

Abstract
Background and purpose:
The effects of veratridine, an alkaloid found in Liliaceae plants, on tetrodotoxin (TTX)-sensitive voltage-gated Na+ channels were investigated in mouse vas deferens.
Experimental approach:
Effects of veratridine on TTX-sensitive Na+ currents (INa) in vas deferens myocytes dispersed from BALB/c mice, homozygous mice with a null allele of NaV1.6 (NaV1.6−/−) and wild-type mice (NaV1.6+/+) were studied using patch-clamp techniques. Tension measurements were also performed to compare the effects of veratridine on phasic contractions in intact tissues.
Key results:
In whole-cell configuration, veratridine had a concentration-dependent dual action on the peak amplitude of INa: INa was enhanced by veratridine (1–10 µM), while higher concentrations (≥30 µM) inhibited INa. Additionally, two membrane current components were evoked by veratridine, namely a sustained inward current during the duration of the depolarizing rectangular pulse and a tail current at the repolarization. Although veratridine caused little shift of the voltage dependence of the steady-state inactivation curve and the activation curve for INa, veratridine enhanced a non-inactivating component of INa. Veratridine caused no detectable contractions in vas deferens from NaV1.6−/− mice, although in tissues from NaV1.6+/+ mice, veratridine (≥3 µM) induced TTX-sensitive contractions. Similarly, no detectable inward currents were evoked by veratridine in NaV1.6−/− vas deferens myocytes, while veratridine elicited both the sustained and tail currents in cells taken from NaV1.6+/+ mice.
Conclusions and implications:
These results suggest that veratridine possesses a dual action on INa and that the veratridine-induced activation of contraction is induced by the activation of NaV1.6 channels.
Keywords: channel enhancer, dual action, NaV1.6, smooth muscle-type Na+ channels, tetrodotoxin, veratridine
Introduction
Veratridine is an alkaloid toxin isolated from the rhizomes of Veratrum album and the sabadilla seeds of Mexican Schoenocaulon officinale. Using electrophysiological techniques, several studies have been performed to clarify the mechanisms underlying the action of veratridine on fast transient inward voltage-gated Na+ currents (INa) in a wide variety of tissues such as skeletal muscle (Campos et al., 2004), neurons (Rando, 1989), cardiac muscle (Nánási et al., 1994); and Xenopus oocytes expressed with cloned Na+ channel (NaV) genes (µ1, namely NaV1.4; Wang et al., 2000; channel nomenclature follows Alexander et al., 2008). These studies show an increase of INa as veratridine concentrations are increased. It has been reported that veratridine binds to the D1 homologous domain of the S6 transmembrane segment in α subunits of voltage-gated Na+ channels (Wang and Wang, 2003b). When veratridine binds to these sites, Na+ channels are shifted towards long-lasting open states with a reduction in single-channel conductance, thereby altering voltage dependency and Na+ permeability (Wang and Wang, 2003a). It is well known that veratridine enhances the channel activity of most NaV1.X isoforms including NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6 and NaV1.7 (reviewed by Catterall et al., 2005).
Recently, we reported the biophysical and molecular properties of tetrodotoxin (TTX)-sensitive INa recorded in smooth muscle cells, freshly dispersed from mouse vas deferens, showing that the molecular identity of the only voltage-gated Na+ channel functional in these cells is the NaV1.6 channel, encoded by the Scn8a gene (Zhu et al., 2008). This system therefore provides an ideal opportunity to investigate the actions of veratridine on a single functional TTX-sensitive voltage-gated Na+ channel, NaV1.6, in native smooth muscle cells.
In the present study, we have shown that veratridine caused a dual action on the peak amplitude of INa, depending on its concentrations, and that veratridine induced a sustained inward current during the depolarizing rectangular pulse. We also found a tail current at the repolarization of the pulse, exhibiting a Na+ permeability and sensitivity to TTX. Furthermore, we have demonstrated that veratridine induces TTX-sensitive phasic contractions in the isolated vas deferens from wild-type littermates (NaV1.6+/+), and that veratridine caused no contractions in tissues from NaV1.6-null mice (NaV1.6−/−). Finally, using NaV1.6-null mice and wild-type littermates, we show that the detectable inward currents evoked by veratridine were due to INa mediated by NaV1.6 channels.
Methods
Preparation of the vasa deferentia
All animal experiments were approved by the animal care and use committee of Faculty of Medicine, Kyushu University (Fukuoka, Japan). Male BALB/c mice (8–10 weeks) or C3HeB/FeJ mice (2–3 weeks) were killed by cervical dislocation. Efforts were made to minimize the number of animals used and their suffering; several types of experiment were carried out on aliquots of cells from any one animal.
Contraction studies
For tension measurements, a modified Krebs solution was used (in mM): Na+ 137, K+ 5.9, Mg2+ 1.2, Ca2+ 2.5, Cl– 133.7, HCO3– 15.4, H2PO4– 1.2 and glucose 11.5 which was bubbled with 97% O2 and 3% CO2. Tissue strips were prepared as described previously (see Teramoto and Brading, 1996; Cuprian et al., 2005). The mucosa was removed from the tissues and isometric contraction of the longitudinal smooth muscle layer was measured using a force transducer (Minebea Co. Ltd., Nagano, Japan). An initial tension equivalent to 0.5 g weight was applied to the tissues and they were allowed to equilibrate for approximately 2-3 h at 37°C. Isometric contractions of the longitudinal muscle layer were sampled (100 Hz) and digitized using Chart 5.0 software (AD Instruments, Castle Hill, Australia) and an IBM computer (VAIO PCV-HX51B, SONY, Tokyo, Japan). The area under the tension trace was measured (expressed as mN min mg−1) and compared with control values.
Electrophysiology
Mouse vas deferens single smooth muscle cells isolated from the intact tissue were dissociated freshly by the gentle tapping method (described previously in Teramoto and Brading, 1996; Zhu et al., 2008) and stored in cold physiological salt solution (see below) at 4–5°C. After isolation, relaxed spindle-shaped cells were stored on ice in the room, and used within 3–4 h. Patch-clamp experiments (conventional whole-cell configuration) were performed at room temperature (21–23°C) and the data recording system used was essentially the same as that described previously (Zhu et al., 2008). The whole-cell current data were low-pass filtered at 5 kHz by an eight-pole Bessel filter, sampled at 0.1 ms and analysed on a PowerMac G4 computer (Apple Computer Japan, Tokyo, Japan) using Chart v5.5.6 (AD Instruments Pty Ltd., Castle Hill, Australia). A method for pharmacological subtraction of the leak and the capacitive currents was performed. By subtracting the current in the presence of TTX (1 µM) and Cd2+ (100 µM) from that in the presence of Cd2+, a TTX-sensitive component was obtained (see Zhu et al., 2008). No electronic compensation for either series resistance or membrane capacitance was applied for the pharmacological subtraction of the membrane currents. However, both series resistance and membrane capacitance were measured in BALB/c mice vas deferens myocytes (series resistance 19.4 ± 6.6 MΩ; membrane capacitance, 49.3 ± 10.6 pF; n= 53 cells, 23 different animals) and C3HeB/FeJ mice vas deferens myocytes (series resistance, 15.4 ± 5.8 MΩ; membrane capacitance, 13.1 ± 2.8 pF; n= 9 cells, 6 different animals). Due to the different ages between BALB/c mice (8–10 weeks) and C3HeB/FeJ mice (2–3 weeks), the size of the smooth muscle cells was different.
