

Abstract
Background and purpose:
Obesity is often accompanied by hypercholesterolemia characterized by elevated levels of plasma low-density lipoprotein (LDL) and associated with non-alcoholic steatohepatitis, which could progress to hepatic fibrosis. Hepatic stellate cells (HSCs) are the major effectors of hepatic fibrogenesis. This study aims to clarify effects of LDL on activation of HSC, to evaluate roles of curcumin in suppressing these effects and to further elucidate the underlying molecular mechanisms.
Experimental approaches:
HSCs were prepared from rats and cell proliferation was measured by cell proliferation assays (MTS assays). Transient transfection assays were performed to evaluate gene promoter activities. Real-time polymerase chain reaction and Western blotting were used to analyse the expression of genes.
Key results:
LDL stimulated HSC activation in vitro, which was attenuated by curcumin. Curcumin reduced the abundance of LDL receptor (LDLR) in activated HSCs, decreasing cellular cholesterol. Curcumin-dependent activation of peroxisome proliferator-activated receptor-γ (PPARγ) differentially regulated the expression of the transcription factors, sterol regulatory element-binding proteins (SREBPs), in activated HSCs, resulting in the suppression of LDLR gene expression.
Conclusions and implications:
Curcumin suppressed LDLR gene expression in activated HSCs in vitro by activating PPARγ and differentially regulating gene expression of SREBPs, reducing cellular cholesterol and attenuating the stimulatory effects of LDL on HSC activation. These results provide novel insights into the roles and mechanisms of curcumin in the inhibition of LDL-induced HSC activation. This curcumin, a constituent of turmeric, may be useful in preventing hypercholesterolemia-associated hepatic fibrogenesis.
Keywords: hepatic fibrosis, obesity, low-density lipoprotein, hypercholesterolemia, hepatic stellate cell, gene expression, non-alcoholic steatohepatitis
Introduction
Obesity is an increasingly serious public health problem in the United States. Approximately 300,000 deaths per year are related to obesity in the United States (O'Brien and Dixon, 2002). Obesity is often accompanied by hypercholesterolemia characterized by elevated levels of plasma low-density lipoprotein (LDL), non-alcoholic steatohepatitis (NASH) is an advanced form of non-alcoholic fatty liver disease (NAFLD) and is commonly found in patients with obesity and/or type 2 diabetes. The estimated prevalence of NAFLD in the general population ranges from 6 to 14% (Clark, 2006). Approximately 15–40% of NASH patients develop hepatic fibrosis. However, the roles of hypercholesterolemia in NASH-associated hepatic fibrosis are largely undefined (Neuschwander-Tetri, 2001; Clark, 2006; Cave et al., 2007).
Plasma cholesterol circulates as a constituent of lipoproteins, among which is LDL. Plasma LDL enters cells via endocytosis mediated by the cell-surface LDL receptor (LDLR), and cholesterol is subsequently released from lipoproteins (Goldstein et al., 2006). The LDLR is a glycoprotein that plays a critical role in the homeostatic control of blood cholesterol by mediating the removal of cholesterol-containing lipoprotein particles from circulation (Goldstein et al., 2006). The liver contains about 70% of total LDLR, most of which are in hepatocytes, making the liver the major organ responsible for the turnover of LDL in the circulation (Bilheimer et al., 1984). Lipid homeostasis is tightly controlled, via biosynthesis and cellular uptake, by a group of proteins regulated by the transcription factors sterol regulatory element-binding proteins (SREBPs) (Osborne, 2000), which are encoded by two genes, srebp-1 and srebp-2 (Brown and Goldstein, 1997). SREBP-2 predominantly regulates sterol metabolism and SREBP-1 controls fatty acid (FA) biosynthesis. SREBPs are synthesized as inactive, membrane-bound precursors and are proteolytically activated when cellular sterol concentration is low. The active SREBPs are subsequently transported into the nucleus and bind to sterol regulatory elements (SREs) in promoter regions of target genes, facilitating transcription of the target genes, including ldlr (Brown and Goldstein, 1997).
Hepatic fibrogenesis is a pathophysiological process caused by chronic liver injury, regardless of etiology. Hepatic stellate cells (HSCs), previously called fat- or vitamin A-storing cells, lipocytes or Ito cells, are the effector cells in hepatic fibrogenesis (Kisseleva and Brenner, 2006; Friedman, 2008). HSCs are quiescent and non-proliferative in the normal liver. Upon liver injury, quiescent HSCs become activated, characterized by enhanced cell growth and overproduction of extracellular matrix (ECM) components. Culturing quiescent HSCs on plastic plates induces spontaneous activation, mimicking the process seen in vivo, which provides a good model for elucidating underlying mechanisms of HSC activation and for studying possible therapeutic interventions in the process (Kisseleva and Brenner, 2006; Friedman, 2008).
Accumulating evidence has indicated the hypocholesterolemic properties of curcumin, the yellow pigment in curry from turmeric, in animal models (Ramirez-Tortosa et al., 1999; Zhang et al., 1999; Asai and Miyazawa, 2001; Kapoor et al., 2002; Arafa, 2005). Curcumin attenuates diet-induced hypercholesterolemia (Arafa, 2005) by increasing the rate of cholesterol catabolism (Babu and Srinivasan, 1997), increasing fecal excretion (Rao et al., 1970) and inducing changes in the expression of genes involved in cholesterol homeostasis (Peschel et al., 2007). In addition, curcumin inhibits LDL oxidation (Ramirez-Tortosa et al., 1999). We have demonstrated that curcumin inhibits HSC activation by reducing cell proliferation, inducing apoptosis and suppressing gene expression of ECM components in HSCs in vitro (Xu et al., 2003; Zheng and Chen, 2004; 2006;). Further studies reveal that curcumin induces gene expression of peroxisome proliferator-activated receptor-γ (PPARγ) (Xu et al., 2003), interrupting signalling cascades for platelet-derived growth factor (PDGF) (Zhou et al., 2007), epidermal growth factor (EGF) (Zhou et al., 2007) and transforming growth factor-beta (TGF-β) (Zheng and Chen, 2004; 2006;), and attenuating oxidative stress in activated HSCs (Zheng et al., 2007). This phytochemical protects the liver from injury and fibrogenesis in animal models (Park et al., 2000; Nanji et al., 2003; Fu et al., 2008).
It remains largely unknown whether elevated levels of LDL make a direct contribution to HSC activation and hepatic fibrogenesis in NASH patients. The current study aimed to clarify effects of LDL on activation of HSC, to evaluate roles of curcumin in attenuating these effects and to further elucidate underlying molecular mechanisms. Results from this study supported our hypothesis that LDL stimulated the activation of HSC, which could be attenuated by curcumin by suppressing gene expression of LDLR. Our results provide novel insights into the roles and mechanisms of curcumin in the inhibition of LDL-induced HSC activation and its possible use for therapeutic interventions in hypercholesterolemia-associated hepatic fibrogenesis.
Methods
Isolation and culture of rat HSCs
Rat HSCs were isolated from male Sprague-Dawley rats (200–250 g) as described previously (Xu et al., 2003). Passaged HSCs were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v·v−1) of fetal bovine serum (FBS). Cultured HSCs were used at Passage 4∼9. In some experiments, cells were cultured in serum-depleted DMEM, containing 0.5% (v·v−1) of FBS, for 24 h prior to treatment. The process of serum-starvation made HSCs more sensitive to stimuli, including exogenous LDL.
Evaluation of cell proliferation in vitro
Semi-confluent, serum-starved HSC were stimulated with exogenous LDL at indicated concentrations with or without curcumin at indicated doses for 24 h. Cell proliferation assays were conducted by using the CellTiter 96 aqueous non-radioactive cell proliferation assay kit (MTS assays) purchased from Promega (Madison, WI, USA) following the protocol provided by the manufacturer. Each group had triplicates. Experiments were repeated three times. The MTS assay is a colorimetric method for determining the number of viable cells.
Quantitation of intracellular cholesterol, fatty acids (FA) and triglyceride (TG)
Contents of intracellular cholesterol, FA and TG were, respectively, determined by using the cholesterol quantitation kit (Catalogue no. K603-100), the free FA quantification kit (Catalogue no. K612-100) and the TG quantification kit (Catalogue no. K622-100) from Biovision, Inc. (Mountain View, CA, USA), following the protocols provided by the manufacturer. Cellular cholesterol or FA were extracted from cell lysates by chloroform and dried under a stream of nitrogen to remove chloroform. The whole process was conducted on ice. For all assays, one portion of the lysates was used for determining protein concentrations. Contents of cholesterol, FA and TG were measured spectrophotometrically at 570 nm. Protein concentrations were determined by using the BCA™ Protein Assay Kit according to the protocol provided by the manufacturer (Pierce, Rockford, IL, USA). The values of intracellular cholesterol were calculated as µg cholesterol (mg·cellular protein)−1, whereas the concentrations of FA and TG were calculated as nmol (mg·cellular protein)−1.
