

Abstract
Background and purpose:
It is postulated that nitrite requires reduction to nitric oxide in order to exert its relaxant effect upon isolated hypoxic vessels. Herein, we evaluate the relative contribution of nitric oxide and characterize the downstream mechanisms of nitrite-induced vasorelaxation.
Experimental approach:
Aortic rings were treated with pharmacological agents and exposed to hypoxia (<1% O2). Following pre-constriction, nitrite (10 µM final) was added to appropriate baths; isometric tension was recorded throughout.
Key results:
Nitrite (under hypoxic conditions at physiological pH) is capable of exerting physiological effects that cannot be completely inhibited by the inhibitor of soluble guanylate cyclase (sGC), 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one or a nitric oxide scavenger (carboxy-2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide). Simultaneous blockade of both sGC and cyclooxygenase (COX) completely inhibited the response to nitrite. With regard to the nitric oxide-dependent component, we confirm that aldehyde oxidase, but not xanthine oxidase or endothelial nitric oxide synthase, was important for the actions of nitrite in our model.
Conclusions and implications:
Nitric oxide generated from nitrite is not exclusively responsible for the physiological actions observed in isolated hypoxic vessels. Nitrite operates via different pathways dependent on the presence or absence of endothelium to produce vasorelaxation. In intact vessels, both sGC and COX enzymes appear to be important. Irrespective of this difference in relaxation mechanism, nitrite is capable of producing the same maximum relaxation, regardless of the presence of endothelium. Having investigated possible nitrite reduction sites, we confirm that aldehyde oxidase is important for the actions of nitrite.
Keywords: nitrite, nitric oxide, vasodilatation, hypoxia, ischaemia, cyclooxygenase, soluble guanylate cyclase
Introduction
The nitrite anion (NO2-) represents an interesting constituent of the nitrogen oxide(s) (NOx) family in pharmacological terms. The real-time study of nitric oxide (NO) has always posed a problem due to its highly reactive nature; nitrite, although significantly less potent than NO, has been utilized as a more stable and practical solution to this experimental problem. However, nitrite has its own caveats, most notably contamination in in vitro systems and alternative sources in vivo (e.g. diet) where levels can be oxygen dependent. Nitrite itself has been known to be vasoactive for a number of years (Furchgott and Bhadrakom, 1953). Although relatively less potent than pharmaceutical nitrodilators, interest in its mode of action has recently revived after recognition that it may represent a circulating source of bioavailable ‘NO-like’ activity and may have an important role clinically (see Lundberg et al. (2008)). Nitrite was previously thought of as a relatively inert NO metabolite, although current studies now allow us to appreciate the dynamic ‘interplay’ between the metabolites of NO and their potential as physiological effectors (Rogers et al., 2007). In vivo, nitrite undergoes a number of variable reactions leading to direct NO signalling pathways and to S-nitrosylation which may in turn regulate key pathways (Angelo et al., 2006; Gladwin et al., 2006).
A key point of nitrite pharmacology is that its vasodilator properties are greatly enhanced in hypoxia when compared to normoxia (Maher et al., 2008). Indeed, we recently demonstrated selective and enhanced venous versus arterial dilatation in vitro and in healthy human subjects under hypoxic and to a lesser extent normoxic conditions (Maher et al., 2008). This enhanced activity can be attributable not only to the oxygen gradient observed from artery to vein, but also to a vessel-specific sensitivity to nitrite. There are a number of theories as to how nitrite exerts its effects at a tissue level, most notably reduction back to NO. In blood, deoxyhaemoglobin is proposed as an effective nitrite reductase that matches oxygen requirement with NO availability (Isbell et al., 2007). Several issues remain with this concept, including the effective entry of nitrite into erythrocytes at physiological concentrations, and subsequent escape of bio-available NO from haemoglobin recapture. Perhaps of equal importance, how does this process occur to alter vessel tone within the artery to vein transit of a tissue bed (Allen and Piantadosi, 2006)? However, our study does not attempt to address possible roles for haemoglobin saturation. A number of publications now cite oxidoreductase enzymes to be capable of converting significant amounts of nitrite to NO, in particular xanthine oxidase and aldehyde oxidase (Godber et al., 2000; Li et al., 2001; 2008; Baker et al., 2007). These tissue-derived nitrite reduction routes appear to take precedence under conditions of hypoxia and are not reliant on haemoglobin as we have shown previously (Maher et al., 2008).
Downstream, previous studies have suggested that the nitrite hypoxic vasodilatation is independent of endothelial NO synthase (eNOS), but is dependent on soluble guanylate cyclase (sGC) (Dalsgaard et al., 2007).