Current–voltage relationships were obtained from measurement of the peak amplitude of voltage-gated Na+ currents (INa) within the first 10 ms after application of depolarizing pulses (see Figure 3 and see Figure 7).
Figure 3.
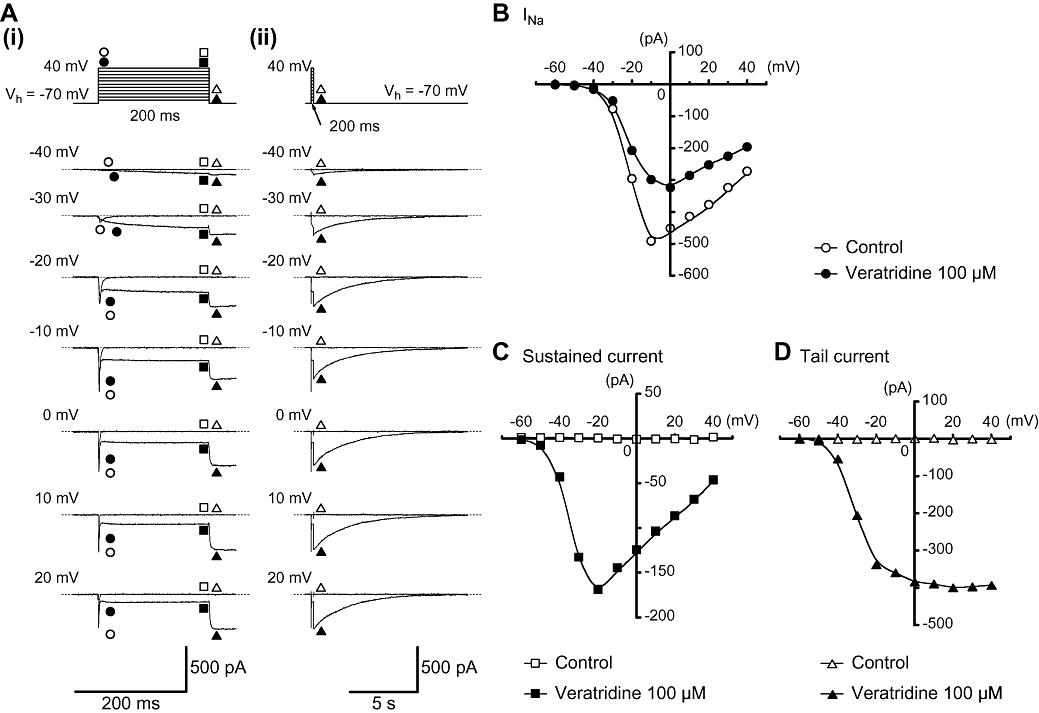
Effects of veratridine (100 µM) on INa in myocytes from mouse vas deferens. (A) (i) Original current traces were superimposed before (control) and after application of 100 µM veratridine at indicated pulse potentials. (ii) The decay of the tail current traces in the absence and presence of 100 µM veratridine superimposed at a slower time frame of (i). (B) Current–voltage relationships of the peak amplitude of transient INa obtained in the absence (control) or presence of 100 µM veratridine. The peak amplitude of INa was measured as the largest amplitude of the first 10 ms of the test pulse. (C and D) Current–voltage relationships of the 100 µM veratridine-induced sustained current (measured mean amplitude of the last 20 ms of the test pulse) and tail current (the peak amplitude). All curves were drawn by eye.
Figure 7.
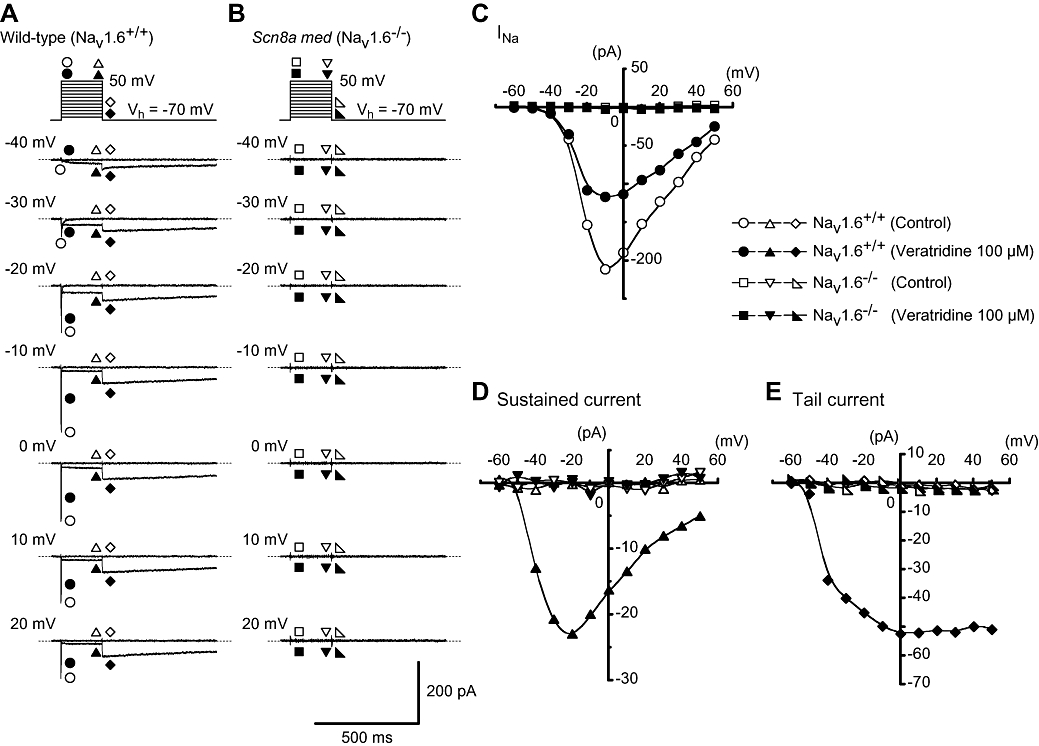
Comparative studies of the effects of veratridine (100 µM) on membrane currents in myocytes from vasa deferentia of NaV1.6+/+ and NaV1.6−/− mice. (A) In NaV1.6+/+ cells, representative traces of the membrane currents in the absence and presence of veratridine at the indicated membrane potentials. (B) In NaV1.6−/− cells, representative traces of the membrane currents in the absence and presence of veratridine at the indicated membrane potentials. (C) The current–voltage relationships in NaV1.6+/+ and NaV1.6−/− myocytes. The current amplitude was measured as the peak amplitude of INa (within the first 10 ms of the depolarizing test pulse) and the peak amplitude of the tail current at each membrane potential. The mean amplitude of the veratridine-induced sustained currents was measured at the last 20 ms of the rectangular pulse. (D and E) Current–voltage relationships of the veratridine-induced sustained current (measured mean amplitude of the last 20 ms of the test pulse) and tail current (the peak amplitude) in NaV1.6+/+ and NaV1.6−/− myocytes. The curves were drawn by eye.