Plasmids and transient transfection assays
The SREBP trans-activation activity luciferase reporter plasmid (p6 × SRE-Luc) was a gift from Dr. Andrew J. Brown (Wong et al., 2006). The LDLR promoter luciferase reporter plasmids pLDLR-Luc, containing a fragment (1500 or 211 bp) of the ldlr promoter, were gifts from Dr. Dayami Lopez (Lopez and Ness, 2006). The luciferase reporter plasmids pLDLR-(wt)-Luc and pLDLR-(m)-Luc respectively contained a 335 bp fragment of the ldlr promoter with or without site-directed mutagenesis at SRE (Castoreno et al., 2005). They were purchased from Addgene Inc. (Cambridge, MA, USA). The luciferase reporter plasmids pSREBP-1-Luc with a 2600 bp length of the srebp-1 promoter was kindly provided by Dr. Hitoshi Shimano (Amemiya-Kudo et al., 2000). The luciferase reporter plasmid pSREBP-2-Luc with a 4316 bp length of the srebp-2 promoter was kindly provided by Dr. Timothy Osborne (Shin and Osborne, 2003). The cDNA expression plasmids pSREBP-1 and pdn-SREBP-1 were purchased from Addgene Inc. pSREBP-1 contained a full fragment of wild-type SREBP-1 cDNA, whereas pdn-SREBP-1 contained a dominant negative SREBP-1 cDNA. The PPARγ cDNA expression plasmid pPPARγ, containing a full size of PPARγ cDNA, was a gift from Dr. Reed Graves (Department of Medicine, the University of Chicago). The PPARγ promoter luciferase reporter plasmid p PPARγ-Luc contained the 5′-flanking region (−2776 bp) of the PPARγ gene promoter (Fajas et al., 1999). The PPARγtrans-activation activity reporter plasmid pPPRE-TK-Luc contained three copies of the PPARγ response elements from acyl-CoA oxidase gene linked to the herpes virus thymidine kinase promoter (−105/+51) and a luciferase vector, which was a gift from Dr. Kevin J. McCarthy (Louisiana State University Health Sciences Center in Shreveport). Transient transfection of semi-confluent HSCs in six-well plates was performed using the LipofectAMINE® reagent (Invitrogen Corp. Carlsbad, CA, USA), as we previously described (Xu et al., 2003). Each condition was assayed in triplicate in every experiment and each experiment was repeated at least three times. Luciferase assays were conducted as previously described (Xu et al., 2003). Transfection efficiency was determined by co-transfection of the β-galactosidase reporter plasmid pSV-β-gal (Promega). β-galactosidase assays were performed using an assay kit from Promega Corp., according to the manufacturer's instruction. Luciferase activities were expressed as relative units after normalization with β-galactosidase activities. Results were combined from at least three independent experiments.
Preparation of nuclear protein extracts
Nuclear extracts were prepared as we previously described (Chen and Davis, 1999). In brief, after washing twice with phosphate-buffered saline, cells were evenly re-suspended in Buffer A [10 mmol·L−1 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES)-KOH pH 7.9, 1.5 mmol·L−1 MgCl2, 10 mmol·L−1 KCl, 1 mmol·L−1 dithiothreitol (DTT), 1 mmol·L−1 phenylmethylsulphonyl fluoride (PMSF)]. After incubation on ice for 10 min, cells were mixed by vortex for 30 s and centrifuged at 8500×g for 10 s. Pellets were re-suspended in Buffer C (20 mmol·L−1 HEPES-KOH pH 7.9, 25% glycerol, 420 mmol·L−1 NaCl, 1.5 mmol·L−1 MgCl2, 0.2 mmol·L−1 ethylenediaminetetraacetic acid, 1 mmol·L−1 DTT and 1 mmol·L−1 PMSF) and incubated in ice for 15 min before vortexing. The lysates were centrifuged at 8500×g at 4°C for 2 min, and the resulting supernatants were taken as nuclear protein extracts, and stored at −80°C until use.
Western blotting analyses
Whole cell lysates were prepared using radio-immunoprecipitation analysis buffer supplemented with protease inhibitors. Protein samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and electro-blotted onto polyvinylidene fluoride membranes. Target proteins were detected by primary antibodies and secondary antibodies conjugated with horseradish peroxidase. Antibodies used in Western blotting analyses were presented in Table 1. β-actin was used as an invariant control for equal loading. Protein bands were visualized by using chemiluminescence reagent (Amersham, Piscataway, NJ, USA). Densities of bands in Western blotting analyses were normalized with the internal invariable control β-actin. Levels of target protein bands were densitometrically determined by using Quantity One® 4.4.1 (Bio-Rad, Foster City, CA, USA). Variations in the density were expressed as fold changes compared with the control in the blot.
Table 1.
Antibodies use for Western blotting analyses
| Company | Description | Catalog number |
|---|---|---|
| Santa Cruz Biotech. Inc. | Rabbit α-PDGF-βR antibody | sc-432 |
| Rabbit α-Tβ-RII antibody | sc-400 | |
| Rabbit α-Tβ-RI antibody | sc-399 | |
| Rabbit α-p27 antibody | sc-528 | |
| Rabbit α-Bax antibody | sc-493 | |
| Rabbit α-SREBP-1 antibody | sc-898 | |
| Goat α-CTGF antibody | sc-14939 | |
| Goat α-Pro-αI(I)col antibody | sc-25974 | |
| Bovine α-goat-IgG-HRP | sc-2350 | |
| Goat α-mouse-IgG-HRP | sc-2005 | |
| Goat α-rabbit-IgG-HRP | Sc-2004 | |
| Cayman Chemical Company | Rabbit α-SREBP-2 antibody | 10007663 |
| Abcam Inc. | Chicken α-LDLR antibody | ab14056 |
| Goat α-chicken-IgY-HRP | ab6877 | |
| Sigma Company | Mouse α-alpha-SMA monoclonal antibody | A2547 |
| Rabbit α-β-actin antibody | A2066 |
HRP, horseradish peroxidase; IgG, immunoglobulin G; LDLR, low-density lipoprotein receptor; SREBP, sterol regulatory element-binding protein; PDGF, platelet-derived growth factor; SMA, smooth muscle actin.
RNA isolation and real-time polymerase chain reaction (real-time PCR)
Total RNA was extracted by using TRI-Reagent® (Sigma, St. Louis, MO, USA), following the protocol provided by the manufacturer. Real-time PCR was carried out using SYBR Green as we described previously (Chen et al., 2002; Yumei et al., 2006). mRNA fold changes of target genes relative to the endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control were calculated as suggested by Schmittgen et al. (Schmittgen et al., 2000). Real-time PCR primers were shown in Table 2.
Table 2.
Primers for real-time PCR
| Genes | Primers |
|---|---|
| α-SMA | (F) 5′-CCG ACC GAA TGC AGA AGG-3′ |
| (R) 5′-ACA GAG TAT TTG CGC TCC GGA-3′ | |
| αI(I) collagen | (F) 5′-CCT CAA GGG CTC CAA CGA G-3′ |
| (R) 5′-TCA ATC ACT GTC TTG CCC CA-3′ | |
| CTGF | (F) 5′-TGTGTG ATG AGC CCA AGG AC-3′ |
| (R) 5′-AGT TGG CTC GCA TCA TAG TTG-3′ | |
| PDGF-βR | (F) 5′-ATC CTC ATC ATG CTG TGG CAG-3′; |
| (R) 5′-GCC AAG ATG GCT GAG ATC ACC AC-3′ | |
| Tβ-RI | (F) 5′-ATC CAT GAA GAC TAT CAG TTG CCT-3′; |
| (R) 5′-CAT TTT GAT GCC TTC CTG TTG GCT-3′ | |
| Tβ-RII | (F) 5′-TGT GCT CCT GTA ACA CAG AG-3′; |
| (R) 5′-GAT CTT GAC AGC CAC GGT CT-3′ | |
| LDLR | (F) 5′-CAG ACC CAG AGC CAT CGT AGT G-3′ |
| (R) 5′-GTC ACC AGA GAG TAG ATG TCT AC-3′ | |
| SREBP-1 | (F) 5″-TGC CCT AAG GGT CAA AAC CA-3′ |
| (R) 5′-TGG CGG GCA CTA CTT AGG AA-3′ | |
| SREBP-2 | (F) 5′-GCT GCA GAT CCC GCA GTA C-3′ |
| (R) 5′-GGT GGA TGA GGG AGA GAA GGT-3′ | |
| GAPDH | (F) 5′-GGC AAA TTC AAC GGC ACA GT-3′ |
| (R) 5′-AGA TGG TGATGG GCT TCC C-3′ |
α-SMA, α-smooth muscle actin; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HRP, horseradish peroxidase; IgG, immunoglobulin G; LDLR, low-density lipoprotein receptor; SREBP, sterol regulatory element-binding protein; PDGF-βR, platelet-derived growth factor-β receptor; Tβ-RI, type I transforming growth factor-β receptor; Tβ-RII, type II transforming growth factor-β receptor.
Statistical analyses
Differences between means were evaluated by an unpaired two-sided Student's t-test (P < 0.05 considered as significant). Where appropriate, comparisons of multiple treatment conditions with controls were analyzed by analysis of variance with Dunnett's test for post hoc analysis.
Materials
The PPARγ antagonist PD68235 was provided by Pfizer (Ann Arbor, MI, USA). Curcumin (purity >94%) and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) were purchased from Sigma (St. Louis, MO, USA). The authentic human plasma LDL (7.57 mg·mL−1) was purchased from Athens Research & Technology, Inc (Athens, GE, USA) and used before expiration date. The presence of endotoxin in LDL was <0.1 units·mL−1, as measured by Limulus Amoebocyte lysate assay kit (Whittaker M.A. Bioproducts, Walkersville, MD, USA).
Results
LDL stimulated activation of HSCs in vitro
To evaluate the role of LDL in HSC activation, passaged rat HSCs were serum-starved for 24 h in DMEM with 0.5% of FBS prior to the addition of exogenous LDL at various concentrations (0–200 µg·mL−1) for an additional 24 h. Serum-starvation made HSCs more sensitive to stimuli, including exogenous LDL. Cell proliferation was analyzed by MTS assays. As shown in Figure 1A, exogenous LDL dose-dependently increased the density of viable HSCs in culture. For instance, LDL at 100 µg·mL−1 increased the cell density by 64%, suggesting an increase in cell proliferation, confirmed by cell counts (data not shown). Total RNA and whole cell proteins were, respectively, prepared from the LDL-treated HSCs for real-time PCR and Western blotting analyses. As shown in Figure 1B, exogenous LDL dose-dependently increased the levels of mRNA of αI(I) collagen and α-SMA, the markers of activated HSCs, as well as other genes closely relevant to HSC activation, including pro-mitogenic PDGF-β receptor (PDGF-βR), and pro-fibrogenic type I and type II TGF-β receptors (Tβ-RI and Tβ-RII) and connective tissue growth factor (CTGF). Western blotting analyses verified that LDL elevated the abundance of the protein product of these genes (Figure 1C). Taken together, these results collectively demonstrated that LDL stimulated the activation of HSC in vitro by stimulating cell proliferation and inducing the production of type I collagen, as well as expression of genes closely relevant to HSC activation.
