

Abstract
Background and purpose:
Mildronate [3-(2,2,2-trimethylhydrazinium) propionate] is an anti-ischaemic drug whose mechanism of action is based on its inhibition of L-carnitine biosynthesis and uptake. As L-carnitine plays a pivotal role in the balanced metabolism of fatty acids and carbohydrates, this study was carried out to investigate whether long-term mildronate treatment could influence glucose levels and prevent diabetic complications in an experimental model of type 2 diabetes in Goto-Kakizaki (GK) rats.
Experimental approach:
GK rats were treated orally with mildronate at doses of 100 and 200 mg·kg−1 daily for 8 weeks. Plasma metabolites reflecting glucose and lipids, as well as fructosamine and β-hydroxybutyrate, were assessed. L-carnitine concentrations were measured by ultra performance liquid chromatography with tandem mass spectrometry. An isolated rat heart ischaemia-reperfusion model was used to investigate possible cardioprotective effects. Pain sensitivity was measured with a tail-flick latency test.
Key results:
Mildronate treatment significantly decreased L-carnitine concentrations in rat plasma and gradually decreased both the fed- and fasted-state blood glucose. Mildronate strongly inhibited fructosamine accumulation and loss of pain sensitivity and also ameliorated the enhanced contractile responsiveness of GK rat aortic rings to phenylephrine. In addition, in mildronate-treated hearts, the necrosis zone following coronary occlusion was significantly decreased by 30%.
Conclusions and implications:
These results demonstrate for the first time that in GK rats, an experimental model of type 2 diabetes, mildronate decreased L-carnitine contents and exhibited cardioprotective effects, decreased blood glucose concentrations and prevented the loss of pain sensitivity. These findings indicate that mildronate treatment could be beneficial in diabetes patients with cardiovascular problems.
Keywords: mildronate, Goto-Kakizaki rat, diabetes, L-carnitine, glucose, pain sensitivity, myocardial infarction
Introduction
Diabetes mellitus is a widespread metabolic disease characterized by hyperglycaemia and associated with severe complications, including cardiovascular disease, dyslipidaemia and increased predisposition to peripheral neuropathy. Risk of cardiovascular disease is considerably increased in diabetic patients and novel cardioprotective strategies are being sought that could target both diabetic and cardiovascular outcomes (Taegtmeyer et al., 2002; Choy et al., 2008). Among others, L-carnitine (3-hydroxy-4-N-trimethylaminobutyrate) was shown to play a pivotal role in both fatty acid and carbohydrate metabolism (Stephens et al., 2007). In addition to being an important cofactor in the L-carnitine-mediated transport of long-chain fatty acids into the mitochondrial matrix, L-carnitine as a pharmacological agent enhances the glycolytic processing of carbohydrates (Foster, 2004; Mingrone, 2004; Stephens et al., 2007). L-carnitine was shown to increase pyruvate dehydrogenase complex activity in human mitochondria and this effect was related to the modulation of the intramitochondrial acetyl CoA/CoA ratio (Uziel et al., 1988). L-carnitine also stimulated glucose oxidation in intact fatty acid-perfused rat heart (Broderick et al., 1992). Moreover, L-carnitine supplementation is believed to be beneficial for diabetic patients (Mingrone, 2004). More recently, systemic L-carnitine deficiency caused severe hypoglycaemia in mice (Kuwajima et al., 2007). Similarly, after treatment with sodium pivalate, glucose oxidation was increased in L-carnitine-deficient hearts (Broderick, 2006). Thus, there is experimental evidence that the stimulation of glucose metabolism might be regulated differentially by the availability of L-carnitine under certain physiological or pathological conditions and overall metabolic states.
Mildronate (3-(2,2,2-trimethylhydrazinium) propionate) is a cardioprotective drug whose mechanism of action is based on inhibition of the availability of L-carnitine (Dambrova et al., 2002). Mildronate has been used in combination therapy for post-infarction chronic heart failure in patients with type 2 diabetes mellitus (Statsenko et al., 2007). The cardioprotective effect of mildronate was shown experimentally in vitro in an isolated rat heart infarction model (Liepinsh et al., 2006), and in vivo in a rat heart infarction model (Sesti et al., 2006). In addition, mildronate treatment also improved functional parameters of the heart in rats with heart failure induced by myocardial infarction (Hayashi et al., 2000). Recently, we reported that mildronate decreased L-carnitine and glucose concentrations in blood plasma, stimulated glucose uptake and glucose metabolism-related gene expression in mouse heart (Liepinsh et al., 2008). It was concluded that mildronate induced a decrease in L-carnitine content, leading to stimulated glucose uptake (Liepinsh et al., 2008).
The present study was carried out to investigate whether the decrease in L-carnitine induced by long-term mildronate treatment could influence glucose levels and prevent diabetic complications in the experimental model of type 2 diabetes in Goto-Kakizaki (GK) rats. These rats exhibit mild hyperglycaemia, impaired glucose-induced insulin secretion, marked glucose intolerance, hepatic glucose overproduction and peripheral insulin resistance (Bisbis et al., 1993; Gauguier et al., 1996). Despite the mild hyperglycaemia, both functional and morphological manifestations of several diabetes complications have been demonstrated in this model (Suzuki et al., 1990; Bisbis et al., 1993; Ueta et al., 2005; Azuma et al., 2006). The present study aimed to evaluate the effects of long-term mildronate treatment in this model of type 2 diabetes by measuring the fed- and fasted-state blood glucose levels and insulin concentrations. To test the effect of mildronate treatment on the fatty acid oxidation rate, we measured plasma β-hydroxybutyric acid (β-ketone) and free fatty acid (FFA) concentrations. An isolated GK rat heart ischaemia-reperfusion model and the constriction of aortic rings were used to investigate the cardioprotective effects of mildronate in GK rats after 8 weeks of treatment. In addition, a tail-flick test was performed to determine the effect of mildronate on the loss of thermal pain sensitivity as a measure of the functional manifestation of neurological complications of diabetes.