Steady-state inactivation of the current was assessed using a two-step protocol in which the cells were stepped from −70 mV to a range of voltages between −110 and 30 mV for a period of 20 s before a 200 ms step to −10 mV. The peak amplitude of INa evoked by each test pulse was measured. The peak amplitude of INa with application of the conditioning pulse at −110 mV was normalized to unity. The lines were drawn by fitting the data to the Boltzmann's equation in the least-squares method;
where I, I1, I2, V, Vhalf and k are the relative amplitude of INa observed at various amplitudes of the conditioning pulse (I) and the amplitude of the inactivating component of current (I1), the amplitude of the non-inactivating component (I2), the amplitude of the conditioning pulse (V), and where the amplitude of INa was reduced to half (Vhalf), and the slope factor (k) respectively (see Figure 5A).
Figure 5.
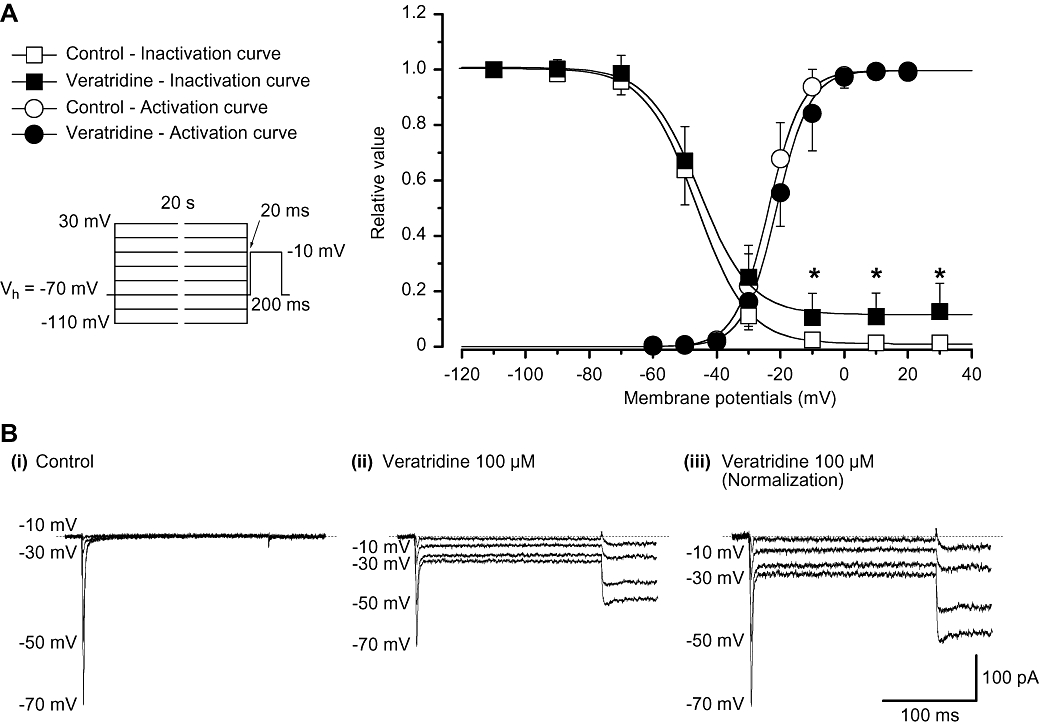
Effects of veratridine on the voltage-dependent activation and inactivation of INa in myocytes from mouse vas deferens. (A) The steady-state inactivation curves in the absence or presence of veratridine were drawn using the following values: I1= 0.99, I2= 0.01, Vhalf=−45.8 mV and k = 8.0 in control; I1= 0.88, I2= 0.12, Vhalf=−45.5 mV and k = 8.0 in the presence of veratridine for the peak amplitude of INa. The activation curves in the absence or presence of veratridine were drawn using the following values: Imax= 1.0, Vhalf=−23.7 mV and k = 5.2 in control; Imax= 1.0, Vhalf=−21.2 mV and k = 5.2 in the presence of veratridine for the peak amplitude of INa. Each symbol indicates the mean of five observations with ±SD shown by vertical lines. Some of the SD bars are less than the size of the symbol. (B) Original current traces before (control, (i)) and after application of 100 µM veratridine (ii) at the indicated conditioning pulse potentials. (iii) INa from (ii) scaled to match the peak amplitude of the current with no conditioning pulse in the absence of veratridine (i.e. control) are shown.
Activation curves were derived from the current–voltage relationships. Conductance (G) was calculated from the equation G = INa/(Em− ERev-Na), where INa is the peak current elicited by depolarizing test pulses to 40 mV from a holding membrane potential of −70 mV and ERev-Na is the reversal potential for Na+. ERev-Na was estimated by extrapolation of each current–voltage relationship to the current axis. Gmax is the maximal Na+ conductance (calculated at potentials above −10 mV). Values for G/Gmax were plotted against membrane potential as relative amplitudes.
Solutions and drugs
The composition of the bath solution (physiological salt solution) for patch-clamp experiments was (mM): Na+ 140, K+ 5, Mg2+ 1.2, Ca2+ 2, glucose 5, Cl– 151.4, HEPES 10, titrated with Tris base to pH 7.35–7.40. The composition of the pipette solution was (mM): Cs+ 130, tetraethylammonium (TEA+) 10, Mg2+ 2, Cl– 144, glucose 5, EGTA 5, ATP 5, HEPES 10, titrated with Tris base to pH 7.35–7.40. Occasionally, 20 mM NaCl was added to the above pipette solution to estimate the reversal potential of tail currents (i.e. see results in Figure 4). Cells were allowed to settle in the small experimental chamber (80 µL in volume) which was superfused by gravity throughout the experiments at a rate of 2 mL min−1 from a solution reservoir. TTX citrate (Sankyo Co. Ltd., Tokyo, Japan) was dissolved in deionized water to produce a stock solution of 1 mM, aliquoted and stored frozen (−20°C) until required. All other drugs were purchased from Sigma-Aldrich (Sigma-Aldrich Japan K.K., Tokyo, Japan).
Figure 4.
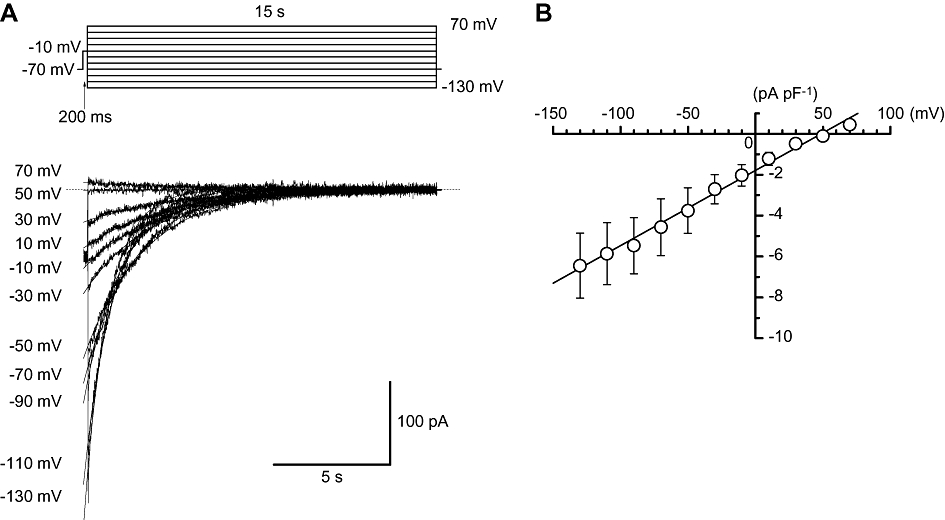
Ion selectivity of the veratridine-induced tail currents in myocytes from mouse vas deferens. (A) Current traces at the indicated membrane potentials. After an initial depolarization to −10 mV from a holding potential of −70 mV, the cell was repolarized to different test potentials (from −130 to 70 mV). (B) The peak amplitude of the veratridine-induced tail currents was plotted against the test potentials. The reversal potential was estimated to be 48.5 ± 1.6 mV.
