

Abstract
Background and purpose:
The macrophage-derived chemokine (MDC/CCL22) is a prototypic Th2-type chemokine intimately involved in Th2-skewed allergic diseases, such as atopic dermatitis and asthma. The statins (3-hydroxy-3-methyl glutaryl coenzyme A reductase inhibitors) have been demonstrated to relieve allergic inflammation. However, the immunological effects and mechanisms of statins against atopic dermatitis remain unknown, at least in vitro. This study aimed to define how different statins affect MDC expression in HaCaT cells, a human keratinocyte cell line.
Experimental approach:
To measure the effects of statins on MDC expression in HaCaT cells, we used a cell viability assay, reverse transcription-polymerase chain reaction, enzyme-linked immunosorbent assay and Western blotting analyses.
Key results:
Fluvastatin, but not atorvastatin or simvastatin, inhibited MDC expression induced by interferon (IFN)-γ and NF-κB activation. A NF-κB inhibitor, but not a STAT1 inhibitor, suppressed MDC expression in HaCaT cells. Further, inhibition of p38 mitogen-activated protein kinases (MAPKs) significantly suppressed IFN-γ-induced MDC expression and NF-κB activation. Interestingly, fluvastatin suppressed IFN-γ-induced NF-κB activation in parallel with p38 MAPK phosphorylation.
Conclusions and implications:
These results indicate that fluvastatin inhibited expression of the CC chemokine MDC induced by IFN-γ in HaCaT cells, by inhibiting NF-κB activation via the p38 MAPK pathway. This blockade of a Th2 chemokine by fluvastatin may suppress the infiltration of Th2 cells into skin lesions and lessen the skin inflammation seen in atopic dermatitis, suggesting a potential therapeutic use of fluvastatin for this condition.
Keywords: fluvastatin, MDC/CCL22, HaCaT cells, atopic dermatitis (AD)
Introduction
Macrophage-derived chemokine (MDC/CCL22) is one of the CC chemokines that binds to the CC chemokine receptor 4 (CCR4), expressed selectively on Th2 cells. The interaction between MDC and CCR4 plays an important role in the migration of Th2 cells into inflamed tissue (Imai et al., 1998; Yoshie et al., 2001). Previous studies showed that serum or plasma levels of MDC were increased significantly in patients with atopic dermatitis (AD), and the increased levels of MDC correlated with the severity of AD (Kakinuma et al., 2002; Shimada et al., 2004; Hashimoto et al., 2006). Accordingly, keratinocytes express MDC in lesioned skin, but not in normal skin, of AD patients (Horikawa et al., 2002). The HaCaT (human adult low-calcium high-temperature) cell, a spontaneously transformed human keratinocyte cell line (Boukamp et al., 1988), has been commonly used as an in vitro model for proliferative epidermis (Lehmann, 1997). Cultured normal human keratinocytes and human HaCaT keratinocytes secrete MDC upon stimulation with interferon (IFN)-γ (Horikawa et al., 2002; Fujii-Maeda et al., 2004). Together, these data hinted that MDC released from keratinocytes might be importantly involved in the pathogenesis of AD, suggesting a promising therapeutic target in AD. Although immunosuppressive drugs such as glucocorticoids are temporarily effective against AD, prolonged use of these drugs incurs many adverse effects because of non-specific immune modulation (Kakinuma et al., 2001; Furukawa et al., 2004). With the increasing incidence of AD, a better targeted therapeutic agent for AD is urgently needed.
Of many possible treatments, statins [inhibitors of 3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) reductase] have shown some signs of immune modulation (Zamvil and Steinman, 2002). For instance, statins inhibit expression of class II major histocompatibility complex (MHC II) and modulate macrophage, monocyte and lymphocyte functions, implying that statins possess anti-inflammatory effects (Rothe et al., 1999; Kwak et al., 2000; Musial et al., 2001; Hillyard et al., 2002). Furthermore, simvastatin exerts anti-inflammatory effects in a murine model of allergic asthma (McKay et al., 2004). Several lines of in vitro evidence showed that statins significantly inhibited chemokine release from endothelial cells (Mach, 2002) and that fluvastatin suppressed the production of interleukin-8 in human vascular smooth muscle cells (Ito et al., 2002) as well as tumour necrosis factor (TNF)-α in human endothelial cells stimulated by C-reactive protein (Wang et al., 2005). Emerging data (Samson et al., 2006) show that fluvastatin suppressed the production of Th2-related chemokine TARC and Th1-related chemokine IP-10 in peripheral blood mononuclear cells from patients with allergic asthma. Furthermore, statins down-regulated the expression of Th1-type chemokine receptors, CCR5 and CXCR3, on T cells (Neuhaus et al., 2002). These preclinical and clinical data strongly suggest that statins could affect the immune response in Th1- or Th2-skewed diseases respectively. However, the immunological effects and mechanisms of statins against AD have not been previously elucidated. While the Th2 regulatory action of statins has been sparsely reported in some cell types, the effects of statins on the Th2-related chemokine MDC in keratinocytes remain unknown.
The aim of this study was to define how different statins (atorvastatin, fluvastatin and simvastain) affect MDC expression in HaCaT cells by using a cell viability assay, reverse transcription-polymerase chain reaction, enzyme-linked immunosorbent assay (ELISA) and Western blotting analyses. Here we describe how, of the statins tested, only fluvastatin inhibited IFN-γ-induced MDC expression in HaCaT cells by inhibiting NF-κB activation via the p38 mitogen-activated protein kinase (MAPK) pathway.
Methods
Cell culture
The human keratinocyte cell line, HaCaT (ATCC, Manassas, VA, USA), was cultured with RPMI 1640 medium (BioWhittaker Inc., Walkersville, MD, USA) containing 10% fetal bovine serum (BioWhittaker Inc., Walkersville, MD, USA), 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin (BioWhittaker Inc., Walkersville, MD, USA) at 37°C in a 5% CO2 incubator. Before performing experiments described below, the growth medium was exchanged for serum-free RPMI 1640 medium.
Cell viability assay
Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, HaCaT cells were seeded into 96-well microtiter plates at a density of 1 × 104 cells per well (200 µL) and incubated in complete medium. After 24 h, the medium was replaced with serum-free medium containing various concentrations of statins, with RPMI 1640 as the negative control. After incubating for 24 h, cells were treated with 20 µL MTT (5 mg·mL−1) (Sigma-Aldrich Co.) for 4 h. The formazan precipitate was dissolved in 200 µL of dimethyl sulphoxide (DMSO), and the absorbance at 570 nm was detected with an automated microplate reader. The percentage of cell viability was calculated by the following formula: cell viability (%) = (mean absorbency in test wells)/(mean absorbency in control wells) × 100.
ELISA
The HaCaT cells (1 × 106 cells per well) were seeded into six-well plates in 1 mL medium with 10% fetal bovine serum. After reaching confluence, the cells were washed and incubated with 1 mL serum-free medium containing the indicated concentration of IFN-γ for 24 h. The culture supernatants were analysed for MDC by ELISA (R&D Systems) according to the manufacturer's instructions. In some experiments, the HaCaT cells were incubated with statins or pharmacological inhibitors together with IFN-γ.