Using an aortic ring bioassay, we have attempted to elucidate the pharmacology of nitrite at an isolated vessel level. We show that nitrite does produce marked vasodilatation and operates via different signalling pathways which are dependent on the presence or absence of endothelium. The major pathways involved are those governed by aldehyde oxidase reduction and operated through sGC and cyclooxygenase (COX). In addition, we demonstrate that the effects of nitrite are not completely dependent upon nitrite reduction back to NO.
Methods
Test system
Care of animals and all procedures were carried out under the Animals (Scientific Procedures) Act 1986, and were approved by the local ethical committee. Male New Zealand White rabbits (2–2.5 kg) were terminally anaesthetized with sodium pentobarbitone (120 mg·kg−1, intravenously). The rib cage was then opened, and the aorta was carefully excised and placed in fresh Krebs buffer (NaCl 109.0 mM, KCl 5.36 mM, KH2PO4 1.17 mM, MgSO4·7H2O 1.21 mM, NaHCO3 24.99 mM, glucose 10.99 mM, CaCl2·2H2O 1.49 mM) (all Fisher Scientific, UK Ltd, Loughborough, UK). The aorta was then cleaned of minor vessels and adipose tissue, and cut into eight equally divided 2 mm rings, discarding the tissue most distal and proximal to the heart (aortic arch and start of abdominal aorta). Rings were mounted on matched stainless steel hooks for isometric tension recording in 8 mL baths containing 5 mL of Krebs buffer, 37°C. Output transducer (AD Instruments, Chalgrove, UK) signals were amplified and converted for visualization on ‘chart for Windows’ (version 4.1.2, ADINSTRUMENTS). Aortic rings were maintained at 2 g resting tension in all experiments.
Experimental protocol
After a 1 h equilibration period, ring tension was reset (allowing for stretch-induced relaxation). All tissue was then exposed to phenylephrine (1 µM) (Sigma-Aldrich, Dorset, UK). Once constriction had reached a plateau, acetylcholine (10 µM) (Sigma) was added to demonstrate endothelium viability or its absence. The pre-conditioning process was then repeated until consistent phenylephrine-induced tensions were achieved. Pre-conditioning was carried out at 95% O2/5% CO2.
Hypoxic experiments were carried out at ∼1% O2 (gassed with 95% N2/5% CO2) as utilized previously by us (James et al., 2004). The tissue was kept under hypoxic conditions for 10 min before exposure to phenylephrine [3 µM, used to achieve the same tension (in g) observed at 95% O2, ensuring a true % relaxation as opposed to a contractile artefact]. All pharmacological blocking agents were pre-incubated at respective concentrations and time periods. Hypoxia always caused a degree of relaxation in phenylephrine pre-constricted aortic rings, above that seen in normoxia.
Model characteristics
The slow and continuous relaxation of isolated vessels during hypoxia, as observed in the controls of all our experiments, is an established phenomenon. The precise cause of vasorelaxation remains unknown. Both energy limitation and interruption of excitation contraction coupling have been suggested as possible causes (Shimizu et al., 2000). In our model, hypoxia-induced relaxation proceeds to the same extent independently of endothelium or NOS activity [denudation/l-NG-monomethyl arginine (l-NMMA) data]. It is important to note that the smooth muscle cells are not simply dying in hypoxia. After a period of hypoxia, tissue could be re-equilibrated in oxygenated buffer, and showed comparable contraction and relaxation to exogenous agents. Of related interest, relaxation induced in arteries and veins by NO donors (such as S-nitrosoglutathione; GSNO) and nitrite is enhanced under hypoxic compared to normoxic conditions (James et al., 2004; Maher et al., 2008).
Tension recordings
Maximum relaxation was measured at 20 min after nitrite addition, and expressed as percentage of the maximum constriction to phenylephrine. The 20 min time-point is used to reflect total nitrite-induced relaxation, as nitrite was observed to reach a sustained plateau and have had its maximum effect within this time.
When using pharmacological blockers, percentage inhibition was always expressed with respect to the appropriate control tissue or agent control, as some inhibitors altered basal tone.
Data analysis
Data were compared using a one-way analysis of variance with Bonferroni's multiple comparison post hoc test. All analyses were carried out using Graphpad Prism software (version 4.0); n= 1 represents data averaged from a pair of matched aortic rings. Data showing time course profiles were compared statistically over the entire profile. The 20 min time-point was picked for a single time-point comparison of total relaxation induced.