NaV1.6-null mice
Heterozygous Scn8a-motor end-plate disease (med) mice maintained on a C3HeB/FeJ background were obtained from The Jackson Laboratories (Bar Harbor, ME, USA; see Zhu et al., 2008). The med mutation produces complete loss of NaV1.6 (Ensembl ID: ENSG00000196876) expression (Alexander et al., 2008). In order to compare animals homozygous for med (NaV1.6−/−) with wild-type (NaV1.6+/+) littermates, genotyping was performed with individual pups from the intercrosses of heterozygote (NaV1.6+/−) mice, and then NaV1.6−/− or NaV1.6+/+ mice were selected for further examination. Genotyping of NaV1.6 was performed by PCR as described previously (Kohrman et al., 1996; Zhu et al., 2008). PCR amplification was achieved with 35 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 30 s using the following specific primers: for the wild-type allele, 5′-GGA GCA AGG TTC TAG GCA GCT TTA AGT GTG-3′ and 5′-GTC AAA GCC CCG GAC GTG CAC ACT CAT TCC-3′ and for the mutant allele, 5′-TCC AAT GCT ATA CCA AAA GTC CC-3′ and 5′-GGA CGT GCA CAC TCA TTC CC-3′. PCR products were separated on a 1.5% agarose gel, allowing the resolution of a 230-base-pair product for the wild-type allele and a 194-base-pair product for the mutant allele.
Data analysis
Statistical comparisons were performed using Student's t-test for paired observations, taking P < 0.05 as significant. Statistical analyses were also performed with anova tests (two-way with replication) unless otherwise stated. Measurements are presented as mean ± standard deviation (SD).
Results
Dual action of veratridine on the peak amplitude of voltage-gated Na+ currents in murine vas deferens myocytes
Patch-clamp experiments were performed to investigate the effects of veratridine on TTX-sensitive Na+ channels in myocytes freshly dissociated from BALB/c mice vas deferens. Using a conventional whole-cell configuration, a rectangular depolarizing voltage step pulse to −10 mV from a holding potential of −70 mV elicited a fast transient inward Na+ current (INa) in the presence of 100 µM Cd2+ (described previously in Zhu et al., 2008). Figure 1A shows the representative current traces of the membrane currents in the absence (i.e. control) and presence of veratridine (1–100 µM). Veratridine (1–10 µM) enhanced the peak amplitude of INa (P < 0.05) although higher concentrations of veratridine (30–100 µM) reduced the peak amplitude of INa (30 µM, 91 ± 12%; n= 5 cells, 2 different animals, P < 0.05; 100 µM, 61 ± 10%; n= 17 cells, 7 different animals, P < 0.05), showing a dual action when the peak amplitude of INa was normalized as one just before the application of veratridine (Figure 1B).
Figure 1.
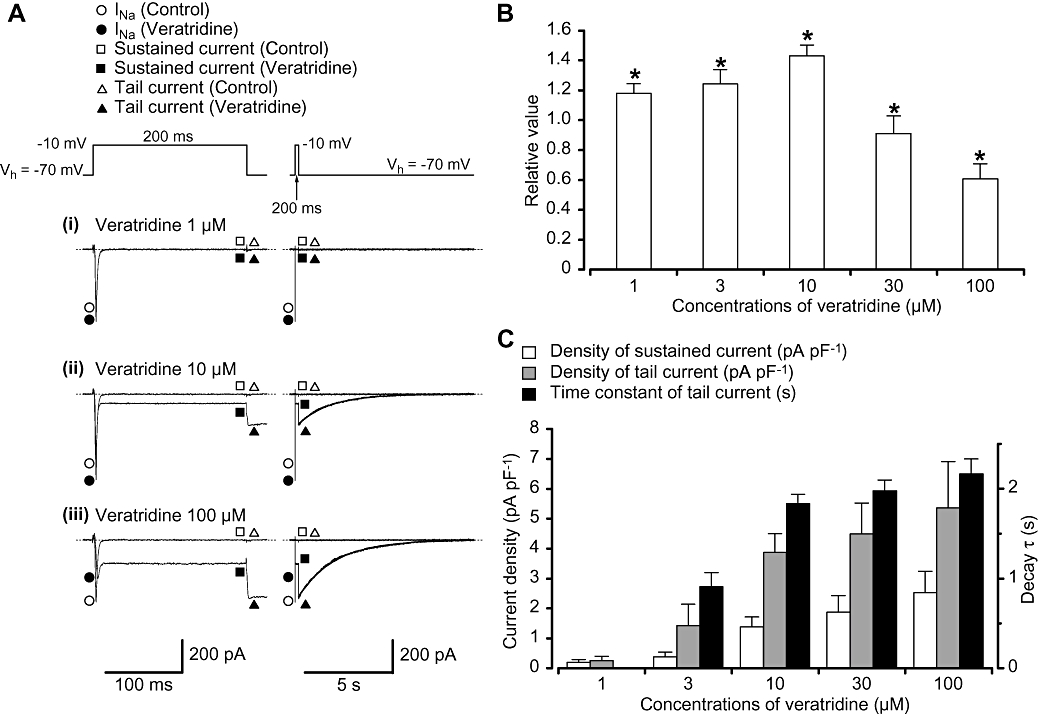
Effects of veratridine on the membrane currents in myocytes from mouse vas deferens using a conventional whole-cell configuration. (A) Representative current traces in the absence (control) and presence of veratridine (1 (i), 10 (ii) and 100 µM (iii)) using the same cell are shown at the left panel. The tail currents are also shown as an expanded trace at the right panel. The tail currents were well fitted by an exponential (10 µM, τ= 1.8 s; 100 µM, τ= 2.1 s). Membrane currents were evoked by a depolarizing rectangular pulse (200 ms duration; 20 s interval) from a holding potential of −70 to −10 mV. (B) Dual action of veratridine (1–100 µM) on the peak amplitude of INa. The relative values of the peak amplitude were expressed when the peak amplitude of INa in the absence of veratridine (control) was normalized as one. (C) Veratridine (1–100 µM) increased the amplitude of the sustained current (measuring the mean value of the last 20 ms of the test pulse) and the peak amplitude of the tail current in a concentration-dependent manner. The time constant of tail current decay (τ) was prolonged when the concentrations of veratridine were increased. The ordinate at the left side shows the current density of INa (pA pF−1). The time constant of the tail current decay (τ) was also indicated at the right side. Each column shows the mean of 5–17 cells (13 different animals) with +SD shown by vertical lines. Asterisk indicates a statistically significant difference and changes were considered significant at P < 0.05 (anova test).