Reverse transcription-polymerase chain reaction
The HaCaT cells were incubated as above for 12 h to analyse the mRNA expression of MDC. Total RNA was isolated using the TRI reagent (Molecular Research Center Inc., Cincinnati, USA). The quantity and purity of RNA was verified by measuring A260 and A280. cDNA was synthesized from total RNA (2 µg) and oligo (dT)18 primers (0.5 µg) using Prime RT Premix (GeNet Bio, Korea) according to the manufacturer's instructions. The cDNA obtained was then amplified. The primers used in this study were as follows: MDC (forward) 5′-AGG ACA GAG CAT GGC TCG CCT ACA GA-3′, (reverse) 5′-TAA TGG CAG GGA GGT AGG GCT CCT GA-3′; and GAPDH (forward) 5′-ACC ACA GTC CAT GCC ATC AC-3′, (reverse) 5′-TCC ACC ACC CTG TTG CTG TA-3′. GAPDH primers were used as an internal control. All samples were pre-denatured for 5 min at 94°C. Conditions of polymerase chain reaction amplification were as follows: MDC, 94°C for 30 s, 65°C for 30 s, 72°C for 30 s for a total of 32 cycles; GAPDH, 94°C for 30 s, 56°C for 30 s, 72°C for 30 s for a total of 30 cycles. Following these cycles of polymerase chain reaction amplifications, the amplified cDNAs were further extended by additional extension at 72°C for 7 min. Amplified products were subjected to electrophoresis on 2% agarose gels and visualized by staining with ethidium bromide.
Whole-cell and nuclear fractionation
The preparation of whole-cell and nuclear extracts were performed using the Nuclear Extract Kit (Active Motif, Carlsbad, CA). Briefly, HaCaT cells (2 × 107) were washed twice with 3 mL ice-cold phosphate-buffered saline containing phosphatase inhibitors, centrifuged 5 min at 500×g. at 4°C, lysed in 300 µL complete lysis buffer, mixed completely and centrifuged at 14 000×g. for 20 min at 4°C, and then supernatants (whole-cell extracts) were stored at −80°C until further use. For nuclear extracts, after washing, cells were lysed in 500 µL hypotonic buffer and then centrifuged at 14 000×g for 30 s at 4°C. After removing the supernatant, pellets were resuspended in 50 µL complete lysis buffer and centrifuged at 14 000×g for 10 min at 4°C. Supernatants (nuclear fraction) were then stored at −80°C until further use. Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories, CA).
Western blotting analysis
HaCaT cells were treated with medium alone or with IFN-γ in the presence or absence of drugs for the indicated time. Proteins (40 µg) were separated on SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were then blocked with 5% non-fat milk, washed briefly, incubated with primary antibodies at 4°C overnight, and then incubated with corresponding horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized by incubating membranes with chemiluminescence reagents before exposure to X-ray film. The quantitation of the chemiluminescent signal was analysed using Image-Pro Plus version 6.0 (Media Cybernetics, MD, USA).
Statistical analysis
Comparisons between two groups were analysed using the Student's t-test. All data are presented as the mean ± SEM of three separate experiments. A P value less than 0.05 was considered to be statistically significant.
Materials
Atorvastatin [calcium salt, (C33H34FN2O5)2Ca•3H2O] was obtained from Pfizer (Groton, CT, USA). Fluvastatin (sodium salt, C24H25FNNaO4), simvastatin (sodium salt, C25H39O6•Na), Bay11-7082, AG490, Janus-activated kinase (JAK) inhibitor I, SB203580, PD98059 and SP600125 were purchased from Calbiochem (La Jolla, CA, USA). Mevalonic acid and 5′-deoxy-5′-(methylthio)-adenosine (MTA) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Recombinant human IFN-γ was from Abcam Inc. (Cambridge, MA, USA). ELISA reagent for human MDC/CCL22 was obtained from R&D Ssystems (Minneapolis, MN, USA). Antibodies for p38 MAPK, phospho-specific p38 MAPK, NF-κB p65, poly(ADP-ribose) polymerase, and horseradish peroxidase-conjugated goat anti-rabbit IgG were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An enhanced-chemiluminescence Western blotting detection system was obtained from Amersham Pharmacia Biotech (Tokyo, Japan). The stock solution of atorvastatin was made in methanol, while stock solutions of fluvastatin, simvastatin and pharmacological inhibitors were made in DMSO. Before use, these stock solutions were diluted in culture medium at the indicated concentrations, and the maximum concentrations of methanol or DMSO were not more than 0.1%, and did not affect cell viability.
Results
Cell viability
The effects of statins on the viability of HaCaT cells were assessed by MTT assay. As shown in Figure 1, low concentrations of atorvastatin (0.1–2 µM), fluvastatin (0.1–4 µM) and simvastatin (0.1–2 µM) had minimal effects on the viability of HaCaT cells, although all these statins significantly reduced the viability at higher concentrations of 4–16 µM (atorvastatin and simvastatin) or 8–16 µM (fluvastatin) after 24 h incubation. Therefore, 2 µM was chosen as the maximum concentration of statins for use throughout the following experiments.
Figure 1.
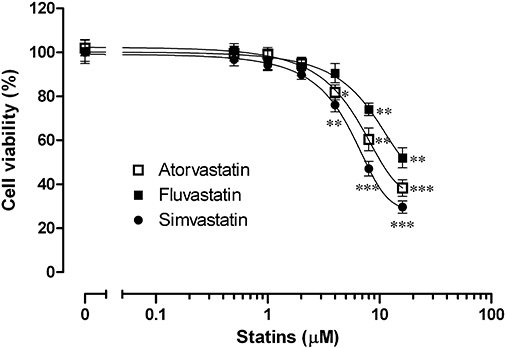
Effect of statins (atorvastatin, fluvastatin and simvastatin) on the viability of HaCaT cells. Cells were incubated with various concentrations of statins for 24 h and RPMI 1640 media were used as a negative control. Cell viability was then determined by an MTT assay. All values are presented as mean ± SEM of three experiments conducted in duplicate, *P < 0.05, **P < 0.01, ***P < 0.001 versus control cells incubated with medium alone.
Fluvastatin, but not atorvastatin or simvastatin, inhibited IFN-γ-induced MDC expression in HaCaT cells
We compared the effects of statins on MDC expression (after 24 h incubation) in HaCaT cells stimulated with IFN-γ (10 ng·mL−1). Figure 2B shows that IFN-γ markedly stimulated MDC release from HaCaT cells and that this release was reduced by treatment of fluvastatin at 1 and 2 µM. Interestingly, the inhibition of MDC release by treatment with fluvastatin (2 µM) was significantly reversed by treatment with mevalonic acid (100 µM) (P < 0.01). Similarly, fluvastatin (2 µM) markedly suppressed IFN-γ-induced mRNA expression of MDC as compared with control cells stimulated by IFN-γ alone (Figure 2D). Conversely, neither atorvastatin (0.1–2 µM) nor simvastatin (0.1–2 µM) affected IFN-γ-induced MDC expression in the presence or absence of mevalonic acid (Figure 2A and C). Consequently, we next explored the signalling pathway by which fluvastatin inhibited MDC expression in HaCaT cells.