Inhibitors
Concentration–effect curves were obtained for the inhibitors to establish the appropriate concentration for maximum effect. The final concentration of nitrite in all experiments was 10 µM, an effective concentration previously established in our laboratory in vitro, but also a concentration that is achievable in vivo, in plasma with pharmacological nitrite dosing (Dejam et al., 2007; Mack et al., 2008; Maher et al., 2008). As this final nitrite concentration induced approximately 80% relaxation, it was ideal to test agents which either inhibited or augmented the response. The agents were incubated for 30 min at normoxia before commencing the hypoxic stage of the experimental protocol. The final concentrations of inhibitor used are shown in Table 1. All inhibitors were obtained from Sigma, apart from 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), carboxy-2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide (CPTIO) and U-51605 which were obtained from Axxora, Nottingham, UK.
Table 1.
Concentrations and actions of pharmacological tools used
| Agent | Action | Final concentration |
|---|---|---|
| ODQ | Soluble guanylate cyclase inhibitor | 10 µM |
| Indomethacin | Cyclooxygenase inhibitor | 5 µM |
| CPTIO | Nitric oxide scavenger | 1 mM |
| U-51605 | Prostacyclin synthase inhibitor (PGISi) | 10 µM |
| Raloxifene | Aldehyde oxidase inhibitor | 50 nM |
| Oxypurinol | Xanthine oxidase inhibitor | 100 µM |
| l-NMMA | eNOS inhibitor | 300 µM |
These represent final bath concentrations required or established for maximum effect on nitrite-induced vasodilatation.
ODQ, 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; CPTIO, carboxy-2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide; l-NMMA, l-NG-monomethyl arginine.
Results
Nitrite consistently induced significant relaxation in hypoxic vascular rings, with or without endothelium (Figures 1A and 2A). Note that the time course of this relaxation is minutes rather than seconds, as is seen with more potent NO donors, native NO or erythrocyte-induced hypoxic vasodilatations (James et al., 2004).
Figure 1.
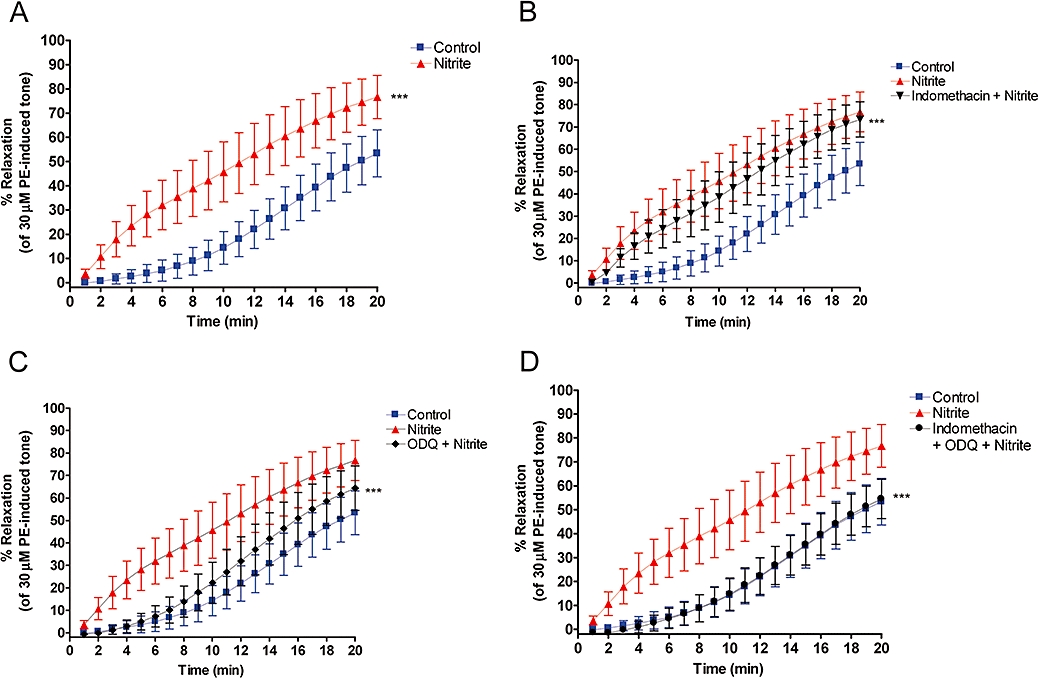
Relaxation profiles of endothelium-intact aortic rings over 20 min. (A) Nitrite (10 µM) produces a marked relaxation compared to control *** (P < 0.001). (B) Partial inhibition of nitrite-induced relaxation by indomethacin (5 µM). *** (P < 0.001) significantly different from nitrite alone over the course of the profile. (C) Substantial inhibition of nitrite-induced relaxation by 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (10 µM) *** (P < 0.001) compared to nitrite alone. (D) Complete inhibition of nitrite-induced relaxation by indomethacin (5 µM) and ODQ (10 µM) in combination *** (P < 0.001) compared to nitrite alone. ODQ relaxation is significantly different from indomethacin and ODQ in combination (P < 0.001) (n= 5 in all cases).