Higher concentrations of veratridine (≥3 µM) promoted the appearance of both a sustained inward component during the test depolarization, and a slowly deactivating tail current at repolarization. The amplitude of the veratridine-induced sustained inward current became stable during the depolarizing step pulse and increased in a concentration-dependent manner (Figure 1C). The decay of the veratridine-induced tail current was well fitted by a single exponential. The time constant of the current decay (τ) was prolonged from 0.9 ± 0.2 s (veratridine 3 µM, n= 6 cells, 5 different animals) to 2.2 ± 0.2 s (veratridine 100 µM, n= 17 cells, 7 different animals) in a concentration-dependent manner (Figure 1C). Note that the veratridine-induced sustained inward current and the veratridine-induced tail current were reversibly abolished by additional application of TTX (1 µM).
Figure 2A shows the time course of the effects of veratridine (100 µM) on the peak amplitude of INa, the mean amplitude of the sustained inward current during the last 20 ms, and the peak amplitude of the tail current respectively. Application of veratridine (100 µM) gradually induced the sustained inward current and the tail current, while, in contrast, the peak amplitude of INa was inhibited by veratridine (100 µM). In order to identify the veratridine-induced membrane currents as Na+ currents, extracellular Na+ concentration was changed to an iso-osmotic solution of TEA+ in the presence of veratridine (100 µM). When extracellular Na+ was replaced with TEA+, the peak amplitude of INa was readily suppressed (see Figure 2A, (iii)). Similarly, the sustained inward current and the tail current markedly decreased, and 2 min later, both veratridine-induced membrane currents were abolished. On reintroduction of Na+ and removal of TEA+, the membrane currents recovered to the control level (Figure 2, n= 5 cells, 3 different animals). Similar results were obtained when the same amount of Na+ was replaced with Tris+ or N-methyl-D-glucamine (NMDG+) (data not shown).
Figure 2.

The time course of the effects of veratridine (100 µM) on three different parameters of the membrane currents (the peak amplitude of INa, the mean value of the sustained current (the mean value of the last 20 ms of the test pulse) and the peak amplitude of the tail current) elicited by repetitive depolarizing pulses (200 ms duration; 20 s interval) from −70 to −10 mV. (A) In the presence of 100 µM veratridine, the extracellular Na+ was reversibly replaced with TEA+. (B) Original current traces before (control, (i)) and after (ii) application of veratridine (100 µM), as indicated in (A). (iii) indicates a current trace after extracellular Na+ was replaced with several iso-osmotic solutions (TEA+) in the presence of veratridine. On removal of Na+-free solution, the membrane current was recovered (iv). (v) indicates a current trace after the removal of veratridine (100 µM).
Effects of veratridine on INa
As shown in Figure 3A, at potentials more positive than −40 mV, the evoked INa reached a peak and then gradually decayed. The peak amplitude was obtained at approximately −10 mV and the amplitude was reduced at more positive potentials, demonstrating a voltage dependency. In the absence of veratridine (control), no tail currents were evoked after the rectangular depolarization pulses (Figure 3A). In the presence of 100 µM veratridine, the peak amplitude of INa was reduced, shifting the maximum value to approximately 0 mV. Figure 3B shows the current–voltage relationships in the absence and presence of 100 µM veratridine. The maximum amplitude of the veratridine-induced sustained inward current was obtained approximately at −20 mV, reducing the peak value at more positive potentials (Figure 3C). In contrast, the peak amplitude of the veratridine-induced tail current gradually increased and the amplitude became stable at 0 mV (Figure 3D).
Ion selectivity of veratridine-induced tail currents
In order to make a rough estimation of the ion selectivity of the slow veratridine-induced tail currents, a double pulse protocol [conditioning pulse, rectangular pulse (200 ms duration) from −70 mV to −10 mV; test pulse, 15 s duration, see inset, Figure 4A] was performed when the external concentration of Na+ was 140 mM and the pipette Na+ concentration was 20 mM. Figure 4A shows the current traces of the veratridine-induced tail currents at the indicated membrane potentials (from −130 mV to 70 mV). As shown in Figure 4B, the peak amplitude of the veratridine-induced tail currents was plotted against the test potentials, estimating the reversal potential. As the current voltage relationship was linear, the reversal potential was estimated to be 48.5 ± 1.6 mV (n= 5 cells, 2 different animals). This value was close to the theoretical Na+ equilibrium potential calculated on the basis of the Nernst equation (ENa= 50.0 mV).
Effects of veratridine on inactivation and activation curves of INa
The voltage dependency of inactivation was investigated before and after application of veratridine (100 µM) using the experimental protocol shown in Figure 5A (conditioning pulse duration, 20 s; holding membrane potential, −70 mV). In the absence of veratridine (control), inactivation of INa occurred with conditioning pulses positive to −50 mV [the 50% inactivation potentials (Vhalf); control, Vhalf=−45.9 ± 4.0 mV, n= 5 cells, 2 different animals]. In the presence of veratridine (approximately 5 min later), the voltage-dependent inactivation curve for the peak amplitude of INa was not shifted significantly (Vhalf=−45.3 ± 3.7 mV; n= 5 cells, 2 different animals; P > 0.05) although the non-inactivated component was enhanced [0.99 ± 0.02 (n= 5 cells, 2 different animals) of the peak INa inactivated in control whereas 0.89 ± 0.09 (n= 5 cells, 2 different animals) of peak INa inactivated in the presence of veratridine; P < 0.05, anova test]. The activation curves obtained from the current–voltage relationships in Figure 3B, fitted to the Boltzmann equation, are shown in Figure 5A. In the presence of veratridine (100 µM), the activation curve for the peak amplitude of INa was not significantly shifted (the 50% activation potentials (Vhalf); control, Vhalf=−23.7 ± 2.7 mV, n= 5 cells, 3 different animals; 100 µM veratridine, Vhalf=−21.9 ± 2.9 mV, n= 5 cells, 3 different animals; P > 0.05). As shown in Figure 5B, current traces of the test pulses at the indicated conditioning pulse potentials were detected in the absence and presence of 100 µM veratridine. In the presence of veratridine, the peak-matched traces are also demonstrated at the indicated conditioning pulse potentials when the peak amplitude of INa evoked by the test pulse with no conditioning pulse was superimposed on that in the absence of veratridine (i.e. control).