Figure 2.
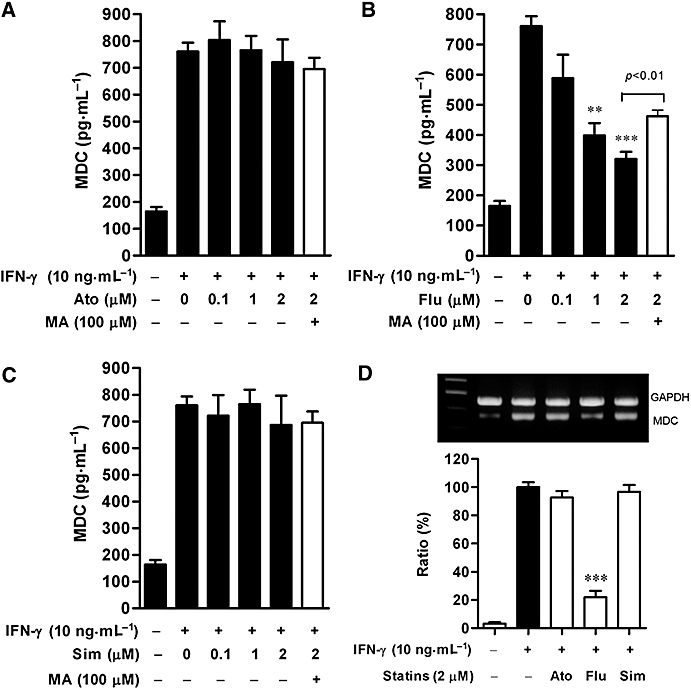
Effects of statins on release and mRNA expression of macrophage-derived chemokine (MDC) in HaCaT cells. Cultured cells were incubated with medium alone or with interferon (IFN)-γ (10 ng·mL−1) in the presence or absence of indicated concentrations of atorvastatin (A), fluvastatin (B) or simvastatin (C). In some experiments, mevalonic acid (100 µM) was added. At 24 h, MDC release was analysed by ELISA. Data are presented as mean ± SEM of three experiments conducted in duplicate, **P < 0.01, ***P < 0.001 versus IFN-γ alone. (D) Cells were incubated with medium alone or with IFN-γ (10 ng·mL−1) in the presence or absence of 2 µM statins – atorvastatin (Ato), fluvastatin (Flu) and simvastatin (Sim). At 12 h, MDC and GAPDH mRNA levels were analysed. Representative results from three separate experiments are shown. The lower panel shows the signal intensity ratio between MDC and GAPDH. The ratio in IFN-γ-stimulated cells was set as 100%. Data are presented as mean ± SEM of three separate experiments, ***P < 0.001 versus IFN-γ alone.
Involvement of NF-κB in IFN-γ-induced MDC expression in HaCaT cells
The MDC promoter contains STAT- and NF-κB-binding sequences and these transcription factors may mediate the transcription of this gene (Nakayama et al., 2004). Moreover, IFN-γ increases the transcriptional activities of STAT1 and NF-κB in keratinocytes (Kanda et al., 2007). We thus examined whether these transcription factors were involved in IFN-γ-induced MDC release by using a STAT1 inhibitor, MTA (Mowen et al., 2001; Shen and Lentsch, 2004) or a NF-κB inhibitor, Bay11-7082. As shown in Figure 3A, IFN-γ (10 ng·mL−1) induced an increase in the basal MDC release after 24 h incubation and this increase was markedly reduced following treatment with Bay11-7082 (10 µM) (P < 0.01). In contrast, MTA (100 µM) had minimal effect on MDC release as compared with control cells stimulated with IFN-γ alone. We next examined whether treatment with Bay11-7082 would reduce MDC expression at the transcriptional level. The results showed that treatment with 10 µM Bay11-7082 strongly suppressed mRNA expression of MDC after 12 h treatment with IFN-γ (Figure 3B).
Figure 3.
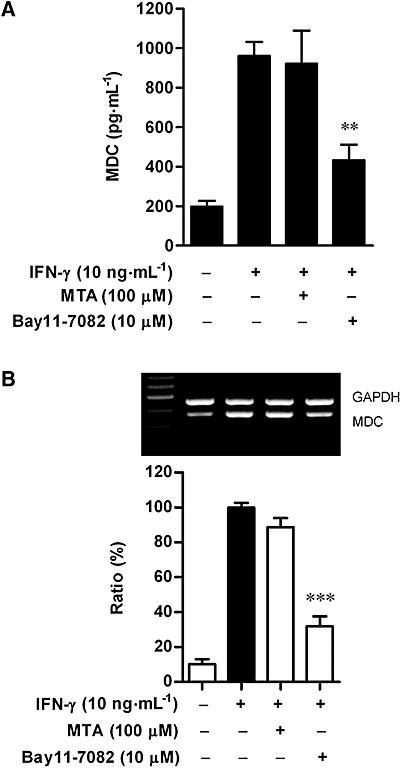
Effects of MTA and Bay11-7082 on MDC release (A) and mRNA expression (B) in HaCaT cells. (A) Cultured cells were incubated with medium alone or with interferon (IFN)-γ (10 ng·mL−1) in the presence or absence of MTA (100 µM) or Bay11-7082 (10 µM) for 24 h. MDC release into culture medium was analysed by ELISA. Data are presented as mean ± SEM of three experiments conducted in duplicate, **P < 0.01 versus IFN-γ alone. (B) Cells were incubated as above. At 12 h, MDC and GAPDH mRNA levels were analysed. Representative results from three separate experiments are shown. The lower panel shows the signal intensity ratio between MDC and GAPDH. The ratio in IFN-γ-stimulated cells was set as 100%. Data are presented as mean ± SEM of three separate experiments, ***P < 0.001 versus IFN-γ alone. MDC, macrophage-derived chemokine; MTA, 5′-deoxy-5′-(methylthio)-adenosine.
Fluvastatin inhibits IFN-γ-induced NF-kB activation in HaCaT cells
Because fluvastatin blocked MDC expression in HaCaT cells (Figure 2), we next examined whether fluvastatin affects NF-κB activation that is involved in MDC expression (Figure 3). As the p65 subunit is the primary component of activated NF-κB, the activation of NF-κB in IFN-γ-stimulated HaCaT cells was studied by measuring the nuclear translocation of NF-κB subunit p65 by Western blot. IFN-γ significantly increased the nuclear translocation of NF-κB p65 at 30 min. Fluvastatin (2 µM) decreased the nuclear levels of NF-κB p65 by approximately 70% in IFN-γ-stimulated HaCaT cells (P < 0.01) (Figure 4A), which indicated the inhibition of IFN-γ-induced activation of NF-κB by fluvastatin in HaCaT cells. In a manner similar to that of fluvastatin, the NF-κB inhibitor, Bay11-7082 (10 µM), suppressed IFN-γ-induced nuclear translocation of NF-κB by approximately 65% (Figure 4B).
Figure 4.