Figure 1.
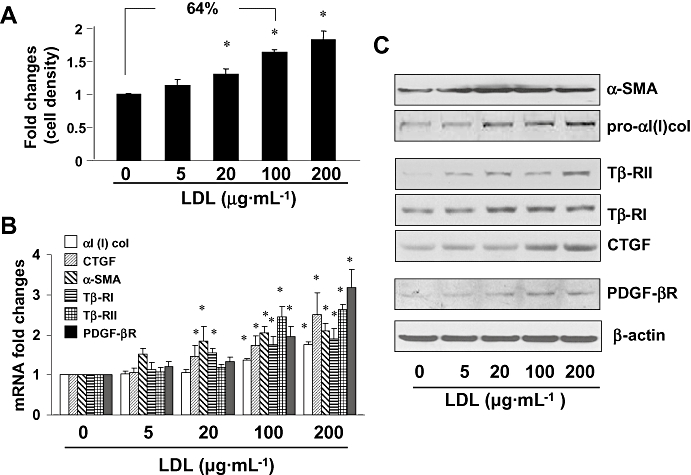
Low-density lipoprotein (LDL) stimulated HSC activation. Semi-confluent hepatic stellate cells (HSCs) were serum-starved for 24 h, followed by the addition of LDL at various concentrations as indicated for another 24 h. (A) MTS assays of cell proliferation. Results were expressed as cell density (%), compared with the untreated control [means ± standard deviation (SD)] (n= 3). *P < 0.05 versus cells with no treatment. (B) Real-time PCR analyses of the steady state levels of mRNA of genes relevant to HSC activation. The levels of target mRNAs were normalized with glyceraldehyde-3-phosphate dehydrogenase mRNA and were presented as mean ± SD (n= 3). *P < 0.05, compared with the untreated corresponding control (the first columns on the left side). (C) Western blotting analyses of proteins relevant to HSC activation. β-actin was used as an internal control for equal protein loading. Representative results from one of three independent experiments. α-SMA, α-smooth muscle actin; CTGF, connective tissue growth factor; PDGF-βR, platelet-derived growth factor-β receptor.
Curcumin attenuated the stimulatory effects of LDL on expression of genes related to HSC activation
Curcumin inhibits the activation of HSCs both in vitro and in vivo (Shishodia et al., 2005; Fu et al., 2008; O'Connell and Rushworth, 2008) and we therefore assumed that curcumin might inhibit LDL-induced HSC activation. To test our assumption, serum-starved HSCs were stimulated with or without LDL (100 µg·mL−1) in the presence of curcumin at various concentrations (0–30 µmol·L−1) for 24 h. MTS assays revealed that curcumin dose-dependently reduced the increased proliferation induced by exogenous LDL (Figure 2A). Further experiments were then performed to evaluate the effect of curcumin on expression of genes closely related to HSC activation. Results from real-time PCR (Figure 2B) and Western blotting analyses (Figure 2C) demonstrated that curcumin dose-dependently reduced the levels of transcripts and proteins of genes, including αI(I) collagen and α-SMA, pro-fibrogenic CTGF, Tβ-RI and Tβ-RII, as well as pro-mitogenic PDGF-βR. In addition, curcumin significantly increased the contents of the cyclin-dependent kinase inhibitor p27Kip1 and pro-apoptotic protein Bax. These results collectively indicated that curcumin attenuated the stimulatory effects of LDL on the activation of HSC in vitro by reducing cell proliferation and suppressing expression of genes related to HSC activation.
Figure 2.
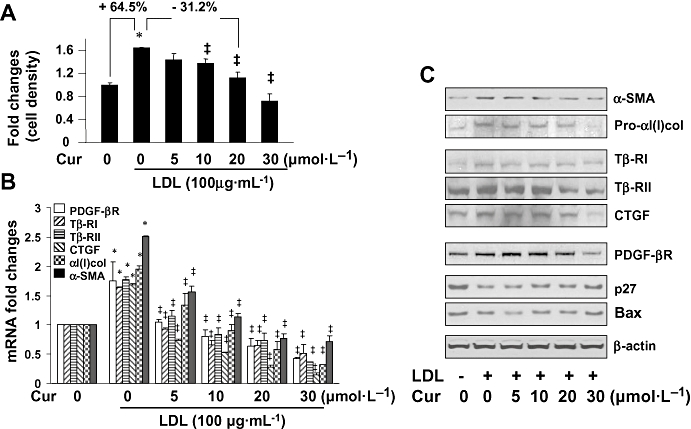
Curcumin attenuated the stimulatory effects of low-density lipoprotein (LDL) on hepatic stellate cell (HSC) activation. Serum-starved HSC were stimulated with or without exogenous LDL (100 µg·mL−1) in the presence of curcumin as shown for 24 h. (A) MTS assays of cell proliferation. Results were expressed as cell density (%), compared with the untreated control [means ± standard deviation (SD)] (n= 3). *P < 0.05 versus cells with no treatment. ‡P < 0.05 versus cells treated with exogenous LDL only. (B) Real-time PCR analyses of the steady state levels of mRNA of genes relevant to HSC activation. The levels of target mRNAs were normalized with glyceraldehyde-3-phosphate dehydrogenase mRNA and were presented as mean ± SD (n= 3). *P < 0.05, compared to the untreated corresponding control. ‡P < 0.05 versus the cells treated with exogenous LDL only. (C) Western blotting analyses of proteins relevant to HSC activation. β-actin was used as an internal control for equal protein loading. Representative results from one of three independent experiments. α-SMA, α-smooth muscle actin; CTGF, connective tissue growth factor; PDGF-βR, platelet-derived growth factor-β receptor.
Curcumin reduced cellular cholesterol and increased cellular FA and TG in cultured HSCs
To understand the mechanism by which curcumin attenuated HSC activation stimulated by LDL, experiments were conducted to measure the effect of curcumin on intracellular cholesterol and on lipogenesis in HSCs. Cultured HSCs were divided into two groups. One group was treated with curcumin at indicated concentrations in DMEM with FBS (10%) for 24 h. The other group was serum-starved for 24 h. Cells were then treated with or without exogenous LDL (100 µg·mL−1) and curcumin at indicated concentrations in serum-depleted media (0.5% FBS) for another 24 h. Cell extracts were prepared for quantitating cellular cholesterol, FA and TG. As shown in Figure 3A, curcumin dose-dependently reduced intracellular cholesterol in cultured HSCs (left panel). On the other hand, compared to the untreated control, exogenous LDL at 100 µg·mL−1 significantly increased intracellular cholesterol in cultured HSCs (right panel), and in these cells, curcumin reversed the stimulation by exogenous LDL and dose-dependently reduced intracellular cholesterol (right panel).
Figure 3.
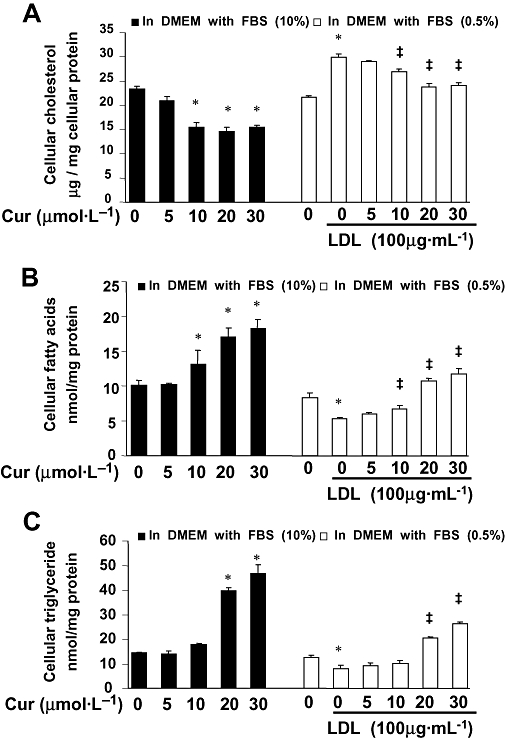
Curcumin significantly reduced intracellular cholesterol and increased the levels of fatty acids and triglyceride (TG) in cultured hepatic stellate cells (HSCs). Cultured HSCs were divided into two groups. One group was treated with curcumin as shown in Dulbecco's modified Eagle's medium (DMEM) with fetal bovine serum (FBS) (10%) for 24 h. The other group was serum-starved for 24 h. Cells were then treated with or without exogenous low-density lipoprotein (LDL) (100 µg·mL−1) and curcumin at indicated concentrations in serum-depleted DMEM containing 0.5% FBS for another 24 h. Cell extracts were prepared as stated in ‘Methods’ for quantitating cellular cholesterol, fatty acids and TG. Each treatment had triplicates. Experiments were repeated three times. *P < 0.05 versus the untreated control. ‡P < 0.05 versus the cells treated with exogenous LDL only. Experiments were repeated three times. Results were expressed as mean ± standard deviatin (n= 3). (A) Quantitation of cellular cholesterol. Cholesterol concentrations were expressed as µg per mg cellular proteins. (B) Determination of cellular fatty acids. Levels of fatty acids were presented as nmol·mg−1 protein. (C) Quantitation of cellular TG. Levels of TG were presented as nmol·mg−1 protein.
Curcumin also dose-dependently increased the levels of cellular FA and TG in HSCs cultured with 10% FBS (left panel in Figure 3B,C respectively). In the other group of cells (0.5% FBS; right panels in Figure 3B,C), compared with the untreated control, LDL reduced the levels of cellular FA (Figure 3B) and TG (Figure 3C) in HSCs. Curcumin dose-dependently reversed the inhibitory effects of LDL and elevated the levels of FA (Figure 3B) and TG (Figure 3C). Taken together, these results demonstrated that curcumin reduced intracellular cholesterol and increased cellular FA and TG in cultured HSCs. These results were further investigated to analyse the underlying mechanisms.