Figure 2.
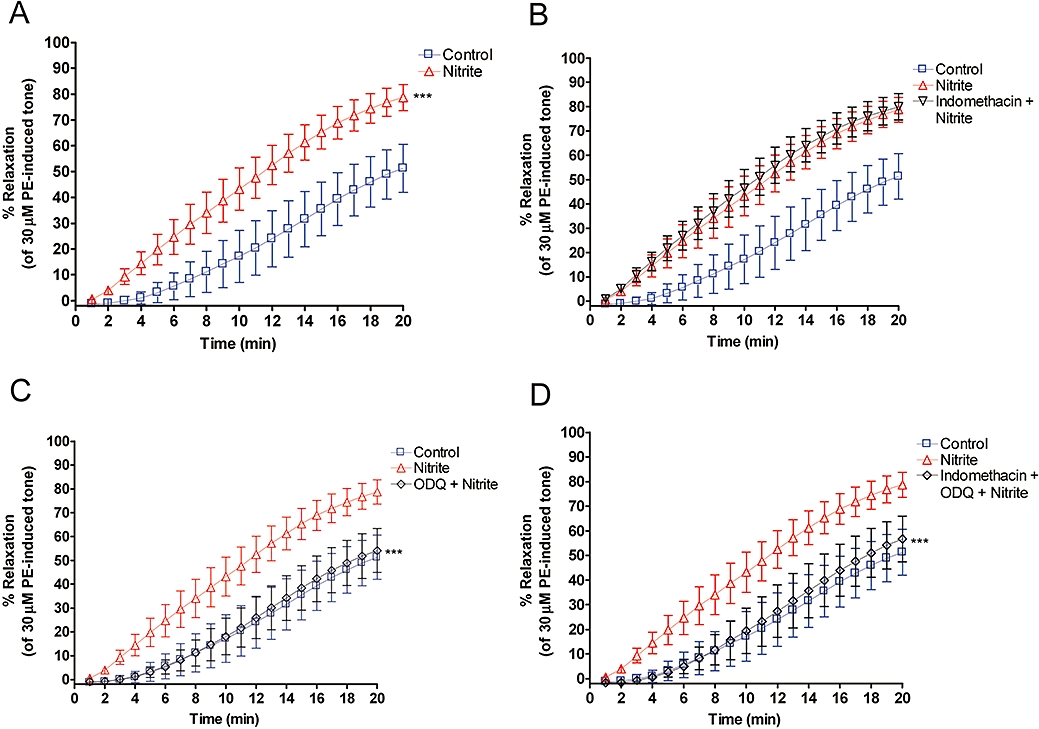
Relaxation profiles of endothelium-denuded aortic rings over 20 min. (A) Nitrite (10 µM) produces a marked relaxation compared to control *** (P < 0.001). (B) No inhibition of nitrite-induced relaxation by indomethacin (5 µM). (C) Complete inhibition of nitrite-induced relaxation by 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (10 µM) *** (P < 0.001) compared to nitrite alone. (D) Complete inhibition of nitrite-induced relaxation by indomethacin (5 µM) and ODQ (10 µM) in combination *** (P < 0.001) compared to nitrite alone (n= 5 in all cases).
Endothelium-denuded vessels
In vessels that were denuded of endothelium, blockade of sGC with ODQ (10 µM) completely inhibited the nitrite-induced relaxation (Figure 2C). This inhibition was unaffected by the addition of indomethacin (5 µM) (Figures 2D and 3).
Figure 3.