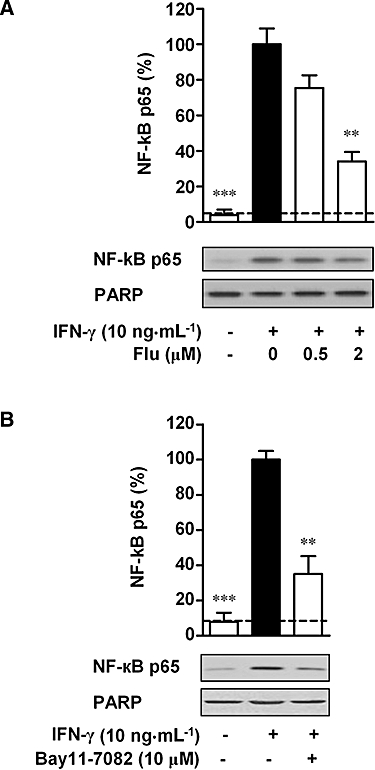
Effect of fluvastatin and Bay11-7082 on nuclear translocation of NF-κB in interferon (IFN)-γ-stimulated HaCaT cells. Cells were incubated with medium alone or with IFN-γ (10 ng·mL−1) in the presence or absence of fluvastatin (Flu, 0–2 µM) (A), or Bay11-7082 (10 µM) (B). After 30 min, the nuclear proteins were extracted. Equal amounts of protein (40 µg per lane) were subjected to Western blotting using an antibody against NF-κB p65. As a nuclear protein loading control, the membrane was blotted with anti-poly(ADP-ribose) polymerase (PARP) antibody. Nuclear levels of NF-κB p65 in IFN-γ-stimulated cells were set as 100%. The dashed line represents the nuclear level of NF-κB p65 in resting cells. Data are presented as mean ± SEM of three separate experiments, **P < 0.01, ***P < 0.001 versus IFN-γ alone.
Involvement of the p38 MAPK pathway in IFN-γ-induced MDC expression and NF-κB activation in HaCaT cells
IFN-γ activates receptor-associated JAKs or MAPKs depending on the cell type (Goh et al., 1999; Nguyen et al., 2000). We thus examined the involvement of these signalling kinases in IFN-γ-increased MDC expression in HaCaT cells. As illustrated in Figure 5A. the IFN-γ-induced MDC release was significantly reduced by treatment of cells with the p38 MAPK inhibitor, SB203580 (10 µM) (P < 0.01), but not by treatment with JAK inhibitor I, AG490, PD98059 or SP600125, inhibitors of JAK1/2/3, JAK2, ERK or JNK respectively. Consistent with this finding, IFN-γ-induced mRNA expression of MDC was also greatly suppressed by SB203580 (Figure 5B).
Figure 5.
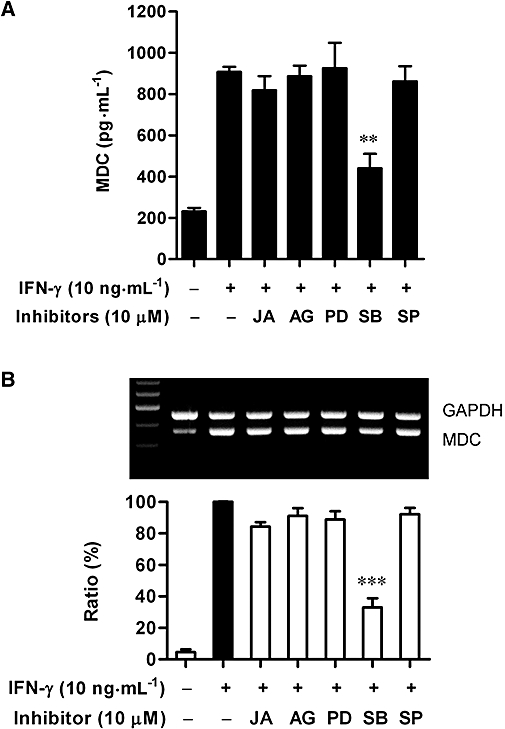
Effects of inhibiting various signalling pathways on the release and mRNA expression of macrophage-derived chemokine (MDC) in interferon (IFN)-γ-stimulated HaCaT cells. (A) Cultured cells were incubated with medium alone or with IFN-γ (10 ng·mL−1) in the presence or absence of JAK inhibitor I (JA, 10 µM), AG490 (AG, 10 µM), PD98059 (PD, 10 µM), SB203580 (SB, 10 µM) or SP600125 (SP, 10 µM) for 24 h. MDC release into culture medium was analysed by ELISA. Data are presented as mean ± SEM of three experiments conducted in duplicate, **P < 0.01 versus IFN-γ alone. (B) Cells were incubated as above. At 12 h, MDC and GAPDH mRNA levels were analysed. Representative results from three separate experiments are shown. The lower panel showed the signal intensity ratio between MDC and GAPDH. The ratio in IFN-γ-stimulated cells was set as 100%. Data are presented as mean ± SEM of three separate experiments, ***P < 0.001 versus IFN-γ alone.
We subsequently examined whether p38 MAPK is involved in IFN-γ-induced activation of NF-κB. The activation of NF-κB was examined by measuring the nuclear translocation of NF-κB subunit p65 as described above. IFN-γ-induced nuclear translocation of NF-κB p65 was significantly suppressed by treatment with SB203580 at 10 µM (P < 0.01) (Figure 6). Together, both MDC expression and NF-κB activation were inhibited by treatment with SB203580 in IFN-γ-stimulated HaCaT cells.
Figure 6.
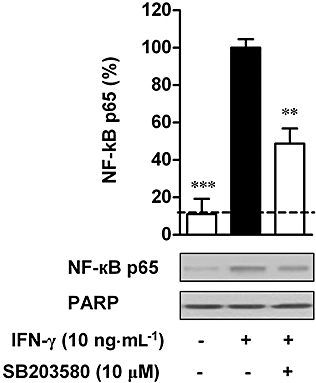
Effect of SB203580 on nuclear translocation of NF-κB in interferon (IFN)-γ-stimulated HaCaT cells. Cells were incubated with medium alone or with IFN-γ (10 ng·mL−1) in the presence or absence of SB203580 (10 µM). After 30 min, the nuclear proteins were extracted. Equal amounts of protein (40 µg per lane) were subjected to Western blotting using an antibody against NF-κB p65. PARP was used as a loading control. Nuclear levels of NF-κB p65 in IFN-γ-stimulated cells were set as 100%. The dashed line represents the nuclear level of NF-κB p65 in resting cells. Data are presented as mean ± SEM of three separate experiments, **P < 0.01, ***P < 0.001 versus IFN-γ alone.
Fluvastatin inhibits activation of p38 MAPK stimulated by IFN-γ in HaCaT cells
Finally, we examined the effect of fluvastatin on the activation of p38 MAPK by IFN-γ stimulation in HaCaT cells. The activation of p38 MAPK results in phosphorylation at threonine and tyrosine residues of this kinase (Morel et al., 2002). In the present study, the phosphorylation of p38 MAPK was induced as early as 10 min following incubation with IFN-γ (10 ng·mL−1), and this phosphorylation was greatly inhibited by fluvastatin (2 µM) treatment. Further, a significant inhibition was also observed at the 30 min time point (Figure 7). The inhibitory effect of fluvastatin on phosphorylation of p38 MAPK was lost after 60 min of stimulation by IFN-γ. Although there was no significant inhibitory effect at this time point, fluvastatin decreased the relative phosphorylation of p38 MAPK after 60 min of stimulation with IFN-γ when compared with control cells incubated with IFN-γ alone (Figure 7B).