Curcumin suppressed gene expression of LDLR in activated HSCs in vitro
Plasma LDL is delivered into cells through endocytosis mediated by cell-surface LDLR and cholesterol is subsequently released from LDL within cells (Brown and Goldstein, 1986; Goldstein et al., 2006). It is, therefore, possible that curcumin might reduce cellular cholesterol by suppressing gene expression of LDLR in HSCs. To test this hypothesis, HSCs were transiently transfected with the ldlr promoter luciferase reporter pLDLR-Luc, which contained a 5′-flanking fragment of the LDLR gene promoter (1.5 kb) (Lopez and Ness, 2006). After overnight recovery from transfection, cells were treated with curcumin at different doses (0–30 µmol·L−1) for 24 h. Luciferase assays revealed (Figure 4A) that curcumin dose-dependently reduced luciferase activities, indicating that curcumin suppressed the ldlr promoter activity in activated HSCs in vitro. To verify this inhibitory effect of curcumin on ldlr expression, HSCs were treated with curcumin at various concentrations (0–30 µmol·L−1) for 24 h. Total RNA and whole cell proteins were, respectively, prepared from these cells for real-time PCR and Western blotting analyses. It was observed that curcumin reduced the steady state mRNA levels (Figure 4B) and the protein abundance (Figure 4C) of LDLR in HSCs. Taken together, these results demonstrated that curcumin dose-dependently suppressed gene expression of LDLR in activated HSCs in vitro, leading to reduced intracellular cholesterol in HSCs treated with LDL.
Figure 4.
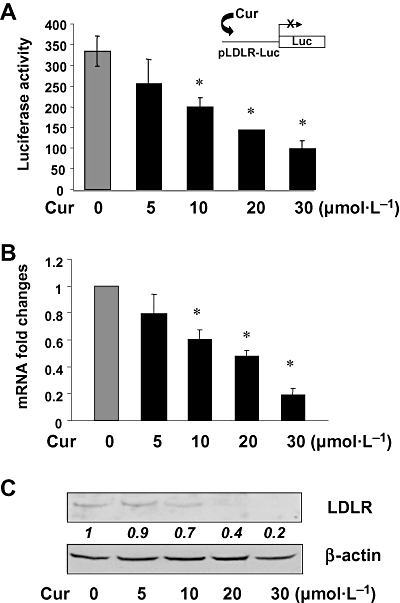
Curcumin dose-dependently suppressed gene expression of low-density lipoprotein (LDL) receptor in activated HSC in vitro. Semi-confluent hepatic stellate cells (HSCs) were treated with curcumin as shown for 24 h. (A) Luciferase assays of cells transiently transfected with the ldlr promoter luciferase reporter pLDLR-Luc. Luciferase activities were expressed as relative units after β-galactosidase normalization [means ± standard deviation (SD); n≥ 6]. *P < 0.05 versus cells with no treatment. The inset denoted the pLDLR-Luc construct in use and the application of curcumin to the system; (B) Real-time PCR analyses of the steady-state levels of LDLR mRNA. Glyceraldehyde-3-phosphate dehydrogenase was used as an invariant control for calculating mRNA fold changes. Values were expressed as means ± SD (n≥ 3). *P < 0.05 versus the untreated control; (C) Western blotting analyses of the abundance of LDLR. β-actin was used as an invariant control for equal loading. Representative results from one of three independent experiments. Italic numbers beneath the blot were fold changes in the densities of the bands compared to the control without treatment in the blot (mean values; n= 3), after normalization with the internal invariable control β-actin. Because of the limited space, standard deviations were not presented. LDLR, low-density lipoprotein receptor.
The SRE within the ldlr promoter is the cis-response element responsible for suppression by curcumin of the promoter activity of LDLR gene in HSCs
To elucidate molecular mechanisms of curcumin in the suppression of ldlr expression in activated HSCs, we, first of all, carried out promoter deletion analyses of LDLR gene to localize curcumin response element(s) in the ldlr promoter. HSCs were transfected with ldlr promoter luciferase reporter plasmids with various lengths of 5′-flanking promoter regions of LDLR gene (i.e. −1500, −335 and −211 bp respectively). Luciferase assays indicated (Figure 5A) that the deletion of the promoter region between −1500 and −335 bp caused a significant increase in luciferase activity, suggesting the existence of a cis-repressor binding element(s) in this region. In contrast, deletion of the fragment from −335 to −211 bp in the ldlr promoter resulted in a dramatic reduction in luciferase activity, which was at the same level in cells transfected with the empty control vector pGL3-basic (Figure 5). This result indicated that this promoter region might be responsible for the basal transcription of ldlr in HSCs. A critical cis-activator binding element(s) might exist within this fragment.
Figure 5.
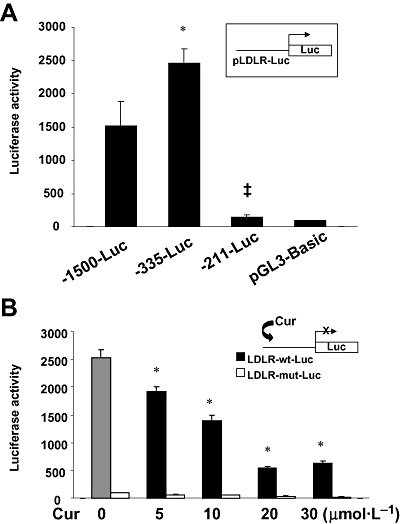
Localization of the curcumin response element(s) in the ldlr promoter. Semi-confluent hepatic stellate cells (HSCs) in six-well plates were transiently transfected with luciferase reporter plasmids. After recovery, cells were treated with or without curcumin as shown for 24 h. Luciferase assays were performed. Luciferase activities were expressed as relative units after β-galactosidase normalization (means ± SD; n≥ 6). The insets denoted the pLDLR-Luc constructs in use and the application of curcumin to the system. (A) Luciferase assays of cells transfected with the ldlr promoter luciferase reporter pLDLR-Luc with various lengths of the 5′-flanking promoter region of the ldlr promoter. *P < 0.05 versus cells transfected with pLDLR-Luc with the −1500 bp promoter region (p-1500-Luc); ‡P < 0.05 versus cells transfected with pLDLR-Luc with the −335 bp promoter region (p-335-Luc). (B) Luciferase assays of cells transfected with pLDLR-wt-Luc or pLDLR-mut-Luc, containing wild-type sterol regulatory element (SRE) or mutated SRE respectively. *P < 0.05 versus cells without curcumin treatment.
Prior studies revealed a binding site for SREBPs, that is, sterol regulatory element (SRE) (ATCACCCCAC), within the promoter region (−335 and −211 bp) of the LDLR gene (Castoreno et al., 2005). SREBPs are adipogenic transcription factors that control the metabolism of cellular cholesterol and FAs in animal cells (Briggs et al., 1993; Castoreno et al., 2005). We, therefore, assumed that the suppression of ldlr expression induced by curcumin might be mediated by the cis-SRE in the promoter of LDLR gene in activated HSCs. To test this assumption, HSCs were, respectively, transfected with the ldlr promoter luciferase reporter plasmids pLDLR-wt-Luc, or pLDLR-mut-Luc, containing the 335 bp fragment of the ldlr promoter with or without site-directed mutagenesis at the SRE. After transfection, cells were treated with various doses of curcumin as indicated for 24 h. Luciferase assays (Figure 5B) demonstrated that cells transfected with the plasmid pLDLR-wt-Luc with the wild-type SRE responded to curcumin and showed a significant reduction in luciferase activities in a dose-dependent manner. However, cells transfected with the plasmid pLDLR-mut-Luc with the mutated SRE showed luciferase activities at very low levels, which did not respond to curcumin. These results collectively suggested that the SRE within the promoter region between −355 and −211 bp might be responsible for the basic transcription activity of the ldlr promoter and be the cis-response element mediating the suppression by curcumin of the promoter activity of LDLR gene in HSCs.
Curcumin reduced the trans-activation activity of SREBPs in activated HSCs
To further elucidate molecular mechanisms of curcumin in the suppression of ldlr expression in activated HSCs and to clarify the roles of the transcription factors SREBPs in the process, HSCs were transfected with the SREBP trans-activation activity luciferase reporter plasmid p6x-SRE-Luc, in which six repeats of SRE (ATCACCCCAC) derived from the ldlr promoter were subcloned in the luciferase reporter vector pGL3-TK (Briggs et al., 1993; Wong et al., 2006). After overnight recovery, cells were treated with curcumin at various concentrations (0–30 µmol·L−1) for 24 h. Luciferase assays (Figure 6) demonstrated that curcumin dose-dependently reduced luciferase activities. This result demonstrated that curcumin reduced the trans-activation activity of SREBPs in activated HSCs in vitro. Additional experiments were performed to explore the underlying mechanisms.
Figure 6.
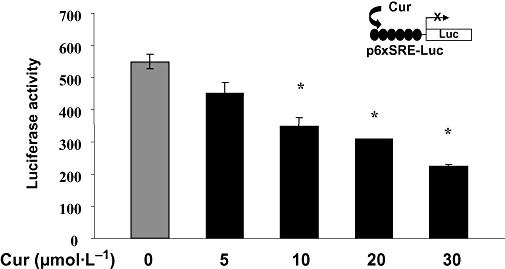
Curcumin reduced the trans-activation activity of sterol regulatory element-binding protein (SREBP) in activated hepatic stellate cells (HSCs). Semi-confluent HSCs were transiently transfected with the SREBP activity luciferase reporter plasmid p6x-SRE-Luc, containing six repeats of SRE (ATCACCCCAC) derived from the ldlr promoter. After recovery, cells were treated with curcumin as shown for 24 h. Luciferase assays were performed. Luciferase activities were expressed as relative units after β-galactosidase normalization (means ± standard deviation; n≥ 6). *P < 0.05 versus cells with no treatment. The inset denoted the p6x-SRE-Luc construct in use and the application of curcumin to the system.