Effects of veratridine on muscle tone of intact vas deferens in NaV1.6-null mice (med mouse) lacking the expression of Na+ channel gene, Scn8a
As described previously in Zhu et al. (2008), the only functional voltage-gated Na+ channel present in mouse vas deferens when depolarizing pulses were applied was the NaV1.6 channel. The effects of veratridine on the muscle tone of vas deferens were thus compared between homozygous mice with a null allele of NaV1.6 (med mice, NaV1.6−/−; Burgess et al., 1995) and wild-type mice (NaV1.6+/+). Under normal conditions, the vas deferens from wild-type mice does not show detectable spontaneous contractions. In vas deferens from NaV1.6+/+ mice, the application of low concentrations of veratridine (≤1 µM) did not cause any change to the isometric tension recorded from the tissue compared with control. However, application of higher concentrations of veratridine (≥3 µM) resulted in a gradual increase in phasic contractions (n= 4, 3 different animals; see Figure 6A). The integrated area of the veratridine-induced spontaneous contractions (the last 1 min duration) increased in a concentration-dependent manner (n= 4, 3 different animals, Figure 6C). In some preparations, these contractions were superimposed upon an increase in the basal tension in the tissue. These phasic contractions were abolished by the additional application of 1 µM TTX (n= 6 tissues, 3 different animals), suggesting that veratridine was having its effects through stimulation of Na+ channels (Figure 6A). Furthermore, in vasa deferentia from NaV1.6−/−, mice, cumulative application of veratridine (1–30 µM) caused no spontaneous contractions (Figure 6B and C). Figure 6C summarizes the difference in the veratridine (3 and 10 µM)-induced contractions between tissues from NaV1.6+/+ and NaV1.6−/− mice (P < 0.05, anova test).
Figure 6.
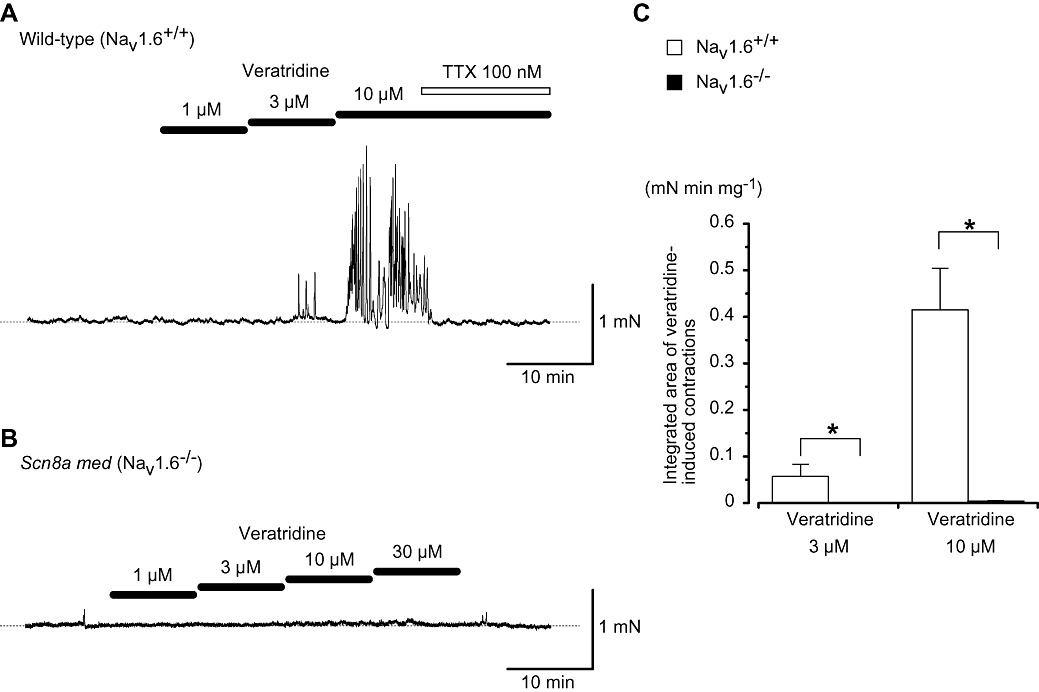
Veratridine-induced contractions involve activation of NaV1.6 channels. (A) Veratridine-induced contractions in NaV1.6+/+ vas deferens. The cumulative application of veratridine resulted in rhythmical contractions which were abolished rapidly by subsequent application of tetrodotoxin (TTX) (100 nM). The dashed line indicates the mean resting tone of vas deferens. (B) Trace showing how the application of veratridine (1–30 µM) resulted in little change to baseline tension of vas deferens from a NaV1.6−/− mouse. The dashed line indicates the mean resting tone of vas deferens. (C) Overall results showing veratridine (3 and 10 µM)-induced contractions in vasa deferentia from NaV1.6+/+ and NaV1.6−/− mice. The integrated areas of the veratridine-induced contractions (the last 1 min duration) were calculated (mN min mg−1). Each column indicates the mean + SD shown by vertical lines. Asterisk indicates a statistically significant difference and changes were considered significant at P < 0.05 (anova test).
The effects of veratridine on the membrane currents in NaV1.6−/− myocytes
To ensure that veratridine was only activating a current mediated by the NaV1.6 channel, membrane currents in smooth muscle cells from the vasa deferentia of NaV1.6−/− mice were investigated (n= 5 cells, 4 different animals). As shown in Figure 7A, in NaV1.6+/+ cells, TTX-sensitive INa were evoked by application of depolarizing rectangular pulses from a holding potential of −70 mV (n= 4 cells, 2 different animals), which showed similar electrophysiological and pharmacological properties with those recorded in BALB/c mouse vas deferens cells. Similarly, in NaV1.6+/+ type cells, veratridine (100 µM) induced both the sustained currents and the tail currents although the peak amplitude of INa was inhibited. Application of veratridine (100 µM) to tissues from NaV1.6−/− mice caused no detectable inward membrane currents in dispersed smooth muscle cells from the vas deferens (Figure 7B). These results imply that the veratridine-induced membrane currents observed in vas deferens smooth muscle cells are closely related to the activation of NaV1.6, and that no other voltage-gated Na+ channels are contributing to the membrane currents evoked by veratridine.
Discussion
In the present study, it has been demonstrated that veratridine possesses a dual action on INa in freshly dispersed smooth muscle cells from mouse vas deferens, depending on its concentrations. The present work also establishes that the veratridine-induced contractions, sustained currents and tail currents are TTX-sensitive. Finally, these currents and contractions appear to link to the activation of NaV1.6 channels.
Veratridine-induced contractions
Veratridine has been used as a potent voltage-gated Na+ channel activator in many excitable tissues, and like the other lipid soluble neurotoxins, batrachotoxin and aconitine, is thought to target the type 2 receptor site (D1-S6) of the α subunit of the voltage-gated Na+ channel, causing depolarization of the resting membrane potential and repetitive action potential firing (see Wang and Wang, 2003b). We have previously shown that smooth muscle cells of the mouse vas deferens express NaV1.6 as the sole functional channel present in these cells, which was activated by veratridine (Zhu et al., 2008), and so the actions of veratridine on the longitudinal layer of the intact vas deferens were investigated. Veratridine (10 µM) induced rhythmical contractions in all preparations of wild-type littermates (i.e. NaV1.6+/+) but not NaV1.6-null mice (i.e. NaV1.6−/−), similar to the contractions induced by veratrine (a mixture of alkaloid esters, including veratridine, cevadine, cevine, cevadilline, and sabadine) detected by Chen et al. (2002). Furthermore, the veratridine-induced contractions were TTX-sensitive, suggesting that NaV1.6 channels were involved.