Figure 7.
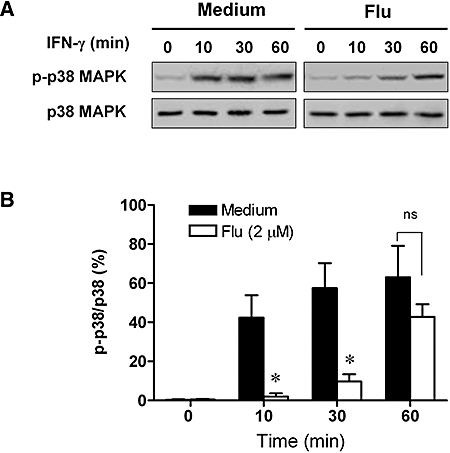
Effect of fluvastatin on the phosphorylation of p38 mitogen-activated protein kinases (MAPKs) in interferon (IFN)-γ-stimulated HaCaT cells. (A) HaCaT cells were untreated or pretreated with 2 µM fluvastatin (Flu) for 12 h and then treated with 10 ng·mL−1 IFN-γ for the indicated times. Whole-cell extracts were prepared and analysed by Western blotting using antibodies against phospho-p38 MAPK or p38 MAPK. (B) The phosphorylation levels of p38 MAPK were evaluated by the ratio between phosphor-p38 MAPK and p38 MAPK. Data are presented as mean ± SEM of three separate experiments, *P < 0.05 versus IFN-γ alone.
Discussion
Convincing evidence from both in vitro and mouse model data suggest that statins could counteract Th2 immune responses against Th2-skewed diseases such as asthma (Samson et al., 2006; Kim et al., 2007), but the effects of statins on the Th2-related chemokine MDC expression in keratinocytes have been less clear. To clarify this issue, we examined whether different statins (atorvastatin, fluvastatin and simvastain) could inhibit MDC expression in HaCaT cells. Prior to the assay of MDC expression, the optimal concentrations of three different statins were determined and 2 µM was established as the maximal concentration for statins used throughout the following experiments (Figure 1). Unexpectedly, we found that fluvastatin, but neither atorvastatin nor simvastatin, suppressed Th2-type chemokine MDC release and mRNA expression in IFN-γ-stimulated HaCaT cells in a dose-dependent manner (Figure 2). We also demonstrated that fluvastatin inhibited IFN-γ-induced MDC expression in HaCaT cells by inhibiting NF-κB activation via the p38 MAPK pathway.
Although all statins effectively lower cholesterol, the pleiotropic effects of individual statins may well differ. In particular, lipophilic statins, such as atorvastatin, fluvastatin and simvastatin, may have different anti-inflammatory functions depending on stimulation and organ/cell types. For example, fluvastatin may induce production of TNF-α, IFN-γ and IL-18 (Rosenson et al., 1999; Takahashi et al., 2005), whereas atorvastatin and simvastatin inhibit production of TNF-α (Ikeda and Shimada, 1999; Ferro et al., 2000; Grip et al., 2002; Wagner et al., 2002) in monocytes stimulated by lipopolysaccharide. Thus, different types of lipophilic statins may have dissimilar anti-inflammatory activities (Hothersall et al., 2006). In light of the potency of lipophilic statins, recent data showed that lipophilic statins inhibited both proliferation and invasion of human saphenous vein smooth muscle cells with the same order of lipophilic potency (fluvastatin > atorvastatin > simvastatin) (Turner et al., 2007). These different potencies of lipophilic statins might also partially explain the greater inhibition of MDC by fluvastatin over other statins in this study.
A growing body of evidence shows that fluvastatin inhibits the production of pro-inflammatory cytokines and chemokines in peripheral blood mononuclear cells in patients with allergic asthma (Samson et al., 2006), and decreases the serum concentration of inflammatory markers including C-reactive protein and TNF-α in patients with acute coronary syndrome (Yang et al., 2006). Furthermore, fluvastatin has been demonstrated to down-regulate signalling by inflammatory mediators in HaCaT keratinocytes (Alaei et al., 1996). These preclinical and clinical studies suggest the possibility of down-regulation of inflammation by fluvastatin. Even though our data validate this hypothesis in an in vitro keratinocyte model, there remain some limitations to this study, including an absence of proof regarding selective inhibition exerted by fluvastatin on the Th2 chemokine profile and use of an in vitro cell line system instead of primary human keratinocytes or an in vivo AD model. Despite these drawbacks, to the best of our knowledge, this study is the first to show that fluvastatin inhibits IFN-γ-induced MDC expression in HaCaT cells.
We subsequently explored the signalling pathways involved in fluvastatin-induced inhibition of MDC expression in HaCaT cells. The MDC promoter contains one STAT site and two NF-κB sites (Nakayama et al., 2004), and the transcriptional activities of STAT1 and NF-κB are increased by IFN-γ in keratinocytes (Kanda and Watanabe, 2007), so these transcription factors may be involved in the mediation of MDC. We found that IFN-γ-induced MDC expression at the transcriptional and post-transcriptional levels were significantly inhibited by the NF-κB-specific inhibitor, Bay11-7082, but not by a STAT1 inhibitor, MTA, strongly suggesting that NF-κB is involved in MDC expression in IFN-γ-stimulated HaCaT cells. Moreover, fluvastatin significantly reduced the nuclear levels of NF-κB p65 in IFN-γ-stimulated HaCaT cells (Figure 4A), indicating that fluvastatin dose-dependently inhibits IFN-γ-induced NF-κB activation in HaCaT cells. Based on these data, we deduced that fluvastatin may inhibit MDC expression by blocking NF-κB activation in IFN-γ-stimulated HaCaT cells. Many studies have documented that statins can block NF-κB activation induced by a diverse array of stimuli, such as angiotensin II, TNF-α, oxidized low-density lipoprotein and C-reactive protein in cell systems in vitro, such as vascular smooth muscle, mononuclear cells, human primary monocytes and human endothelial cells (Bustos et al., 1998; Zelvyte et al., 2002; Wang et al., 2005). A mechanism by which statins inhibit NF-κB activation has previously been suggested. Statins inhibit HMG-CoA reductase and thus inhibit the production of mevalonate and its metabolites, subsequently decreasing the availability of farnesylated metabolites, and thereby lowering efficacy in post-transcriptional modification of Ras or Rho family proteins. The reduced activities of small GTPases, such as Ras or Rho proteins, may result in the inhibition of NF-κB activation via its upstream molecules, such as MAP kinases (Yang et al., 2000; Haloui et al., 2003; Sakoda et al., 2006; Kim et al., 2007). This scheme might partly explain our results that fluvastatin-induced inhibition of MDC release was significantly blocked by addition of mevalonic acid (Figure 2B). However, the actual effect of fluvastatin on GTPase activities remains to be settled as we did not directly measure the Ras or Rho activities.