Curcumin differentially regulated gene expression of SREBPs in HSCs
There are two SREBP genes, that is, srebp-1 and srebp-2, and three major proteins, SREBP-1a, SREBP-1c and SREBP-2 (Brown and Goldstein, 1997). When synthesized, SREBPs are membrane-bound proteins. SREBP-1a and SREBP-1c are produced from srebp-1 gene by use of two different promoters. Once activated, SREBPs are subsequently relocated into nuclei (Brown and Goldstein, 1997). To elucidate mechanisms by which curcumin reduced the trans-activation activity of SREBPs, semi-confluent HSCs were transfected with the luciferase reporter plasmid pSREBP-1-Luc, or pSREBP-2-Luc, containing a fragment of the 5′-flanking promoter region of srebp-1 or srebp-2 respectively. After overnight recovery, cells were treated with curcumin at indicated concentrations for an additional 24 h. Luciferase assays (Figure 7A) demonstrated that curcumin differentially and dose-dependently altered luciferase activities in these cells. Curcumin increased luciferase activities in cells transfected with pSREBP-1-Luc but reduced luciferase activities in cells transfected with pSREBP-2-Luc. These results suggested that curcumin differentially regulated the gene promoter activities of the two SREBPs in activated HSCs in vitro. To confirm these observations, HSCs were treated with curcumin at various concentrations (0–30 µmol·L−1) for 24 h. Total RNA and nuclear proteins were, respectively, prepared from these cells. Real-time PCR in Figure 7B indicated that curcumin dose-dependently increased the steady state levels of SREBP-1 mRNA, while reducing SREBP-2 mRNA. SREBP-1c and SREBP-2 are predominant SREBP proteins in the liver (Brown and Goldstein, 1997; Shimomura et al., 1997; Horton et al., 2002), and SREBP-1c is 10-fold more abundant than SREBP-1a (Brown and Goldstein, 1997; Shimomura et al., 1997; Horton et al., 2002). We, therefore, focused on SREBP-1c and SREBP-2, not SREBP-1a. Western blotting analyses in Figure 7C confirmed that curcumin dose-dependently increased the abundance of nuclear SREBP-1c, and reduced that of nuclear SREBP-2. These results collectively demonstrated that curcumin differentially regulated gene expression of SREBPs in activated HSCs in vitro, that is, stimulating srebp-1 expression and suppressing srebp-2 expression.
Figure 7.
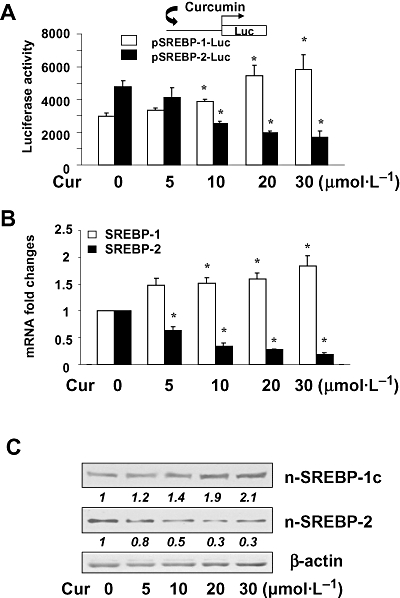
Curcumin differentially regulated gene expression of sterol regulatory element-binding proteins (SREBPs) in hepatic stellate cells (HSCs). Semi-confluent HSC were treated with curcumin as shown for 24 h. (A) Luciferase assays of cells transfected with the luciferase reporter plasmid pSREBP-1-Luc, or pSREBP-2-Luc, containing a fragment of the 5′-flanking promoter region of srebp-1 or srebp-2 respectively. Luciferase activities were expressed as relative units after β-galactosidase normalization [means ± standard deviation (SD); n≥ 6]. *P < 0.05 versus cells with no treatment. The inset denoted the pSREBP-1-Luc and pSREBP-2-Luc constructs in use and the application of curcumin to the system. (B) Real-time PCR analyses of the steady-state mRNA levels of SREBPs. GAPDH was used as an invariant control for calculating mRNA fold changes. Values were expressed as means ± SD (n≥ 3). *P < 0.05 versus the untreated control (the first columns on the left). (C) Western blotting analyses of the abundance of SREBPs. β-actin was used as an invariant control for equal loading. Representative results from one of three independent experiments. Italic numbers beneath the blots are fold changes in the densities of the bands compared to the control without treatment in the blot (mean; n= 3), after normalization with the internal invariable control β-actin. Because of the limited space, standard deviations were not presented.
Differential regulation of gene expression of SREBPs induced by curcumin might be mediated by PPARγ
Activation of PPARγ plays a crucial role in the suppression of HSC activation and hepatic fibrogenesis (Galli et al., 2000; Marra et al., 2000; Miyahara et al., 2000). We previously demonstrated that curcumin induced gene expression of endogenous PPARγ and stimulated its activity, which was a prerequisite for curcumin to inhibit HSC activation (Xu et al., 2003; Zheng and Chen, 2004; Fu et al., 2008). We hypothesized that the activation of PPARγ by curcumin might mediate the differential regulation of SREBPs in activated HSCs. To test the hypothesis, HSCs were pretreated with the PPARγ antagonist PD68235 (20 µmol·L−1) for 30 min prior to the addition of curcumin (20 µmol·L−1) for an additional 24 h. Whole cell proteins were prepared for Western blotting analyses of SREBPs. Compared with the controls, curcumin, as expected, increased the abundance of SREBP-1 and reduced the content of SREBP-2 (Figure 8). Blockade of PPARγ by PPARγ antagonist PD68235 attenuated the effects of curcumin on the levels of SREBPs in activated HSCs, suggesting an involvement of PPARγ in this process.
Figure 8.

Activation of peroxisome proliferator-activated receptor-γ (PPARγ) is required for curcumin to differentially regulate gene expression of sterol regulatory element-binding proteins (SREBPs) in activated hepatic stellate cells (HSCs) in vitro. Passaged HSC were pretreated with the PPARγ antagonist PD68235 (20 µmol·L−1) for 30 min prior to the addition of curcumin (20 µmol·L−1) for an additional 24 h. Whole cell proteins were prepared for Western blotting analyses of SREBPs. β-actin was used as an invariant control for equal loading. Representative results from one of three independent experiments. Italic numbers beneath the blots were fold changes in the densities of the bands compared to the control without treatment in the blot (mean; n= 3), after normalization with the internal invariable control β-actin. Because of the limited space, standard deviations were not presented. PD, PD68235.
To confirm the role of PPARγ in the differential regulation of SREBPs gene expression, HSCs were co-transfected with the PPARγ cDNA expression plasmid pPPARγ plus pSREBP-1-Luc, or pSREBP-2-Luc. The latter luciferase reporter plasmids, respectively, contained the 5′-flanking fragment of the srebp-1 or srebp-2 promoter (Amemiya-Kudo et al., 2000; Shin and Osborne, 2003). A total of 3.8 µg of plasmid DNA per well was used for co-transfection of HSCs in six-well culture plates. It included 2 µg of pSREBP-1-Luc, or pSREBP-2-Luc, 0.8 µg of pSV-β-gal and 1.0 µg of the PPARγ cDNA expression plasmid at indicated doses plus the empty vector pcDNA. The latter was used to ensure an equal amount of total DNA in transfection assays. After recovery, cells were cultured for 24 h. Prior experiments have suggested that 10% of FBS in the medium contains enough agonists to activate PPARγ in HSCs (Miyahara et al., 2000; Xu et al., 2003; Zheng and Chen, 2004). Results from luciferase assays in Figure 9A demonstrated that the activation of PPARγ by forced expression of PPARγ cDNA dose-dependently and differentially altered luciferase activities in these cells; that is, increasing luciferase activities in cells transfected with pSREBP-1-Luc, and reducing luciferase activities in cells transfected with pSREBP-2-Luc. These results indicated that activation of PPARγ differentially regulated the promoter activity of SREBP-1 and SREBP-2 genes in activated HSCs in vitro.
Figure 9.
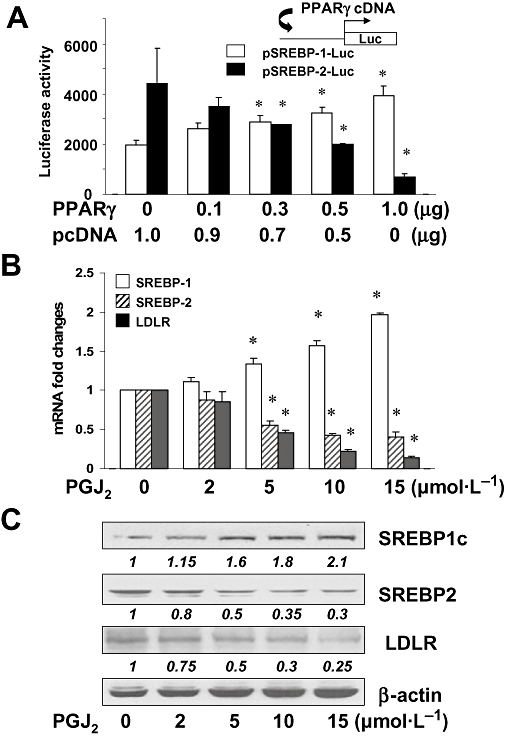
Activation of peroxisome proliferator-activated receptor-γ (PPARγ) differentially regulated gene expression of sterol regulatory element-binding proteins (SREBPs) and low-density lipoprotein receptor (LDLR) in cultured hepatic stellate cells (HSCs). (A). Semi-confluent HSC in six-well plates were co-transfected with the cDNA expression plasmid pPPARγ plus the luciferase reporter plasmid pSREBP-1-Luc, or pSREBP-2-Luc, containing a fragment of the 5′-flanking promoter region of srebp-1 or srebp-2 respectively. After recovery, cells were cultured for 24 h. Luciferase activities were expressed as relative units after β-galactosidase normalization [means ± standard deviation (SD); n≥ 6]; (B). Real-time PCR analyses of the steady-state mRNA levels of SREBPs and LDLR in HSC treated with the PPARγ agonist 15d-PGJ2 (0–15 µmol·L−1) for 24 h. GAPDH was used as an invariant control for calculating mRNA fold changes. Values were expressed as means ± SD (n≥ 3). *P < 0.05 versus the untreated control (the first columns on the left); (C). Western blotting analyses of the abundance of SREBPs and LDLR in HSC treated with 15d-PGJ2 (0–15 µmol·L−1) for 24 h. β-actin was used as an invariant control for equal loading. Representative results from one of three independent experiments. Italic numbers beneath the blots were fold changes in the densities of the bands compared with the control without treatment in the blot (mean; n= 3), after normalization with the internal invariable control β-actin. Because of the limited space, standard deviations are not presented.