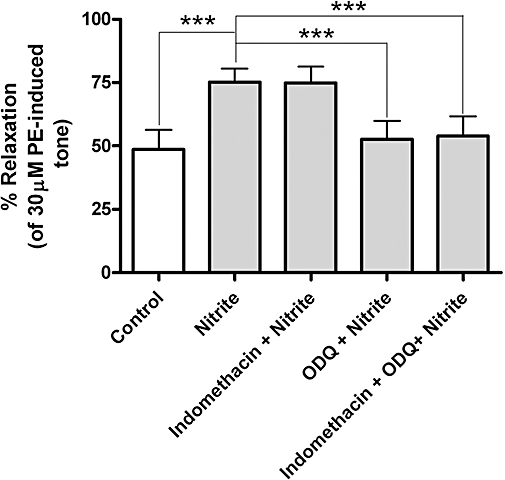
Maximum relaxation [of phenylephrine (PE)-induced tone] induced by nitrite at the 20 min time-point in endothelium-denuded aortic rings. Nitrite produces a marked relaxation compared to control *** (P < 0.001), which is completely inhibited by 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (10 µM) *** (P < 0.001) compared to nitrite alone. Indomethacin produced no inhibition in denuded vessels (n= 5 in all cases).
Endothelium-intact vessels
Pathways
The nitrite-induced vasorelaxation in endothelium-intact vessels was partly prevented by both inhibitors of sGC and of COX used alone (Figures 1B,C and 4). However, when used in combination, these two agents were capable of abolishing the response to nitrite (Figure 1D). The use of indomethacin, the COX inhibitor, in combination with a NO scavenger (CPTIO) also completely inhibited the relaxation to nitrite (Figure 5). However inhibition of protacyclin synthase with U-51605 did not affect nitrite-induced relaxation, although it did alter baseline tension (Figure 5).
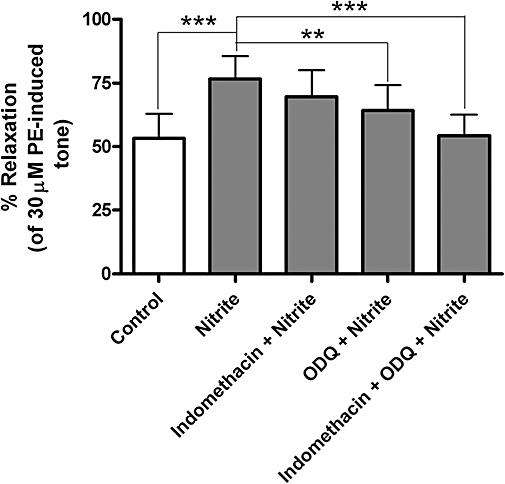
Maximum relaxation [of phenylephrine (PE)-induced tone] induced by nitrite (10 µM) after 20 min, in endothelium-intact aortic rings. Nitrite produces a marked relaxation compared to control *** (P < 0.001), which is inhibited in part by both indomethacin (5 µM) and 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (10 µM), respectively, and completely inhibited by indomethacin (5 µM) and ODQ (10 µM) in combination *** (P < 0.001) compared to nitrite alone (n= 5 in all cases).
Figure 5.
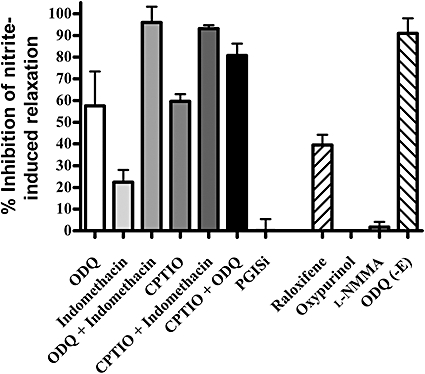
Inhibition of nitrite vasorelaxation in endothelium-intact vessels. Inhibitor concentrations used 1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (10 µM), carboxy-2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide (CPTIO) (1 mM), raloxifene (50 nM), oxypurinol (100 µM), PGI2 synthase inhibitor (10 µM), l-NG-monomethyl arginine (l-NMMA) (300 µM) (n= 4 or 5). Differences between means: ODQ versus CPTIO + ODQ (not significant), CPTIO versus CPTIO + ODQ (not significant). CPTIO versus indomethacin (not significant), CPTIO versus CPTIO + indomethacin (P < 0.0001), indomethacin versus CPTIO + indomethacin (P < 0.0001). Raloxifene versus oxypurinol (P < 0.0008), raloxifene versus l-NMMA (P < 0.0003). CPTIO + indomethacin versus CPTIO + ODQ (P= 0.0705).
NO dependency
In endothelium-intact vessels, a NO-specific scavenger (CPTIO) partly prevented the nitrite-induced response (Figure 5). Inhibition of sGC in combination with the NO-specific scavenger provided a greater block of relaxation than the individual agents alone, but still did not produce a complete inhibition (Figure 5).
Sources of nitrite reduction
The vasorelaxant response was significantly inhibited by the aldehyde oxidase blocker, raloxifene, but not by the xanthine oxidase inhibitor, oxypurinol or an inhibitor of eNOS, l-NMMA. Interestingly, l-NMMA did not alter the basal hypoxia-induced relaxation in control tissue (data not shown).