IFN-γ activates JAK1/2 and the p38 MAPK pathways in keratinocytes (Kanda and Watanabe, 2007; Kanda et al., 2007), the ERK pathway in hepatocytes (Nguyen et al., 2000) or the ERK and JNK pathways in murine macrophages (Rose et al., 1997). As a corollary, these pathways may be involved in the regulation of pro-inflammatory cytokines or chemokines in keratinocytes. We found that the p38 MAPK inhibitor SB203580 significantly inhibited MDC expression in IFN-γ-stimulated HaCaT cells, indicating the involvement of p38 MAPK in mediation of MDC expression, a finding that is in agreement with a previous study using human B cells (Nakayama et al., 2004). In contrast, JAKs, MEK/ERK or JNK had minimal effect on MDC expression induced by IFN-γ in HaCaT keratinocytes.
Consistent with this result, the p38 MAPK inhibitor, SB203580, markedly reduced the activation of NF-κB in HaCaT cells (Figure 6). A role for p38 MAPK in enhancing NF-κB activation, as observed in this study, is supported by a recent report that IFN-γ-induced activation of NF-κB was blocked by SB203580 in human neonatal foreskin keratinocytes (Kanda et al., 2007). Furthermore, fluvastatin (1 µM) completely inhibited p38 MAPK activation induced by hypoxia in pulmonary artery fibroblasts via inhibition of protein geranylgeranylation (Carlin et al., 2007). Despite using a different assay system, our result may reinforce the notion that fluvastatin selectively inhibits the activation of the p38 MAPK pathway in HaCaT cells (Figure 7). Taken together, these data suggest that fluvastatin could inhibit IFN-γ-induced MDC expression by way of inhibiting the activation of NF-κB via the p38 MAPK pathway in HaCaT cells.
Although some reports showed that atorvastatin or simvastatin influences p38 MAPK or NF-κB signalling, the effects might be organ- or cell type-specific. For instance, atorvastatin decreased diabetes-induced p38 MAPK phosphorylation by 3.2-fold (Van Linthout et al., 2007), while it increased the phosphorylation of p38 MAPK in Langendorff-perfused mouse hearts (Efthymiou et al., 2005) and in cardiac myocytes (Nakajima et al., 2006). Simvastatin inhibited p38 MAPK activation in the lung following lipopolysaccharide (LPS) stimulation (Ou et al., 2008) and in the myocardium of bradykinin B2 receptor knockout mice, but did not affect p38 MAPK activation in wild-type mice (Osorio et al., 2008), oesophageal adenocarcinoma cell lines such as OE33 and BIC-1 cells (Ogunwobi and Beales, 2008), and LPS-stimulated U937 mononuclear cells (Sundararaj et al., 2008). On the other hand, atorvastatin and simvastatin both decreased NF-κB activation induced by oxidative stress by around 50% in both vascular smooth muscle cells and monocytic cells (THP-1) (Ortego et al., 2005), whereas another study showed that simvastatin inhibited LPS-stimulated nuclear AP-1, but not NF-κB activity in U937 mononuclear cells (Sundararaj et al., 2008). Taken together, the evidence suggests that individual members of the lipophilic family of statins might have different influences on inflammatory signalling pathways depending on organs or cell types and stimulus. This idea is further supported by our results showing that fluvastatin, but not atorvastatin or simvastatin, suppressed MDC expression in our system. Despite uncovering part of the signalling pathway by which fluvastatin inhibits this expression in HaCaT cells, further assays using siRNA and overexpression techniques will be required to more completely determine the mechanism of drug action.
In summary, our results indicate that fluvastatin inhibits IFN-γ-induced MDC expression in HaCaT cells by inhibiting NF-κB activation via the p38 MAPK pathway. Block of the formation of a Th2 chemokine by fluvastatin may suppress the infiltration of Th2 cells into skin lesions and lessen the skin inflammation, as seen in AD, suggesting that fluvastatin might be used clinically as a therapeutic agent for AD. Further elucidation of the signalling pathways involved in blocking release of this Th2 chemokine by fluvastatin could provide insight into the design of novel targeted therapeutic agents for AD. However, additional studies using an in vivo model of human skin inflammation are necessary to investigate the possibility that fluvastatin could be used for treatment of chronic skin inflammation involving the chemokine MDC/CCL22.
Acknowledgments
We would like to thank Dr Guo-Hua Song, Dr Long-Quan Pi, Mr Jae-Shin Sung and Mr Tae-Eul Jung for their technical assistance.
Glossary
Abbreviations:
- AD
atopic dermatitis
- HMG-CoA
3-hydroxy-3-methyl glutaryl coenzyme A
- MDC/CCL22
macrophage-derived chemokine
- MTA
5′-deoxy-5′-(methylthio)-adenosine
Conflict of interest
There is no declared conflict of interest in this study.
References
- Alaei P, MacNulty EE, Ryder NS. Inhibition of protein prenylation down-regulates signalling by inflammatory mediators in human keratinocytes. Biochem Biophys Res Commun. 1996;222:133–138. doi: 10.1006/bbrc.1996.0710. [DOI] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos C, Hernandez-Presa MA, Ortego M, Tunon J, Ortega L, Perez F, et al. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol. 1998;32:2057–2064. doi: 10.1016/s0735-1097(98)00487-2. [DOI] [PubMed] [Google Scholar]
- Carlin CM, Peacock AJ, Welsh DJ. Fluvastatin inhibits hypoxic proliferation and p38 MAPK activity in pulmonary artery fibroblasts. Am J Respir Cell Mol Biol. 2007;37:447–456. doi: 10.1165/rcmb.2007-0012OC. [DOI] [PubMed] [Google Scholar]
- Efthymiou CA, Mocanu MM, Yellon DM. Atorvastatin and myocardial reperfusion injury: new pleiotropic effect implicating multiple prosurvival signaling. J Cardiovasc Pharmacol. 2005;45:247–252. doi: 10.1097/01.fjc.0000154376.82445.06. [DOI] [PubMed] [Google Scholar]
- Ferro D, Parrotto S, Basili S, Alessandri C, Violi F. Simvastatin inhibits the monocyte expression of proinflammatory cytokines in patients with hypercholesterolemia. J Am Coll Cardiol. 2000;36:427–431. doi: 10.1016/s0735-1097(00)00771-3. [DOI] [PubMed] [Google Scholar]
- Fujii-Maeda S, Kajiwara K, Ikizawa K, Shinazawa M, Yu B, Koga T, et al. Reciprocal regulation of thymus and activation-regulated chemokine/macrophage-derived chemokine production by interleukin (IL)-4/IL-13 and interferon-gamma in HaCaT keratinocytes is mediated by alternations in E-cadherin distribution. J Invest Dermatol. 2004;122:20–28. doi: 10.1046/j.0022-202X.2003.22103.x. [DOI] [PubMed] [Google Scholar]