To further evaluate the role of PPARγ in the differential regulation of SREBPs gene expression and its impact on ldlr expression, HSCs were treated with the endogenous PPARγ agonist 15d-PGJ2 (0–15 µmol·L−1) for 24 h. Total mRNA and whole cell extracts were prepared. As shown by real-time PCR (Figure 9B) and Western blotting analyses (Figure 9C), 15d-PGJ2 differentially regulated gene expression of the two SREBPs in a dose-dependent manner, that is, increasing srebp-1 expression and reducing srebp-2 expression, at levels of transcripts and proteins. In addition, PGJ2 significantly suppressed ldlr expression in the cells. Taken together, these results revealed that the activation of PPARγ differentially regulated gene expression of the two SREBPs in activated HSCs in vitro, leading to suppression of ldlr expression.
Forced expression of SREBP-1 stimulated the pparγ promoter activity and increased the trans-activation activity of PPARγ in cultured HSCs
Curcumin-induced inhibition of srebp-2 expression resulted in the suppression of LDLR gene expression (Figures 4–6), as its expression is mainly regulated by SREBP-2 (Castoreno et al., 2005). Additional experiments were performed to understand the role of curcumin in the stimulation of srebp-1 expression. We hypothesized that the induction of srebp-1 expression by curcumin might facilitate gene expression of PPARγ in activated HSCs. To test the hypothesis, semi-confluent passaged HSCs were co-transfected with the PPARγ promoter luciferase reporter plasmid pPPARγ-Luc and the cDNA expression plasmid pSREBP-1, or pdn-SREBP-1. pSREBP-1 contained a full fragment of wild-type SREBP-1 cDNA, whereas pdn-SREBP-1 encoded a dominant negative SREBP-1. A total of 3.8 µg of plasmid DNA per well was used for co-transfection of HSCs in six-well culture plates, including 2 µg of pPPARγ-Luc, 0.8 µg of pSV-β-gal and 1.0 µg of pSREBP-1, or pdn-SREBP-1, plus the empty vector pcDNA. The latter was used to ensure the equal amount of total DNA in co-transfection assays. Cells were treated with or without curcumin at 20 µmol·L−1 for 24 h. As shown in Figure 10A by luciferase assays, compared with the control, forced expression of pSREBP-1 dose-dependently increased luciferase activities. In contrast, forced expression of dominant negative SREBP-1 (dn-SREBP-1) dramatically reduced luciferase activity. Compared with the untreated control, curcumin significantly increased luciferase activity, and this increase was significantly inhibited by dn-SREBP-1 (the last column on the right side). These results collectively suggested that SREBP-1 might stimulate the promoter activity of PPARγ gene in HSCs and thus facilitating curcumin-induced gene expression of PPARγ in activated HSCs.
Figure 10.
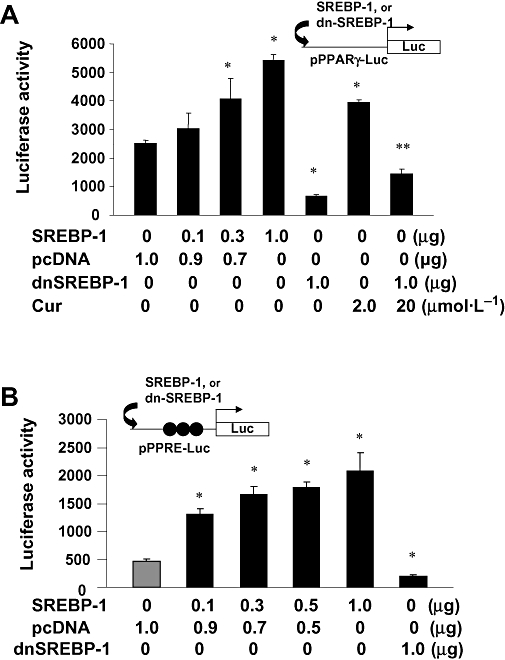
Forced expression of sterol regulatory element-binding protein-1 (SREBP-1) stimulated the pparγ promoter activity and increased the trans-activation activity of peroxisome proliferator-activated receptor-γ (PPARγ) in cultured hepatic stellate cells (HSCs). Semi-confluent HSC in 6-well plates were co-transfected with a luciferase reporter plasmid and the cDNA expression plasmid pSREBP-1, or pdn-SREBP-1, at indicated doses. pSREBP-1 contained a full fragment of wild-type pSREBP-1 cDNA, whereas pdn-SREBP-1 contained a full length of dominant negative pSREBP-1 cDNA. Cells were treated with or without curcumin at 20 µmol·L−1 for 24 h. Luciferase activities were expressed as relative units after β-galactosidase normalization (means ± SD; n≥ 6). *P < 0.05 versus the control cells **P < 0.05 versus the cells treated with curcumin. (A). Luciferase assays of cells co-transfected with the pparγ promoter luciferase reporter plasmid pPPARγ-Luc plus pSREBP-1, or pdn-SREBP-1, at indicated doses; (B). Luciferase assays of cells co-transfected with the PPARγtrans-activity luciferase reporter plasmid pPPRE-Luc, containing three copies of the PPARγ response elements (PPRE), plus pSREBP-1, or pdn-SREBP-1, at indicated doses.
To verify the role of SREBP-1 in stimulating PPARγ activity, passaged HSCs were co-transfected with the plasmid pPPRE-Luc and pSREBP-1, or pdn-SREBP-1. The plasmid pPPRE-Luc was a PPARγtrans-activation activity reporter plasmid, which contained three copies of the PPARγ response elements from acyl-CoA oxidase gene linked to the herpes virus thymidine kinase promoter (−105/+51) and a luciferase vector (Xu et al., 2003; Zheng and Chen, 2007). As shown in Figure 10B by luciferase assays, forced expression of pSREBP-1 dose-dependently increased luciferase activities, whereas dn-SREBP-1 dramatically reduced luciferase activity (the last column on the right side), suggesting that forced expression of SREBP-1 stimulated the PPARγtrans-activation activity in HSCs. Taken together, these data demonstrated the role of SREBP-1 in the stimulation of the pparγ promoter activity and the trans-activation activity of PPARγ in cultured HSCs.
Discussion
In this study, we observed that LDL dose-dependently stimulated the activation of HSCs in vitro. Curcumin suppressed expression of the LDLR gene by activating PPARγ and differentially regulating gene expression of SREBP-1 and SREBP-2, leading to the reduction in the level of intracellular cholesterol and to the attenuation of the stimulatory effects of LDL on HSC activation.
In mammals, LDLR mediate the turnover of LDL and the elimination of plasma LDL. The liver contains about 70% of total LDLR present in the body and is the major organ responsible for the turnover of LDL in circulation (Bilheimer et al., 1984). Accordingly, apparent changes in the abundance of LDLR in the liver will lead to alterations in the level of plasma LDL. Intriguingly, several lines of evidence have showed that curcumin displays differential regulatory effects on ldlr expression based on cell types. Curcumin induces ldlr expression in Xenopus laevis oocytes (Fan et al., 2005), in bovine vascular smooth muscle cells (Liu and Hong, 2006), and in mouse macrophages (Fan et al., 2005). Particularly, curcumin dramatically induces gene expression of LDLR in the human hepatoma cell line HepG2 (Peschel et al., 2007), which might, at least partially, explain the hypocholesterolemic roles of curcumin in increasing endocytosis of plasma LDL by hepatocytes and in reducing plasma cholesterol. Additional experiments are ongoing in our laboratory to explore the underlying mechanisms by which curcumin distinctly regulates ldlr expression in different cell types, including HSCs and hepatocytes.
The level of intracellular cholesterol is mainly determined by biosynthesis, influx, catabolism and efflux of cholesterol in cells. The mechanisms of cholesterol biosynthesis are only partially understood (Espenshade and Hughes, 2007). SREBP-2 governs the genes involving cholesterol biosynthesis and LDL uptake (Yeh et al., 2004). Among them are the genes for LDLR and HMG-CoA reductase. The former scavenges circulating LDL from the bloodstream, whereas HMG-CoA reductase leads to an increase of endogenous production of cholesterol (Brown and Goldstein, 1997). In this report, we focused on the effects of curcumin on LDLR. We demonstrated that curcumin inhibited SREBP-2 gene expression and reduced its activity, which led to the suppression of ldlr expression and the reduction of the uptake of extracellular LDL. As MG-CoA reductase is a target gene of SREBP-2, it is plausible to deduce that curcumin might also reduce biosynthesis of cholesterol in HSCs by suppressing the gene expression of HMG-CoA reductase. Curcumin has been shown to increase the rate of cholesterol catabolism (Babu and Srinivasan, 1997). In addition, we should not exclude a possible impact of curcumin on cholesterol efflux, which is promoted by apolipoprotein (apo) A-I and high-density lipoproteins (HDL) (Rothblat and Phillips, 1982). Curcumin has been shown to elevate the levels of plasma apo A-I and HDL in vivo (Yasni et al., 1993; Jang et al., 2008). Taken together, the reduction in cellular cholesterol in cultured HSCs induced by curcumin might result from the collective effects of the phytochemical on cholesterol biosynthesis, influx, catabolism and efflux, including suppressing ldlr expression, leading to the reduction in the uptake of extracellular LDL into HSCs.