Dual action of veratridine on the peak amplitude of INa
The effects of veratridine on the peak amplitude of INa have been well-examined in a wide variety of excitable cells (reviewed by Wang and Wang, 2003b). Veratridine (30–300 µM) enhanced the peak amplitude of INa in a concentration-dependent manner [frog myelinated nerve (Rando, 1989); rat ventricular myocytes (Zong et al., 1992)], while in contrast, veratridine (100–166 µM) reduced the peak amplitude of INa in other tissues [frog skeletal muscle (Sutro, 1986); N18 neuroblastoma cells (Barnes and Hille, 1988); rat skeletal muscle (Wang and Wang, 2003a)]. Especially, in cultured vascular and visceral smooth muscles, veratridine (100 µM) mainly inhibited the peak amplitude of INa[cultured rat myometrial cells (Martin et al., 1990); cultured human coronary myocytes (Quignard et al., 1996; Boccara et al., 1999); cultured human uterine leiomyosarcoma cell lines (Kusaka and Sperelakis, 1994); descending vasa recta (Zhang et al., 2005)]. Thus, it seems that the effects of veratridine vary between molecular types of voltage-gated Na+ channels, depending on the subtypes of Na+ channels present (neuron, NaV1.2; skeletal muscle, NaV1.4; cardiac myocytes, NaV1.5 etc. reviewed by Catterall et al., 2005). In the present experiments, we have shown a bell-shaped relationship between the concentrations of veratridine and the peak amplitude of INa (acting mainly on NaV1.6; see also Zhu et al., 2008) in murine vas deferens myocytes, reaching maximum enhancement at a concentration of around 10 µM. These results indicate that veratridine possesses a dual effect on the peak amplitude of INa in myocytes from mouse vas deferens (i.e. in native cells expressing the sole functional voltage-gated Na+ channel, NaV1.6), depending on its concentration. As veratridine (100 µM) inhibited the peak amplitude of INa in cultured vascular and visceral smooth muscles, this is the first report to show smooth muscle type Na+ currents which were activated by veratridine at concentrations less than 10 µM. Moreover, as veratridine is a potent agonist for voltage-gated Na+ channels with long-lasting opening times, some reports indicate that veratridine (100 µM) alters the ion selectivity of INa, particularly in skeletal muscle preparations [see, e.g. rat skeletal muscle (Garber and Miller, 1987); frog muscle fibre (Leibowitz et al., 1986)]. However, in the present set of experiments, when extracellular Na+ concentration was changed to an iso-osmotic solution of TEA+ in the presence of 100 µM veratridine, the peak amplitude of INa was abolished, and on reintroduction of Na+ and removal of TEA+, the peak amplitude of INa readily recovered to the control level. These results suggest that INa is carried mainly by Na+, without altering the ion selectivity even in the presence of veratridine. Moreover, it has also been reported that veratridine reduced the size of single-channel conductances to about one quarter of that in control (Barnes and Hille, 1988). It is possible that the reduction of the peak amplitude of INa when high concentrations of veratridine are applied (as observed in this study) is related to a decrease in single-channel conductance.
Veratridine-induced membrane currents in murine vas deferens
In vascular and visceral smooth muscles, two types of the veratridine-induced membrane currents can be detected; namely, the sustained currents during the depolarizing step pulses, which are thought to be due to incomplete fast inactivation, and the tail currents at repolarization, which are thought to be due to the toxin-bound Na+ channels remaining open for a prolonged period at the holding potential (Martin et al., 1990; Kusaka and Sperelakis, 1994; Wang and Wang, 2003a). In murine vas deferens myocytes, application of veratridine (≥3 µM) also induced sustained currents and tail currents. In the present study, both veratridine-induced membrane currents were abolished by additional application of 1 µM TTX in the presence of Cd2+ (showing sensitivity to TTX), and were reversibly suppressed by substitution of Na+ with TEA+, Tris+ and NMDG+ (showing a selective Na+ permeability). After long depolarizing pulses (≥15 s), these currents were inactivated, thereby suggesting that voltage-dependent components were involved. Using NaV1.6−/− mice, neither sustained nor tail veratridine-induced membrane currents were detected in NaV1.6−/− vasa deferentia, while both the tail and sustained veratridine-induced membrane currents were recorded in tissues from NaV1.6+/+ mice. Taken together, these results show unequivocally that the veratridine-induced membrane currents are mediated by TTX-sensitive voltage-gated Na+ channels (i.e. NaV1.6).
Kinetics studies of veratridine-induced INa
As veratridine shifted activation curves to more negative potentials by 40–90 mV, producing the sustained currents and the tail currents, it is generally believed that veratridine modifies the activation gating kinetics of Na+ channels, thereby allowing them to open more easily and to stay open longer (Leibowitz et al., 1986; Hille, 1992; Sunami et al., 1993). However, in the present experiments, the activation and inactivation curves for the peak amplitude of INa in mouse vas deferens were not shifted in the presence of 100 µM veratridine. Similarly, veratridine (100 µM) caused no shift of the activation and inactivation curves for the peak amplitude of INa in cultured smooth muscle cells (human uterine leiomyosarcoma cell lines, Kusaka and Sperelakis, 1994). In contrast, in the presence of veratridine, some INa remained as a sustained component during the depolarization step pulses from −50 mV to 40 mV. Furthermore, veratridine-induced tail currents were evoked after the rectangular step pulses. These results suggest that veratridine may modify the gating of NaV1.6, allowing the channels to stay open longer after depolarization.
Molecular properties of the veratridine-induced INa in NaV1.6+/+ murine vas deferens
Saleh et al. (2005) reported that two different types of voltage-gated Na+ channel proteins (i.e. NaV1.6 and NaV1.7) were localized in murine portal vein by use of immunohistological studies and they concluded that there were multiple types of voltage-gated Na+ channels in vascular smooth muscle. However, recently, we have reported that the molecular identity of the voltage-gated Na+ channels responsible for the TTX-sensitive INa in murine vas deferens myocytes is primarily NaV1.6 (Zhu et al., 2008). Furthermore, using NaV1.6−/− mice, INa were not detected in dispersed smooth muscle cells from the vas deferens, while TTX-sensitive INa were recorded in NaV1.6+/+ (Zhu et al., 2008). As veratridine has been used classically as a voltage-gated Na+ channel agonist, it is generally accepted that veratridine targets several types of voltage-gated Na+ channels, including NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6 and NaV1.7 (Catterall et al., 2005). In the present experiments, application of veratridine caused no further detectable membrane currents in NaV1.6−/− tissues, while veratridine induced the sustained currents and the tail currents in NaV1.6+/+. These results strongly suggest that the sustained currents and the tail currents in mouse vas deferens are primarily due to activation of NaV1.6 channels, confirming the presence of NaV1.6 as the predominant functional channel in mouse vas deferens.
In conclusion, we found that veratridine suppressed the peak amplitude of INa but induced sustained currents and the tail currents through Nav1.6 channels. The present study also provides novel evidence that the Nav1.6 channel is the major TTX-sensitive Na+ channel expressed in mouse vas deferens smooth muscle cells, and that the preparation could provide a unique experimental system for the study of Nav1.6 channel function in native cells.