- Furukawa H, Nakamura K, Zheng X, Tojo M, Oyama N, Akiba H, et al. Enhanced TARC production by dust-mite allergens and its modulation by immunosuppressive drugs in PBMCs from patients with atopic dermatitis. J Dermatol Sci. 2004;35:35–42. doi: 10.1016/j.jdermsci.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grip O, Janciauskiene S, Lindgren S. Atorvastatin activates PPAR-gamma and attenuates the inflammatory response in human monocytes. Inflamm Res. 2002;51:58–62. doi: 10.1007/BF02684000. [DOI] [PubMed] [Google Scholar]
- Haloui M, Meilhac O, Jandrot-Perrus M, Michel JB. Atorvastatin limits the pro-inflammatory response of rat aortic smooth muscle cells to thrombin. Eur J Pharmacol. 2003;474:175–184. doi: 10.1016/s0014-2999(03)02043-0. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Nakamura K, Oyama N, Kaneko F, Tsunemi Y, Saeki H, et al. Macrophage-derived chemokine (MDC)/CCL22 produced by monocyte derived dendritic cells reflects the disease activity in patients with atopic dermatitis. J Dermatol Sci. 2006;44:93–99. doi: 10.1016/j.jdermsci.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Hillyard DZ, Cameron AJ, McIntyre AH, Hadden MH, Marshall HE, Johnston N, et al. Inhibition of proliferation and signalling mechanisms in human lymphocytes by fluvastatin. Clin Exp Pharmacol Physiol. 2002;29:673–678. doi: 10.1046/j.1440-1681.2002.03711.x. [DOI] [PubMed] [Google Scholar]
- Horikawa T, Nakayama T, Hikita I, Yamada H, Fujisawa R, Bito T, et al. IFN-gamma-inducible expression of thymus and activation-regulated chemokine/CCL17 and macrophage-derived chemokine/CCL22 in epidermal keratinocytes and their roles in atopic dermatitis. Int Immunol. 2002;14:767–773. doi: 10.1093/intimm/dxf044. [DOI] [PubMed] [Google Scholar]
- Hothersall E, McSharry C, Thomson NC. Potential therapeutic role for statins in respiratory disease. Thorax. 2006;61:729–734. doi: 10.1136/thx.2005.057976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda U, Shimada K. Statins and monocytes. Lancet. 1999;353:2070. doi: 10.1016/S0140-6736(05)77885-5. [DOI] [PubMed] [Google Scholar]
- Imai T, Chantry D, Raport CJ, Wood CL, Nishimura M, Godiska R, et al. Macrophage-derived chemokine is a functional ligand for the CC chemokine receptor 4. J Biol Chem. 1998;273:1764–1768. doi: 10.1074/jbc.273.3.1764. [DOI] [PubMed] [Google Scholar]
- Ito T, Ikeda U, Yamamoto K, Shimada K. Regulation of interleukin-8 expression by HMG-CoA reductase inhibitors in human vascular smooth muscle cells. Atherosclerosis. 2002;165:51–55. doi: 10.1016/s0021-9150(02)00194-6. [DOI] [PubMed] [Google Scholar]
- Kakinuma T, Nakamura K, Wakugawa M, Mitsui H, Tada Y, Saeki H, et al. Thymus and activation-regulated chemokine in atopic dermatitis: Serum thymus and activation-regulated chemokine level is closely related with disease activity. J Allergy Clin Immunol. 2001;107:535–541. doi: 10.1067/mai.2001.113237. [DOI] [PubMed] [Google Scholar]
- Kakinuma T, Nakamura K, Wakugawa M, Mitsui H, Tada Y, Saeki H, et al. Serum macrophage-derived chemokine (MDC) levels are closely related with the disease activity of atopic dermatitis. Clin Exp Immunol. 2002;127:270–273. doi: 10.1046/j.1365-2249.2002.01727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda N, Watanabe S. Prolactin enhances interferon-gamma-induced production of CXC ligand 9 (CXCL9), CXCL10, and CXCL11 in human keratinocytes. Endocrinology. 2007;148:2317–2325. doi: 10.1210/en.2006-1639. [DOI] [PubMed] [Google Scholar]
- Kanda N, Shimizu T, Tada Y, Watanabe S. IL-18 enhances IFN-gamma-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur J Immunol. 2007;37:338–350. doi: 10.1002/eji.200636420. [DOI] [PubMed] [Google Scholar]
- Kim DY, Ryu SY, Lim JE, Lee YS, Ro JY. Anti-inflammatory mechanism of simvastatin in mouse allergic asthma model. Eur J Pharmacol. 2007;557:76–86. doi: 10.1016/j.ejphar.2006.11.027. [DOI] [PubMed] [Google Scholar]
- Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- Lehmann B. HaCaT cell line as a model system for vitamin D3 metabolism in human skin. J Invest Dermatol. 1997;108:78–82. doi: 10.1111/1523-1747.ep12285640. [DOI] [PubMed] [Google Scholar]
- Mach F. Statins as immunomodulators. Transpl Immunol. 2002;9:197–200. doi: 10.1016/s0966-3274(02)00030-8. [DOI] [PubMed] [Google Scholar]
- McKay A, Leung BP, McInnes IB, Thomson NC, Liew FY. A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. J Immunol. 2004;172:2903–2908. doi: 10.4049/jimmunol.172.5.2903. [DOI] [PubMed] [Google Scholar]
- Morel JC, Park CC, Zhu K, Kumar P, Ruth JH, Koch AE. Signal transduction pathways involved in rheumatoid arthritis synovial fibroblast interleukin-18-induced vascular cell adhesion molecule-1 expression. J Biol Chem. 2002;277:34679–34691. doi: 10.1074/jbc.M206337200. [DOI] [PubMed] [Google Scholar]
- Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, et al. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104:731–741. doi: 10.1016/s0092-8674(01)00269-0. [DOI] [PubMed] [Google Scholar]
- Musial J, Undas A, Gajewski P, Jankowski M, Sydor W, Szczeklik A. Anti-inflammatory effects of simvastatin in subjects with hypercholesterolemia. Int J Cardiol. 2001;77:247–253. doi: 10.1016/s0167-5273(00)00439-3. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Suga H, Matsuno H, Ishisaki A, Hirade K, Kozawa O. Differential roles of MAP kinases in atorvastatin-induced VEGF release in cardiac myocytes. Life Sci. 2006;79:1214–1220. doi: 10.1016/j.lfs.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Hieshima K, Nagakubo D, Sato E, Nakayama M, Kawa K, et al. Selective induction of Th2-attracting chemokines CCL17 and CCL22 in human B cells by latent membrane protein 1 of Epstein-Barr virus. J Virol. 2004;78:1665–1674. doi: 10.1128/JVI.78.4.1665-1674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus O, Strasser-Fuchs S, Fazekas F, Kieseier BC, Niederwieser G, Hartung HP, et al. Statins as immunomodulators: comparison with interferon-beta 1b in MS. Neurology. 2002;59:990–997. doi: 10.1212/wnl.59.7.990. [DOI] [PubMed] [Google Scholar]