Discussion and conclusions
We have shown that nitrite under hypoxic conditions exerts direct vasodilatory effects via different signalling pathways, depending on the presence of endothelium. These are summarized in Figure 6. It has long been assumed that the vascular actions of nitrite in hypoxia were reliant upon it, forming NO as an intermediate. Here, we provide evidence that nitrite (under hypoxic conditions at physiological pH) is also capable of exerting physiological effects independently of a free NO intermediate.
Figure 6.
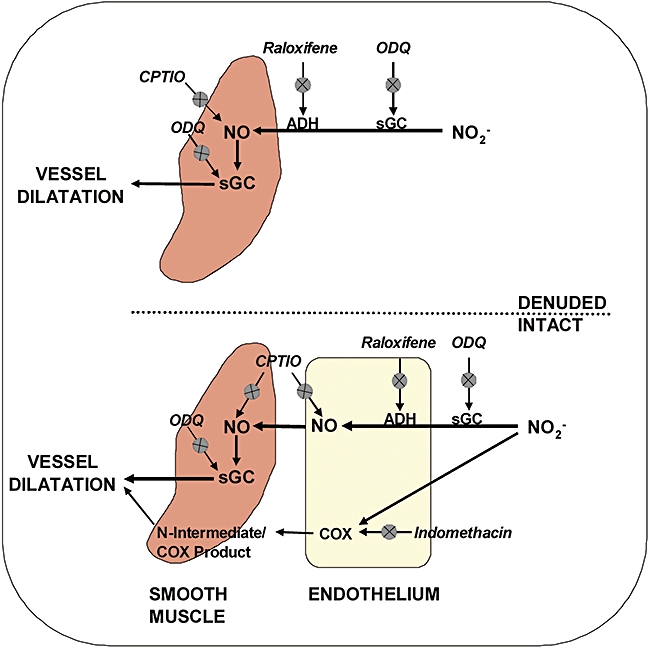
Potential pathways that control the nitrite-mediated relaxation of hypoxic vessels. Upper section of diagram depicts denuded tissue where soluble guanylate cyclase (sGC) appears to control relaxation. The lower section of the diagram attempts to show possible pathways in endothelium-intact vessels.
Component pathways
In vessels with intact endothelium, nitrite at a pharmacological dose operated via two distinct pathways, one controlled by NO/sGC and the other through COX, although the effect appears to be predominantly signalled through the former. It is possible that NO derived from nitrite is not only acting through sGC, but is also generated by sGC in a reductive process (Alzawahra et al., 2008).
Having found that the COX pathway appears to operate independently of the NO intermediate, we attempted to investigate this further. COX is known to be essential for the generation of prostacyclin, a key COX product responsible for vasodilatation in the control of vessel tone (Dusting et al., 1977). To explore the relative contribution of prostacyclin as opposed other COX-derived eicosanoids, we inhibited prostacyclin synthase, which occurs distal to COX in the prostaglandin biosynthetic pathway. Inhibiting prostacyclin synthase did alter baseline tension, but had no effect upon the nitrite-induced hypoxic vasodilatation. This suggests either a possible role for other COX-derived eicosanoids such as PGE2, known to cause vasodilatation or an enzymatic action of COX on nitrite, converting this substrate to other N species. Through utilization of its haem sites under hypoxic conditions, it is possible that COX may convert nitrite to a bioactive NO species, other than free NO. Endothelial COX has been demonstrated to generate free radical species under hypoxic conditions (Rieger et al., 2002). It is also possible that the involvement of COX could cause an inhibition of constriction, as opposed to an enhancement of relaxation. Previous studies have failed to recognize these differential signalling pathways as tissue is often pre-incubated with indomethacin and NOS inhibitors (Dalsgaard et al., 2007).
It is interesting to note the difference in profile between inhibition of sGC and COX (Figure 1). The inhibition of COX appears to be uniform throughout the profile, whereas sGC inhibition is appreciable in the first part of the profile, but starts to tail off towards the end. It would seem that these pathways could be mutually exclusive, but crosstalk between them cannot be ruled out. It is important to remember that inhibition of one pathway may simply cause up-regulation of another, making the relative contribution of each difficult to estimate. This difference in profile also reinforces the idea that nitrite may operate via pathways that are separate both physically and with respect to time. Irrespective of the different inhibition profiles, the combination of sGC and COX inhibition abolished the response to nitrite throughout the entire profile.
