in regions containing these structural variants, particularly when we observe the same pattern of concurrence with the HapMap samples. This insight is important in the evaluation of and the reliance on LD in population genetics and disease scans, since regions of apparent low LD may be attributed to the presence of structural variants like insertions and deletions that confound LD assessment by introducing an overrepresentation of homozygous genotypes. This is certainly true in our observation that structural variants that concur with signals of LD differences are almost entirely composed of insertions and/or deletions. Large-scale genotyping on commercial arrays currently rely on the use of automated and unsupervised calling algorithms to assign genotypes based on the extent of fluorescence produced by allelic hybridization (Teo et al. 2007; The Wellcome Trust Case Control Consortium 2007). These algorithms are typically trained to search for genotype groups with two allelic copies and do not account for SNPs with variable allelic copy numbers. In regions containing insertions and deletions, deviation in allelic copy number from the expected two copies can result in hybridization profiles that either generates greater missingness or more homozygous genotypes (Fig. 7). Calculating the LD involving these SNPs will likely bias the LD statistic.
Figure 7.

Genotype assignment and hybridization intensity profiles of a SNP in a region containing deletions. The two axes represent the fluorescence intensities that indicate the extent of hybridization to the two possible alleles of a biallelic SNP, which have been generically defined as alleles A and B. Solid circles in red, green, blue, and gray indicate samples whose genotypes have been assigned as AA, AB, BB, and NULL (missing), respectively. (Dashed ellipses) Intensity profiles that correspond to homozygous deletion (gray), hemizygous A deletion (light green), hemizygous B deletion (purple), genotype AA (red), genotype AB (dark green), and genotype BB (blue). The figure illustrates that samples with hemizygous deletions have been erroneously assigned to homozygous genotypes, while samples with homozygous deletions have been classified as missing.
One practical concern in our implementation of varLD on comparing patterns of LD between populations is thus the effects of genotyping errors or data fidelity on the identification of genomic regions with apparent LD variations, particularly when the genotype data for one population is of a higher quality (e.g., HapMap data) compared with the second population (e.g., from genome-wide studies using commercial genotyping technologies). The presence of a SNP affected by genotyping error serves to break down LD between itself and surrounding markers, which can introduce artificial signals of LD differences. In a simulation study to investigate the effects of genotyping errors on the sensitivity of varLD (see Supplemental material), we found that the presence of SNPs with confounded genotyping can introduce artificial signals of LD differences, particularly when genotyping problems affect contiguous stretches in the genome (Supplemental Table 6). While genotyping errors at a SNP have minimal effect in yielding large varLD signals, a conservative approach of quality checking the genotype data prior to analysis is strongly recommended. When data sets of multiple populations are available, observing overlapping signals from multiple comparisons across different populations also serve to minimize the possibility of artifacts due to genotyping issues.
While genotyping errors stemming from the presence of common copy number polymorphisms can explain the varLD signals that are consistently present in comparisons between multiple pairs of populations, genomic regions undergoing strong evolutionary pressure of adaptation and selection may also contain population-specific haplotype structure that results in diverse patterns of LD variations between different groups. The identification of the HLA-gene cluster in the MHC region is reassuring, since in the absence of any established methods for addressing LD variation, established genomic regions containing significant haplotype diversity across populations or undergoing population-specific positive natural selection provide useful surrogates for validating the method. Conversely, this may also suggest that the consistent identification of a particular genomic region across multiple comparisons may indicate substantial evolutionary pressure acting differentially across the populations considered. For example, the olfactory receptor gene cluster on chromosome 1 may potentially be attributed to different adaptive pressure to discriminate odors encountered in different diets and environments.
Our method for performing genome-wide comparisons of LD prioritizes the regions that are located at the tail end of the distribution of varLD scores, which is similar to the approach adopted by popular techniques for detecting signatures of natural selection (Sabeti et al. 2006, 2007; Voight et al. 2006). It is important to recognize that such a strategy identifies regions that are more differentiated relative to the rest of the genome, but does not necessarily imply the existence of LD variations that are biologically significant. It is therefore advisable to interpret these genomic regions as candidate regions of LD variations, similar to the candidate interpretation of positive selection signals with the use of the integrated haplotype score (iHS) (Voight et al. 2006) or the extended haplotype homozygosity (EHH) metric (Sabeti et al. 2006, 2007).
Our analysis has investigated the differences in LD patterns between the HapMap populations and two other populations with around half a million polymorphisms. While significant differences in LD are expected to be less prevalent between European populations, comparisons between populations with longer evolutionary history (for example, in the African continent) continue to be an area of great interest, particularly in the use of African cohorts for fine-mapping candidate regions with disease associations. Analytical strategies that localized regional differences in LD across populations will benefit disease studies when extrapolating findings to other populations, especially as more genome-wide data, together with the data from the third phase of the HapMap, become publicly available. Meta-analyses of genome scans for the same diseases across different cohorts, in particular across different ethnic populations, will be greatly enhanced by first understanding the extent of LD differences in candidate regions between these cohorts, since true association signals can be weakened in the presence of significant variations in regional LD when the underlying causal variants exist on different haplotypic backgrounds. The next phase of genetic studies will aim to progress beyond identifying associations to establishing the causal mechanisms of a disease. This is where the ability to quantify LD variation between populations will be particularly relevant when amalgamating findings from multiple genome-wide scans of the same disease.