Our results also indicated that curcumin increased the levels of cellular FA and TG in cultured HSCs. These changes might result from the curcumin-induced activation of PPARγ and SREBP-1 in activated HSCs, which regulate expression of genes involved in lipid synthesis (Sharma and Staels, 2007). These results might raise a concern whether curcumin and its stimulation of PPARγ activity would exacerbate the state of hepatic steatosis in vivo. Hepatocytes are the major cells for fat accumulation during steatosis. Few results are available from direct studies of curcumin in biosynthesis of cellular FA and TG in hepatocytes. It has been, however, demonstrated that curcumin protects the liver from injury and apparently improves the state of hepatic steatosis with a significant reduction in the number of macro- and microvesicular steatosis in hepatocytes in the rat model with hepatic fibrosis induced by CCl4 (Park et al., 2000; Fu et al., 2008). The underlying mechanisms remain largely elusive. HSCs were previously called fat-storing cells, which might reflect one of the major functions of quiescent HSCs in the normal liver. On the other hand, HSCs constitute only 5–8% of the total liver cell populations in the normal liver (Friedman, 2008). Therefore, the overall effect of the curcumin-increased synthesis of FA and TG in HSCs on hepatic steatosis might be negligible.
Our results indicated that curcumin differentially regulated gene expression of SREBPs, that is, up-regulating SREBP-1 and suppressing SREBP-2. SREBP-1 and SREBP-2 play related, but distinct, roles in regulation of cholesterol homeostasis (Brown and Goldstein, 1997; Horton et al., 2002). Although SREBP-1 and SREBP-2 both have a similar protein domain structure and consensus binding sequences (Brown and Goldstein, 1997), they show different preference in regulating expression of target genes. SREBP-1c, the predominant SREBP-1 isoform in adult liver, preferentially activates genes required for FA synthesis, such as ATP citrate lyase (Sato et al., 2000; Moon et al., 2001) and long chain fatty acyl elongase (Moon et al., 2001), whereas SREBP-2 preferentially activates ldlr expression and various genes required for cholesterol biosynthesis (Castoreno et al., 2005). For example, in SREBP-1c knockout and transgenic mice, there is no significant change in ldlr expression (Brown and Goldstein, 1997; Horton et al., 2002). However, in SREBP-2 transgenic mice, ldlr expression is significantly elevated (Brown and Goldstein, 1997; Horton et al., 2002). Germline deletion of SREBP-2 leads to 100% lethality at a later stage of embryonic development (Brown and Goldstein, 1997; Horton et al., 2002). ldlr Expression is mainly regulated by SREBP-2 (Castoreno et al., 2005), and the six repeats of SRE (ATCACCCCAC) in the plasmid p6x-SRE-Luc were originally derived from the ldlr promoter (Briggs et al., 1993; Wong et al., 2006). It is, therefore, not difficult to understand why curcumin significantly reduced luciferase activities in HSCs transfected with p6x-SRE-Luc. It is also plausible to assume that the curcumin-caused suppression of ldlr expression was likely to be mediated by inhibiting srebp-2 expression.
Our results suggested that the differential effect of curcumin might be mediated by activating PPARγ in HSCs. Activation of PPARγ might have distinct roles in regulation of SREBP-1 gene expression in different cell types. For instance, we observed that the activation of PPARγ induced gene expression of SREBP-1 in cultured HSCs. However, a recent report indicated that pioglitazone, an antidiabetic agent and a ligand of PPARγ, activated c-Met, induced lipid retrieval, down-regulated SREBP-1c level and suppressed TG biosynthesis in the liver, suggesting that the activation of PPARγ might serve as a potentially useful method of attenuating hepatic steatosis (Tomita et al., 2004). Additional experiments are necessary to elucidate molecular mechanisms underlying the PPARγ-mediated differential regulation of gene expression of SREBPs in activated HSCs.
Recent studies have indicated the importance of the activation of another nuclear receptor, the farnesoid X receptor (FXR), and its crosstalk with PPARγ in inhibiting HSC activation and protecting against liver fibrogenesis (Fiorucci et al., 2005). FXR is a ligand-activated transcription factor that regulates cholesterol and FA metabolism and functions as an endogenous sensor for bile acids (Forman et al., 1995). FXR deficiency has been found to be a risk factor for NASH development (Kong et al., 2009). The process of HSC activation has little impact on the level of FXR mRNA (Fiorucci et al., 2005). However, FXR ligands up-regulate PPARγ mRNA in HSCs and in rodent models of liver fibrosis. Simultaneous activation of FXR and PPARγ additively reduces the levels of αI(I) collagen in cultured HSCs (Fiorucci et al., 2005). Accumulating evidence has suggested that activation of FXR may be beneficial in treatment of liver fibrosis (Fiorucci et al., 2004). It would be of interest to evaluate the effects of curcumin on FXR activity and its cross-talk with PPARγ in HSCs, which may contribute to the curcumin-induced inhibition of LDL-induced HSC activation.
Curcumin inhibits HSC activation by targeting multiple signalling pathways and utilizing several mechanisms (Xu et al., 2003; Zheng and Chen, 2004; 2006; 2007; Zheng et al., 2007; Zhou et al., 2007). Based on our results, we propose a simplified model of how curcumin inhibits LDL-induced HSC activation (Figure 11). Extracellular LDL is transported into HSCs by LDLR-mediated endocytosis, leading to the stimulation of HSC activation. Curcumin induces gene expression of PPARγ, resulting in the distinct regulation of gene expression of SREBPs, that is, up-regulating srebp-1 expression and down-regulating srebp-2 expression. The down-regulation of SREBP-2 suppresses the gene expression of LDLR, leading to the reduction in endocytosis of plasma LDL and in the level of cellular cholesterol in HSCs. The up-regulation of SREBP-1 provides a positive feedback loop and facilitates, in turn, gene expression of PPARγ. These actions of curcumin collectively attenuate the stimulatory effects of LDL on the activation of HSCs. We would emphasize that our results and the proposed model do not exclude other mechanisms of curcumin in the inhibition of LDL-induced activation of HSCs. Our results in this study present evidence for the stimulatory role of hypercholesterolemia in hepatic fibrogenesis. In addition, our results provide novel insights into the effect of curcumin and its underlying mechanisms in the inhibition of LDL-induced HSC activation and its involvement in therapeutic interventions in hypercholesterolemia-associated hepatic fibrogenesis.
Figure 11.
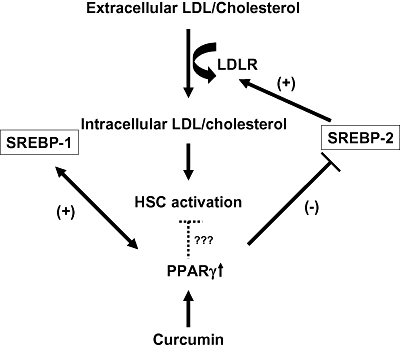
A simplified model for curcumin to inhibit low-density lipoprotein (LDL)-induced hepatic stellate cell (HSC) activation. Extracellular LDL is transported into HSC by LDL receptor (LDLR)-mediated endocytosis, leading to the stimulation of HSC activation. Curcumin induces gene expression of peroxisome proliferator-activated receptor-γ (PPARγ), resulting in the distinct regulation of gene expression of sterol regulatory element-binding proteins (SREBPs), that is, up-regulating srebp-1 expression and down-regulating srebp-2 expression. The down-regulation of SREBP-2 suppresses gene expression of LDLR, leading to the reduction in endocytosis of plasma LDL and in the level of cellular cholesterol in HSC. The up-regulation of SREBP-1 provides a positive feedback loop and facilitates, in turn, gene expression of PPARγ. These actions of curcumin collectively attenuate the stimulatory effects of LDL on the activation of HSC.
Acknowledgments
This work was supported by the grant RO1 DK 047995 from NIH/NIDDK to A. Chen.
Glossary
Abbreviations:
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- α-SMA
α-smooth muscle actin
- DMEM
Dulbecco's modified Eagle's medium
- ECM
extracellular matrix
- EGF
epidermal growth factor
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HSC
hepatic stellate cell
- LDL
low-density lipoprotein
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- PDGF
platelet-derived growth factor
- PPARγ
peroxisome proliferator-activated receptor-γ
- SRE
sterol regulatory element
- SREBP
sterol regulatory element-binding protein
- TGF-β
transforming growth factor-β
This Research Paper is the subject of a Commentary in this issue of BJP entitled Curcumin adds spice to the debate: lipid metabolism in liver disease (Graham, pp. 1352–1353). To view this article visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Conflict of interest
None.