Acknowledgments
This work was supported by a Grant-in-Aid from the Japan Science and Technology Agency (15-B01-2008 to Noriyoshi Teramoto), a Grant-in-Aid for Exploratory Research from the Japanese Society for the Promotion of Science (19650733 to Noriyoshi Teramoto) and a Grant-in-Aid for Scientific Research (B) from the Japanese Society for the Promotion of Science (20300178 to Noriyoshi Teramoto). This work was also supported by the Goho Life Sciences International Fund (2008 to Noriyoshi Teramoto). Dr Noriyoshi Teramoto was also supported by a grant from the Japanese Society for Scientist Exchange Program between the Japan Society for the Promotion of Science and The Royal Society (2006-1-36-RS to Noriyoshi Teramoto). Dr Hai-Lei Zhu was awarded by a Grant-in-Aid from the Japan Society for the Promotion of Science (FY2007 JSPS Postdoctoral Fellowship for Foreign Researcher, P 07196 to Noriyoshi Teramoto). Dr Thomas C. Cunnane is supported by the Wellcome Trust (069768) and Mr Richard D. Wassall is a British Pharmacological Society A.J. Clark Student. Part of this study has been communicated to the 81st Annual Meeting of the Japanese Pharmacological Society in an abstract form (Zhu and Teramoto, 2008).
Glossary
Abbreviations:
- INa
voltage-gated Na+ currents
- med
motor end-plate disease
- NMDG
N-methyl-D-glucamine
- PSS
physiological salt solution
- TEA
tetraethylammonium
- TTX
tetrodotoxin
- Vhalf
50% activation or inactivation potential
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes S, Hille B. Veratridine modifies open sodium channels. J Gen Physiol. 1988;91:421–443. doi: 10.1085/jgp.91.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccara G, Choby C, Frapier JM, Quignard JF, Nargeot J, Dayanithi G, et al. Regulation of Ca2+ homeostasis by atypical Na+ currents in cultured human coronary myocytes. Circ Res. 1999;85:606–613. doi: 10.1161/01.res.85.7.606. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, et al. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Campos FV, Moreira TH, Beirão PS, Cruz JS. Veratridine modifies the TTX-resistant Na+ channels in rat vagal afferent neurons. Toxicon. 2004;43:401–406. doi: 10.1016/j.toxicon.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Chen YF, Lin YT, Tan TW, Tsai HY. Effects of veratrine and paeoniflorin on isolated mouse vas deferens. Phytomedicine. 2002;9:296–301. doi: 10.1078/0944-7113-00122. [DOI] [PubMed] [Google Scholar]
- Cuprian AM, Solanki P, Jackson MV, Cunnane TC. Cholinergic innervation of the mouse isolated vas deferens. Br J Pharmacol. 2005;146:927–934. doi: 10.1038/sj.bjp.0706357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber SS, Miller C. Single Na+ channels activated by veratridine and batrachotoxin. J Gen Physiol. 1987;89:459–480. doi: 10.1085/jgp.89.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Modifiers of gating. In: Hille B, editor. Ionic Channels of Excitable Membranes. 2nd edn. Sunderland, MA: Sinauer Associates; 1992. pp. 445–471. [Google Scholar]
- Kohrman DC, Harris JB, Meisler MH. Mutation detection in the med and medJ alleles of the sodium channel Scn8a. Unusual splicing due to a minor class AT-AC intron. J Biol Chem. 1996;271:17576–17581. doi: 10.1074/jbc.271.29.17576. [DOI] [PubMed] [Google Scholar]
- Kusaka M, Sperelakis N. Veratridine actions on two types of fast Na+ channels in human uterine leiomyosarcoma cells. Eur J Pharmacol. 1994;271:387–393. doi: 10.1016/0014-2999(94)90798-6. [DOI] [PubMed] [Google Scholar]
- Leibowitz MD, Sutro JB, Hille B. Voltage-dependent gating of veratridine-modified Na channels. J Gen Physiol. 1986;87:25–46. doi: 10.1085/jgp.87.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Arnaudeau S, Jmari K, Rakotoarisoa L, Sayet I, Dacquet C, et al. Identification and properties of voltage-sensitive sodium channels in smooth muscle cells from pregnant rat myometrium. Mol Pharmacol. 1990;38:667–673. [PubMed] [Google Scholar]
- Nánási PP, Varró A, Lathrop DA, Bryant SH. Effects of veratridine on Na and Ca currents in frog skeletal muscle. Gen Pharmacol. 1994;25:1661–1666. doi: 10.1016/0306-3623(94)90369-7. [DOI] [PubMed] [Google Scholar]
- Quignard JF, Ryckwaert F, Albat B, Nargeot J, Richard S. A novel tetrodotoxin-sensitive Na+ current in cultured human coronary myocytes. Circ Res. 1997;80:377–382. [PubMed] [Google Scholar]
- Rando TA. Rapid and slow gating of veratridine-modified sodium channels in frog myelinated nerve. J Gen Physiol. 1989;93:43–65. doi: 10.1085/jgp.93.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh S, Yeung SY, Prestwich S, Pucovský V, Greenwood I. Electrophysiological and molecular identification of voltage-gated sodium channels in murine vascular myocytes. J Physiol. 2005;568:155–169. doi: 10.1113/jphysiol.2005.090951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutro JB. Kinetics of veratridine action on Na channels of skeletal muscle. J Gen Physiol. 1986;87:1–24. doi: 10.1085/jgp.87.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami A, Sasano T, Matsunaga A, Fan Z, Swanobori T, Hiraoka M. Properties of veratridine-modified single Na+ channels in guinea pig ventricular myocytes. Am J Physiol. 1993;264:H454–H463. doi: 10.1152/ajpheart.1993.264.2.H454. [DOI] [PubMed] [Google Scholar]
- Teramoto N, Brading AF. Activation by levcromakalim and metabolic inhibition of glibenclamide-sensitive K channels in smooth muscle cells of pig proximal urethra. Br J Pharmacol. 1996;118:635–642. doi: 10.1111/j.1476-5381.1996.tb15448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cao C, Lee-Kwon W, Pallone TL. Descending vasa recta pericytes express voltage operated Na+ conductance in the rat. J Physiol. 2005;567:445–457. doi: 10.1113/jphysiol.2005.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HL, Teramoto N. Modulation of voltage-gated Na+ currents in the murine vas deferens by veratridine. J Pharmacol Sci. 2008;106(Suppl. I):249P, P3J–19. [Google Scholar]
- Zhu HL, Aishima M, Morinaga H, Wassall RD, Shibata A, Iwasa K, et al. Molecular and biophysical properties of voltage-gated Na+ channels in murine vas deferens. Biophys J. 2008;94:3340–3351. doi: 10.1529/biophysj.107.117192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong XG, Dugas M, Honerjäger P. Relation between veratridine reaction dynamics and macroscopic Na current in single cardiac cells. J Gen Physiol. 1992;99:683–697. doi: 10.1085/jgp.99.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Wang SY. Veratridine block of rat skeletal muscle NaV1.4 sodium channels in the inner vestibule. J Physiol. 2003a;548:667–675. doi: 10.1113/jphysiol.2002.035469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Wang GK. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell Signal. 2003b;15:151–159. doi: 10.1016/s0898-6568(02)00085-2. [DOI] [PubMed] [Google Scholar]
- Wang GK, Quan C, Seaver M, Wang SY. Modification of wild-type and batrachotoxin-resistant muscle µ1 Na+ channels by veratridine. Pflügers Arch. 2000;439:705–713. doi: 10.1007/s004249900229. [DOI] [PubMed] [Google Scholar]