Acknowledgments
We thank three anonymous reviewers for their comments, which have resulted in significant improvement of the work. The study used data generated by the Wellcome Trust Case-Control Consortium and MalariaGEN. Full lists of the investigators who contributed to the generation of these data are available at http://www.wtccc.org and http://MalariaGEN.net, respectively. Funding for WTCCC was provided by the Wellcome Trust under award no. 076113. Funding for MalariaGEN was from the Grand Challenges in Global Health initiative. Y.Y.T., K.S.S., D.P.K., and T.G.C. acknowledge support from the Wellcome Trust and from the Bill and Melinda Gates Foundation, through the Foundation for the National Institutes of Health as part of the Grand Challenges in Global Health initiative. T.G.C. and D.P.K. also acknowledge support from the Medical Research Council, UK.
Footnotes
[Supplemental material is available online at http://www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.092189.109.
References
- Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, et al. Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeq T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, et al. Replicating genotype–phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- Clark AG, Li J. Conjuring SNPs to detect associations. Nat Genet. 2007;39:815–816. doi: 10.1038/ng0707-815. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- Daily JP, Sabeti P. A malaria fingerprint in the human genome? N Engl J Med. 2008;358:1855–1856. doi: 10.1056/NEJMe0801414. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006a;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker PI, Burtt NP, Graham RR, Guiducci C, Yelensky R, Drake JA, Bersaglieri T, Penney KL, Butler J, Young S, et al. Transferability of tag SNPs in genetic association studies in multiple populations. Nat Genet. 2006b;38:1298–1303. doi: 10.1038/ng1899. [DOI] [PubMed] [Google Scholar]
- de Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P, Ben-Dor A, Yakhini Z, Ellis RJ, Bruhn L, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: Implications for association studies of complex diseases. Hum Mol Genet. 2007;16:2783–2794. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- Gardner M, González-Neira A, Lao O, Calafell F, Bertranpetit J, Comas D. Extreme population differences across Neuregulin 1 gene, with implications for association studies. Mol Psychiatry. 2006;11:66–75. doi: 10.1038/sj.mp.4001749. [DOI] [PubMed] [Google Scholar]
- Hamblin MT, Di Rienzo A. Detection of the signature of natural selection in humans: Evidence from the Duffy blood group locus. Am J Hum Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin MT, Thompson EE, Di Rienzo A. Complex signatures of natural selection at the Duffy blood group locus. Am J Hum Genet. 2002;70:369–383. doi: 10.1086/338628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchard N, Elzein A, Trafford C, Rockett K, Pinder M, Jallow M, Harding R, Kwiatkowski D, McKenzie C. Classical sickle beta-globin haplotypes exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC Genet. 2007;8:52. doi: 10.1186/1471-2156-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Li Y, Singleton AB, Hardy JA, Abecasis G, Rosenberg NA, Scheet P. Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet. 2009;84:235–250. doi: 10.1016/j.ajhg.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, Kivinen K, Bojang KA, Conway DJ, Pinder M, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzanowski WJ. Permutational tests for correlation matrices. Stat Comput. 1993;3:37–44. [Google Scholar]
- Lamason RL, Mohideen MPK, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005;310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- Li T, Stefansson H, Gudfinnsson E, Cai G, Liu X, Murray RM, Steinthorsdottir V, Januel D, Gudnadottir VG, Petursson H, et al. Identification of a novel neuregulin 1 at-risk haplotype in Han schizophrenia Chinese patients, but no association with the Icelandic/Scottish risk haplotype. Mol Psychiatry. 2004;9:698–704. doi: 10.1038/sj.mp.4001485. [DOI] [PubMed] [Google Scholar]
- Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol. 2005;5:43–57. doi: 10.1038/nri1530. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li J, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Marshall C, Feuk L, Scherer SW. Copy-number variation in control population cohorts. Hum Mol Genet. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Varilla P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servin B, Stephens M. Imputation-based analysis of association studies: Candidate regions and quantitative traits. PLoS Genet. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, Gudfinnsson E, Gunnarsdottir S, Walker N, Petursson H, Crombie C, et al. Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet. 2003;72:83–87. doi: 10.1086/345442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- Teo YY, Inouye M, Small KS, Gwilliam R, Deloukas P, Kwiatkowski DP, Clark TG. A genotype calling algorithm for the Illumina BeadArray platforms. Bioinformatics. 2007;23:2741–2746. doi: 10.1093/bioinformatics/btm443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo YY, Small KS, Fry AE, Wu Y, Kwiatkowski DP, Clark TG. Power consequences of linkage disequilibrium variation between populations. Genet Epidemiol. 2009;33:128–135. doi: 10.1002/gepi.20366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genomewide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Chen Z, Tadesse MG, Glessner J, Grant SFA, Hakonarson H, Bucan M, Li M. Modeling genetic inheritance of copy number variation. Nucleic Acids Res. 2008;36:e138. doi: 10.1093/nar/gkn641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin DV, Meng Z, Ehm MG. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am J Hum Genet. 2006;78:737–746. doi: 10.1086/503710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Shi Y, Tang J, Tang R, Yu L, Gu N, Feng G, Zhu S, Liu H, Xing Y, et al. A case control and family based association study of the neuregulin 1 gene and schizophrenia. J Med Genet. 2004;41:31–34. doi: 10.1136/jmg.2003.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zogopoulos G, Ha KC, Nagib F, Moore S, Kim H, Montpetit A, Robidoux F, Laflamme P, Cotterchio M, Greenwood C, et al. Germ-line DNA copy number variation frequencies in a large North American population. Hum Genet. 2007;122:345–353. doi: 10.1007/s00439-007-0404-5. [DOI] [PubMed] [Google Scholar]