NO dependency
Blockade of sGC or NO scavenging, individually or in combination, was not able to abolish the vascular response to nitrite in hypoxia. However, we did find that the combination of the two agents did give more variable results from experiment to experiment when compared to the effects of other agents. It has been suggested that nitrite may be capable of generating species such as nitrosylated products without the requirement for free NO as an intermediate (Feelisch et al., 2008). These products may be responsible, in part, for the actions of nitrite in an ex vivo tissue model. Interestingly, our data would suggest that these products operate through or are generated by the COX pathway.
Sources of NO reduction
The importance of oxidoreductase enzymes in vascular tissue has recently become apparent, with both xanthine oxidase and aldehyde oxidase being identified as key reductive enzymes (Li et al., 2008). For normal function, these oxidoreductase enzymes rely upon the presence of oxygen. In hypoxia, these enzymes are capable of reducing alternative substrates such as nitrite (Li et al., 2008).
We confirmed the involvement of aldehyde oxidase in nitrite-induced hypoxic vasodilatation, but failed to show a dependence upon xanthine oxidase. This is in agreement with published reports, showing a more significant role for aldehyde oxidase in nitrite reduction to NO at a tissue level (Li et al., 2008). We cannot discount a role for xanthine oxidase as a reductase because although oxypurinol blocks the molybdenum site, the flavin site of the enzyme remains free and could play a role (Li et al., 2004). In addition, we also found that eNOS does not appear to play a role in the nitrite-induced vasorelaxation under hypoxic conditions.
Implications for in vivo conditions
Our findings are directly applicable to conditions in which plasma nitrite is elevated pharmacologically or by diet. A common misconception in the literature is the extrapolation from studies that are performed by infusing nitrite at elevated doses to nitrite as a circulating store of NO in blood under basal physiological conditions (200∼400 nM nitrite). It must be recognized therefore that the component mechanisms of nitrite-induced relaxation at higher doses may not be operational at normal levels, and this is difficult to confirm in vitro and in vivo largely because of assay limitations.
Given the interest in nitrite as a therapeutic agent, the mechanisms we describe are of importance. Our experiments are haemoglobin independent (i.e. nitrite induces vessel relaxation in hypoxia without the presence of haemoglobin or erythrocytes). We cannot discount a role for deoxyhaemoglobin in vivo; indeed, blood-borne NO species derived from nitrite infusion may contribute to long-term vessel tone (Angelo et al., 2006). In an isolated vessel model in vitro, haemoglobin was shown to enhance the relaxation induced by pharmacological nitrite when the tissue was exposed to hypoxia (Cosby et al., 2003). However, a recent work on a model that closely resembles ours confirmed that haemoglobin had no effect across a broad range of nitrite concentrations (Luchsinger et al., 2005). Our results confirmed that nitrite could directly effect relaxation of vessels without interaction with blood, and we elucidated the component mechanisms by which this is achieved at the tissue level.
Although a relatively weak vasodilator compared to pharmacological nitrodilators, the fact that nitrite clearly dilates hypoxic vessels preferentially implies it could act as a targeted vasodilator without causing the global changes in haemodynamics (e.g. fall in blood pressure seen using these agents). We show that the ‘direct’ nitrite-induced relaxation is largely due to nitrite reduction by aldehyde oxidase to NO, but in part is also mediated via COX-dependent processes. These findings now need to be confirmed in vivo following nitrite infusion. The mechanism of action of nitrite in veins is yet to be fully explored, although we have shown relaxation to be enhanced compared to arteries (Maher et al., 2008), and it is interesting to speculate that enhanced effects observed in capacitance vessels in vivo could also be exploited therapeutically.
Acknowledgments
We would like to thank the British Heart Foundation for their continued support.
Glossary
Abbreviations:
- ACh
acetylcholine
- COX
cyclooxygenase
- CPTIO
carboxy-2-phenyl-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide
- eNOS
endothelial nitric oxide synthase
- l-NMMA
l-NG-monomethyl arginine
- ODQ
1H [1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- sGC
soluble guanylate cyclase
- XO
xanthine oxidase
Conflict of interest
This work does not conflict with any other work/activities of the authors.