References
- Amemiya-Kudo M, Shimano H, Yoshikawa T, Yahagi N, Hasty AH, Okazaki H, et al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem. 2000;275:31078–31085. doi: 10.1074/jbc.M005353200. [DOI] [PubMed] [Google Scholar]
- Arafa HM. Curcumin attenuates diet-induced hypercholesterolemia in rats. Med Sci Monit. 2005;11:BR228–BR234. [PubMed] [Google Scholar]
- Asai A, Miyazawa T. Dietary curcuminoids prevent high-fat diet-induced lipid accumulation in rat liver and epididymal adipose tissue. J Nutr. 2001;131:2932–2935. doi: 10.1093/jn/131.11.2932. [DOI] [PubMed] [Google Scholar]
- Babu PS, Srinivasan K. Hypolipidemic action of curcumin, the active principle of turmeric (Curcuma longa) in streptozotocin induced diabetic rats. Mol Cell Biochem. 1997;166:169–175. doi: 10.1023/a:1006819605211. [DOI] [PubMed] [Google Scholar]
- Bilheimer DW, Goldstein JL, Grundy SM, Starzl TE, Brown MS. Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia. N Engl J Med. 1984;311:1658–1664. doi: 10.1056/NEJM198412273112603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs MR, Yokoyama C, Wang X, Brown MS, Goldstein JL. Nuclear protein that binds sterol regulatory element of low density lipoprotein receptor promoter. I. Identification of the protein and delineation of its target nucleotide sequence. J Biol Chem. 1993;268:14490–14496. [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Castoreno AB, Wang Y, Stockinger W, Jarzylo LA, Du H, Pagnon JC, et al. Transcriptional regulation of phagocytosis-induced membrane biogenesis by sterol regulatory element binding proteins. Proc Natl Acad Sci USA. 2005;102:13129–13134. doi: 10.1073/pnas.0506716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, et al. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Chen A, Davis BH. UV irradiation activates JNK and increases alphaI(I) collagen gene expression in rat hepatic stellate cells. J Biol Chem. 1999;274:158–164. doi: 10.1074/jbc.274.1.158. [DOI] [PubMed] [Google Scholar]
- Chen A, Zhang L, Xu J, Tang J. The antioxidant (-)-epigallocatechin-3-gallate inhibits activated hepatic stellate cell growth and suppresses acetaldehyde-induced gene expression. Biochem J. 2002;368:695–704. doi: 10.1042/BJ20020894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(Suppl. 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annu Rev Genet. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- Fajas L, Schoonjans K, Gelman L, Kim JB, Najib J, Martin G, et al. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein 1: implications for adipocyte differentiation and metabolism. Mol Cell Biol. 1999;19:5495–5503. doi: 10.1128/mcb.19.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CL, Qian Y, Wo XD, Yan J, Gao LP. Effect of curcumin on the gene expression of low density lipoprotein receptors. Chin J Integr Med. 2005;11:201–204. doi: 10.1007/BF02836505. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, et al. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther. 2005;315:58–68. doi: 10.1124/jpet.105.085597. [DOI] [PubMed] [Google Scholar]
- Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zheng S, Lin J, Ryerse J, Chen A. Curcumin protects the rat liver from CCl4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol Pharmacol. 2008;73:399–409. doi: 10.1124/mol.107.039818. [DOI] [PubMed] [Google Scholar]
- Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, et al. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology. 2000;31:101–108. doi: 10.1002/hep.510310117. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang EM, Choi MS, Jung UJ, Kim MJ, Kim HJ, Jeon SM, et al. Beneficial effects of curcumin on hyperlipidemia and insulin resistance in high-fat-fed hamsters. Metabolism. 2008;57:1576–1583. doi: 10.1016/j.metabol.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Kapoor GS, Atkins BA, Mehta KD. Activation of Raf-1/MEK-1/2/p42/44(MAPK) cascade alone is sufficient to uncouple LDL receptor expression from cell growth. Mol Cell Biochem. 2002;236:13–22. doi: 10.1023/a:1016185928871. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl. 3):S84–S87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;328:116–122. doi: 10.1124/jpet.108.144600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hong XQ. Effect of three different curcumin pigmens on the prdiferation of vascular smooth muscle cells by ox-LDL and the expression of LDL-R. Zhongguo Zhong Yao Za Zhi. 2006;31:500–503. [PubMed] [Google Scholar]
- Lopez D, Ness GC. Characterization of the rat LDL receptor 5′-flanking region. Biochim Biophys Acta. 2006;1761:492–500. doi: 10.1016/j.bbalip.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Jr, Motomura K, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- Moon YA, Shah NA, Mohapatra S, Warrington JA, Horton JD. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J Biol Chem. 2001;276:45358–45366. doi: 10.1074/jbc.M108413200. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Thomas P, Dannenberg AJ. Curcumin prevents alcohol-induced liver disease in rats by inhibiting the expression of NF-kappa B-dependent genes. Am J Physiol Gastrointest Liver Physiol. 2003;284:G321–G327. doi: 10.1152/ajpgi.00230.2002. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA. Fatty liver and nonalcoholic steatohepatitis. Clin Cornerstone. 2001;3:47–57. doi: 10.1016/s1098-3597(01)90078-x. [DOI] [PubMed] [Google Scholar]
- O'Brien PE, Dixon JB. The extent of the problem of obesity. Am J Surg. 2002;184:4S–8S. doi: 10.1016/s0002-9610(02)01172-8. [DOI] [PubMed] [Google Scholar]
- O'Connell MA, Rushworth SA. Curcumin: potential for hepatic fibrosis therapy? Br J Pharmacol. 2008;153:403–405. doi: 10.1038/sj.bjp.0707580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- Park EJ, Jeon CH, Ko G, Kim J, Sohn DH. Protective effect of curcumin in rat liver injury induced by carbon tetrachloride. J Pharm Pharmacol. 2000;52:437–440. doi: 10.1211/0022357001774048. [DOI] [PubMed] [Google Scholar]
- Peschel D, Koerting R, Nass N. Curcumin induces changes in expression of genes involved in cholesterol homeostasis. J Nutr Biochem. 2007;18:113–119. doi: 10.1016/j.jnutbio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Ramirez-Tortosa MC, Mesa MD, Aguilera MC, Quiles JL, Baro L, Ramirez-Tortosa CL, et al. Oral administration of a turmeric extract inhibits LDL oxidation and has hypocholesterolemic effects in rabbits with experimental atherosclerosis. Atherosclerosis. 1999;147:371–378. doi: 10.1016/s0021-9150(99)00207-5. [DOI] [PubMed] [Google Scholar]
- Rao DS, Sekhara NC, Satyanarayana MN, Srinivasan M. Effect of curcumin on serum and liver cholesterol levels in the rat. J Nutr. 1970;100:1307–1315. doi: 10.1093/jn/100.11.1307. [DOI] [PubMed] [Google Scholar]
- Rothblat GH, Phillips MC. Mechanism of cholesterol efflux from cells. Effects of acceptor structure and concentration. J Biol Chem. 1982;257:4775–4782. [PubMed] [Google Scholar]
- Sato R, Okamoto A, Inoue J, Miyamoto W, Sakai Y, Emoto N, et al. Transcriptional regulation of the ATP citrate-lyase gene by sterol regulatory element-binding proteins. J Biol Chem. 2000;275:12497–12502. doi: 10.1074/jbc.275.17.12497. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- Sharma AM, Staels B. Review: peroxisome proliferator-activated receptor gamma and adipose tissue – understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab. 2007;92:386–395. doi: 10.1210/jc.2006-1268. [DOI] [PubMed] [Google Scholar]
- Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest. 1997;99:838–845. doi: 10.1172/JCI119247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DJ, Osborne TF. Thyroid hormone regulation and cholesterol metabolism are connected through sterol regulatory element-binding protein-2 (SREBP-2) J Biol Chem. 2003;278:34114–34118. doi: 10.1074/jbc.M305417200. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Sethi G, Aggarwal BB. Curcumin: getting back to the roots. Ann N Y Acad Sci. 2005;1056:206–217. doi: 10.1196/annals.1352.010. [DOI] [PubMed] [Google Scholar]
- Tomita K, Azuma T, Kitamura N, Nishida J, Tamiya G, Oka A, et al. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology. 2004;126:873–885. doi: 10.1053/j.gastro.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Wong J, Quinn CM, Brown AJ. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem J. 2006;400:485–491. doi: 10.1042/BJ20060914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Fu Y, Chen A. Activation of peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of curcumin on rat hepatic stellate cell growth. Am J Physiol Gastrointest Liver Physiol. 2003;285:G20–G30. doi: 10.1152/ajpgi.00474.2002. [DOI] [PubMed] [Google Scholar]
- Yasni S, Imaizumi K, Nakamura M, Aimoto J, Sugano M. Effects of Curcuma xanthorrhiza Roxb. and curcuminoids on the level of serum and liver lipids, serum apolipoprotein A-I and lipogenic enzymes in rats. Food Chem Toxicol. 1993;31:213–218. doi: 10.1016/0278-6915(93)90096-h. [DOI] [PubMed] [Google Scholar]
- Yeh M, Cole AL, Choi J, Liu Y, Tulchinsky D, Qiao JH, et al. Role for sterol regulatory element-binding protein in activation of endothelial cells by phospholipid oxidation products. Circ Res. 2004;95:780–788. doi: 10.1161/01.RES.0000146030.53089.18. [DOI] [PubMed] [Google Scholar]
- Yumei F, Zhou Y, Zheng S, Chen A. The antifibrogenic effect of (-)-epigallocatechin gallate results from the induction of de novo synthesis of glutathione in passaged rat hepatic stellate cells. Lab Invest. 2006;86:697–709. doi: 10.1038/labinvest.3700425. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu D, Wo X, Zhang Y, Jin M, Ding Z. Effects of Curcuma Longa on proliferation of cultured bovine smooth muscle cells and on expression of low density lipoprotein receptor in cells. Chin Med J (Engl) 1999;112:308–311. [PubMed] [Google Scholar]
- Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384:149–157. doi: 10.1042/BJ20040928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Chen A. Curcumin suppresses the expression of extracellular matrix genes in activated hepatic stellate cells by inhibiting gene expression of connective tissue growth factor. Am J Physiol Gastrointest Liver Physiol. 2006;290:G883–G893. doi: 10.1152/ajpgi.00450.2005. [DOI] [PubMed] [Google Scholar]
- Zheng S, Chen A. Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-gamma in rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G113–G123. doi: 10.1152/ajpgi.00200.2006. [DOI] [PubMed] [Google Scholar]
- Zheng S, Yumei F, Chen A. De novo synthesis of glutathione is a prerequisite for curcumin to inhibit hepatic stellate cell (HSC) activation. Free Radic Biol Med. 2007;43:444–453. doi: 10.1016/j.freeradbiomed.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zheng S, Lin J, Zhang QJ, Chen A. The interruption of the PDGF and EGF signaling pathways by curcumin stimulates gene expression of PPARgamma in rat activated hepatic stellate cell in vitro. Lab Invest. 2007;87:488–498. doi: 10.1038/labinvest.3700532. [DOI] [PubMed] [Google Scholar]