- Nguyen VA, Chen J, Hong F, Ishac EJ, Gao B. Interferons activate the p42/44 mitogen-activated protein kinase and JAK-STAT (Janus kinase-signal transducer and activator transcription factor) signalling pathways in hepatocytes: differential regulation by acute ethanol via a protein kinase C-dependent mechanism. Biochem J. 2000;349:427–434. doi: 10.1042/0264-6021:3490427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunwobi OO, Beales IL. Statins inhibit proliferation and induce apoptosis in Barrett's esophageal adenocarcinoma cells. Am J Gastroenterol. 2008;103:825–837. doi: 10.1111/j.1572-0241.2007.01773.x. [DOI] [PubMed] [Google Scholar]
- Ortego M, Gomez-Hernandez A, Vidal C, Sanchez-Galan E, Blanco-Colio LM, Martin-Ventura JL, et al. HMG-CoA reductase inhibitors reduce I kappa B kinase activity induced by oxidative stress in monocytes and vascular smooth muscle cells. J Cardiovasc Pharmacol. 2005;45:468–475. doi: 10.1097/01.fjc.0000159042.50488.e5. [DOI] [PubMed] [Google Scholar]
- Osorio JC, Cheema FH, Martens TP, Mahmut N, Kinnear C, Gonzalez AM, et al. Simvastatin reverses cardiac hypertrophy caused by disruption of the bradykinin 2 receptor. Can J Physiol Pharmacol. 2008;86:633–642. doi: 10.1139/y08-068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou XM, Wang BD, Wen FQ, Feng YL, Huang XY, Xiao J. Simvastatin attenuates lipopolysaccharide-induced airway mucus hypersecretion in rats. Chin Med J (Engl) 2008;121:1680–1687. [PubMed] [Google Scholar]
- Rose DM, Winston BW, Chan ED, Riches DW, Henson PM. Interferon-gamma and transforming growth factor-beta modulate the activation of mitogen-activated protein kinases and tumor necrosis factor-alpha production induced by Fc gamma-receptor stimulation in murine macrophages. Biochem Biophys Res Commun. 1997;238:256–260. doi: 10.1006/bbrc.1997.7271. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Tangney CC, Casey LC. Inhibition of proinflammatory cytokine production by pravastatin. Lancet. 1999;353:983–984. doi: 10.1016/S0140-6736(98)05917-0. [DOI] [PubMed] [Google Scholar]
- Rothe G, Herr AS, Stohr J, Abletshauser C, Weidinger G, Schmitz G. A more mature phenotype of blood mononuclear phagocytes is induced by fluvastatin treatment in hypercholesterolemic patients with coronary heart disease. Atherosclerosis. 1999;144:251–261. doi: 10.1016/s0021-9150(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Sakoda K, Yamamoto M, Negishi Y, Liao JK, Node K, Izumi Y. Simvastatin decreases IL-6 and IL-8 production in epithelial cells. J Dent Res. 2006;85:520–523. doi: 10.1177/154405910608500608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson KT, Minoguchi K, Tanaka A, Oda N, Yokoe T, Yamamoto Y, et al. Inhibitory effects of fluvastatin on cytokine and chemokine production by peripheral blood mononuclear cells in patients with allergic asthma. Clin Exp Allergy. 2006;36:475–482. doi: 10.1111/j.1365-2222.2006.02470.x. [DOI] [PubMed] [Google Scholar]
- Shen H, Lentsch AB. Progressive dysregulation of transcription factors NF-kappa B and STAT1 in prostate cancer cells causes proangiogenic production of CXC chemokines. Am J Physiol Cell Physiol. 2004;286:C840–C847. doi: 10.1152/ajpcell.00335.2003. [DOI] [PubMed] [Google Scholar]
- Shimada Y, Takehara K, Sato S. Both Th2 and Th1 chemokines (TARC/CCL17, MDC/CCL22, and Mig/CXCL9) are elevated in sera from patients with atopic dermatitis. J Dermatol Sci. 2004;34:201–208. doi: 10.1016/j.jdermsci.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Sundararaj KP, Samuvel DJ, Li Y, Nareika A, Slate EH, Sanders JJ, et al. Simvastatin suppresses LPS-induced MMP-1 expression in U937 mononuclear cells by inhibiting protein isoprenylation-mediated ERK activation. J Leukoc Biol. 2008;84:1120–1129. doi: 10.1189/jlb.0108064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi HK, Mori S, Iwagaki H, Yoshino T, Tanaka N, Weitz-Schmidt G, et al. Differential effect of LFA703, pravastatin, and fluvastatin on production of IL-18 and expression of ICAM-1 and CD40 in human monocytes. J Leukoc Biol. 2005;77:400–407. doi: 10.1189/jlb.0904510. [DOI] [PubMed] [Google Scholar]
- Turner NA, Midgley L, O'Regan DJ, Porter KE. Comparison of the efficacies of five different statins on inhibition of human saphenous vein smooth muscle cell proliferation and invasion. J Cardiovasc Pharmacol. 2007;50:458–461. doi: 10.1097/FJC.0b013e318123767f. [DOI] [PubMed] [Google Scholar]
- Van Linthout S, Riad A, Dhayat N, Spillmann F, Du J, Dhayat S, et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia. 2007;50:1977–1986. doi: 10.1007/s00125-007-0719-8. [DOI] [PubMed] [Google Scholar]
- Wagner AH, Schwabe O, Hecker M. Atorvastatin inhibition of cytokine-inducible nitric oxide synthase expression in native endothelial cells in situ. Br J Pharmacol. 2002;136:143–149. doi: 10.1038/sj.bjp.0704678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HR, Li JJ, Huang CX, Jiang H. Fluvastatin inhibits the expression of tumor necrosis factor-alpha and activation of nuclear factor-kappaB in human endothelial cells stimulated by C-reactive protein. Clin Chim Acta. 2005;353:53–60. doi: 10.1016/j.cccn.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Yang BC, Wang YS, Liu HS, Lin SJ. Ras signaling is involved in the expression of Fas-L in glioma. Lab Invest. 2000;80:529–537. doi: 10.1038/labinvest.3780058. [DOI] [PubMed] [Google Scholar]
- Yang J, Li XP, Zhao SP, Li J, Li JD, Xie XM. The effect of different doses of fluvastatin on inflammatory markers in the early phase of acute coronary syndrome. Clin Chim Acta. 2006;368:183–187. doi: 10.1016/j.cca.2005.12.029. [DOI] [PubMed] [Google Scholar]
- Yoshie O, Imai T, Nomiyama H. Chemokines in immunity. Adv Immunol. 2001;78:57–110. doi: 10.1016/s0065-2776(01)78002-9. [DOI] [PubMed] [Google Scholar]
- Zamvil SS, Steinman L. Cholesterol-lowering statins possess anti-inflammatory activity that might be useful for treatment of MS. Neurology. 2002;59:970–971. doi: 10.1212/wnl.59.7.970. [DOI] [PubMed] [Google Scholar]
- Zelvyte I, Dominaitiene R, Crisby M, Janciauskiene S. Modulation of inflammatory mediators and PPARgamma and NFkappaB expression by pravastatin in response to lipoproteins in human monocytes in vitro. Pharmacol Res. 2002;45:147–154. doi: 10.1006/phrs.2001.0922. [DOI] [PubMed] [Google Scholar]