References
- Figure 4.Allen BW, Piantadosi CA. How do red blood cells cause hypoxic vasodilation? The SNO–hemoglobin paradigm. Am J Physiol Heart Circ Physiol. 2006;291(4):H1507–H1512. doi: 10.1152/ajpheart.00310.2006. [DOI] [PubMed] [Google Scholar]
- Alzawahra WF, Talukder MA, Liu X, Samouilov A, Zweier JL. Heme proteins mediate the conversion of nitrite to nitric oxide in the vascular wall. Am J Physiol Heart Circ Physiol. 2008;295(2):H499–H508. doi: 10.1152/ajpheart.00374.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci USA. 2006;103(22):8366–8371. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JE, Su J, Fu X, Hsu A, Gross GJ, Tweddell JS, et al. Nitrite confers protection against myocardial infarction: role of xanthine oxidoreductase, NADPH oxidase and K(ATP) channels. J Mol Cell Cardiol. 2007;43(4):437–444. doi: 10.1016/j.yjmcc.2007.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9(12):1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- Dalsgaard T, Simonsen U, Fago A. Nitrite-dependent vasodilation is facilitated by hypoxia and is independent of known NO-generating nitrite reductase activities. Am J Physiol Heart Circ Physiol. 2007;292(6):H3072–H3078. doi: 10.1152/ajpheart.01298.2006. [DOI] [PubMed] [Google Scholar]
- Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, et al. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116(16):1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- Dusting GJ, Moncada S, Vane JR. Prostacyclin (PGX) is the endogenous metabolite responsible for relaxation of coronary arteries induced by arachidonic acid. Prostaglandins. 1977;13(1):3–15. doi: 10.1016/0090-6980(77)90037-5. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Fernandez BO, Bryan NS, Garcia-Saura MF, Bauer S, Whitlock DR, et al. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem. 2008;283:33927–33934. doi: 10.1074/jbc.M806654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF, Bhadrakom S. Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharmacol Exp Ther. 1953;108(2):129–143. [PubMed] [Google Scholar]
- Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, et al. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006;291(5):H2026–H2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, et al. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem. 2000;275(11):7757–7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- Isbell TS, Gladwin MT, Patel RP. Hemoglobin oxygen fractional saturation regulates nitrite-dependent vasodilation of aortic ring bioassays. Am J Physiol Heart Circ Physiol. 2007;293(4):H2565–H2572. doi: 10.1152/ajpheart.00759.2007. [DOI] [PubMed] [Google Scholar]
- James PE, Lang D, Tufnell-Barret T, Milsom AB, Frenneaux MP. Vasorelaxation by red blood cells and impairment in diabetes: reduced nitric oxide and oxygen delivery by glycated hemoglobin. Circ Res. 2004;94(7):976–983. doi: 10.1161/01.RES.0000122044.21787.01. [DOI] [PubMed] [Google Scholar]
- Li H, Samouilov A, Liu X, Zweier JL. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide generation in anoxic tissues. J Biol Chem. 2001;276(27):24482–24489. doi: 10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- Li H, Samouilov A, Liu X, Zweier JL. Characterization of the effects of oxygen on xanthine oxidase-mediated nitric oxide formation. J Biol Chem. 2004;279(17):16939–16946. doi: 10.1074/jbc.M314336200. [DOI] [PubMed] [Google Scholar]
- Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem. 2008;283(26):17855–17863. doi: 10.1074/jbc.M801785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger BP, Rich EN, Yan Y, Williams EM, Stamler JS, Singel DJ. Assessments of the chemistry and vasodilatory activity of nitrite with hemoglobin under physiologically relevant conditions. J Inorg Biochem. 2005;99(4):912–921. doi: 10.1016/j.jinorgbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- Mack AK, McGowan Ii VR, Tremonti CK, Ackah D, Barnett C, Machado RF, et al. Sodium nitrite promotes regional blood flow in patients with sickle cell disease: a phase I/II study. Br J Haematol. 2008;142(6):971–978. doi: 10.1111/j.1365-2141.2008.07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, et al. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117(5):670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- Rieger JM, Shah AR, Gidday JM. Ischemia–reperfusion injury of retinal endothelium by cyclooxygenase- and xanthine oxidase-derived superoxide. Exp Eye Res. 2002;74(4):493–501. doi: 10.1006/exer.2001.1156. [DOI] [PubMed] [Google Scholar]
- Rogers SC, Khalatbari A, Datta BN, Ellery S, Paul V, Frenneaux MP, et al. NO metabolite flux across the human coronary circulation. Cardiovasc Res. 2007;75(2):434–441. doi: 10.1016/j.cardiores.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Bowman PS, Thorne G, 3rd, Paul RJ. Effects of hypoxia on isometric force, intracellular Ca(2+), pH, and energetics in porcine coronary artery. Circ Res. 2000;86(8):862–870. doi: 10.1161/01.res.86.8.862. [DOI] [PubMed] [Google Scholar]