Methods
Animals and treatment
All animal care and experimental procedures were carried out in accordance with the guidelines of the European Community, local laws and policies and were approved by the Latvian Animal Protection Ethical Committee, Food and Veterinary Service, Riga, Latvia. Thirty male GK (10 weeks old, Charles River Laboratories) and 10 age-matched Wistar rats weighing 200–250 g were housed under standard conditions (21–23°C, 12-h light–dark cycle) with unlimited access to food (R3 diet, Lactamin AB, Sweden) and water. Wistar rats were used as the non-diabetic control to evaluate the progression of diabetes in GK rats.
Rats were adapted to local conditions for 2 weeks before the start of treatment. Mildronate (100 and 200 mg·kg−1) was administered p.o. daily for 8 weeks. Control rats received water. Rats were decapitated 24 h after the last administration. Blood was collected into EDTA/diamide- and protease inhibitor (phenylmethylsulphonylfluoride, pepstatin, leupeptin, aprotinin)-containing tubes and centrifuged. The obtained plasma was stored frozen (−80°C) until analysis. The hearts were excised, washed in cold PBS and used for isolated heart experiments.
Determination of L-carnitine, γ-butyrobetaine and mildronate by ultra performance liquid chromatography with tandem mass spectrometry
The determination of L-carnitine, γ-butyrobetaine (GBB) and mildronate concentrations in blood plasma samples was performed by ultra performance liquid chromatography-tandem mass spectrometry in positive ion electrospray mode as described previously (Dambrova et al., 2008). In brief, sample preparation was performed by deproteinization with a methanol/acetonitrile mixture. Ultra performance liquid chromatography was carried out using a Waters Acquity HILIC BEH 1.7 µm 2.1 × 100 mm column. Chromatographic separation was performed in a 10 mM ammonium acetate (pH 4) and acetonitrile gradient at a flow rate of 0.2 mL·min−1. Tandem mass spectrometry analysis was performed on a Micromass Quatro Micro tandem mass spectrometer in positive ion electrospray mode using multiple reaction monitoring mode with the precursor-to-product ion transition m/z 162.4→m/z 103.0 for L-carnitine, m/z 146.3→m/z 87.9 for GBB and m/z 147.3 → m/z 58.4 for mildronate. MassLynx 4.1. software with a QuanLynx 4.1. module (Waters) was used for data acquisition and processing.
Lipid profile, blood glucose, insulin concentrations and oral glucose tolerance test
Lipid profile parameters [FFA, low-density lipoprotein (LDL) and high-density lipoprotein (HDL)-cholesterol] were measured by IL Laboratories (USA) enzymatic kits. Blood glucose and β-hydroxybutyrate concentrations were measured using a MediSense Optium Xceed blood glucose meter and strips. Plasma insulin concentrations were determined with a RIA kit (Biotrend, Germany). Fructosamine concentrations were determined in sodium carbonate buffer (100 mM, pH 10.35) containing nitro blue tetrazolium chloride (0.25 mM). Changes in absorption at 550 nm were detected and 1-deoxy-1-morpholinofructose in bovine serum albumin (40 mg·mL−1) was used as a standard.
GK rats were subjected to fasting overnight, and then the oral glucose tolerance test was performed. For oral glucose tolerance test experiments, rats were given a glucose solution (2 g·kg−1 of body weight) orally and then blood samples were drawn at −10 (10 min before test), 15, 30, 45, 60, 120 and 240 min.
Tail-flick test
Thermal pain sensitivity was determined as changes in reaction latency in control Wistar rats and GK rats, as well as mildronate-treated GK rats 8 weeks after the p.o. treatment with mildronate at doses of 100 and 200 mg·kg−1. The spinal tail-flick response to noxious thermal stimuli was assessed by a tail-flick apparatus (Model DS20, Ugo Basile, Italy). Briefly, the rat tail was placed in a groove that contained a slit under which was located a photoelectric cell. When the heat source of noxious stimulus was turned on, the heat focused on the tail, and the animal responded by flicking its tail out of the groove. Light then passed through the slit and activated the photocell which, in turn, stopped the recording timer. The cut-off time was fixed at 20 s in order to avoid damage to the tail.
Haemodynamic measurements in vivo
Twenty-four hours after the last drug administration, rats were placed in rodent restrainers on the heating pad (37°C). To monitor arterial blood pressure and heart rate, the rat tail was placed in specialized tail cuff with pulse transducer MLT125/R and the blood pressure was recorded using a Non Invasive Blood Pressure Controller ML125 connected to PowerLab8 /30 system (ADInstruments) and PC. The pulse was recorded continuously but blood pressure was sampled periodically for 30 min.
Isolated aortic ring experiments
The experiment was performed as described by Rosen and Wiernsperger (2006). After removal of the heart and lungs, the thoracic aorta was rapidly excised, immersed in oxygenated Krebs-Henseleit buffer (pH 7.4, 4°C) and cleaned of connective tissue. The aorta was cut into 3-mm-long aortic rings that were mounted between two stainless steel hooks in oxygenated Krebs-Henseleit buffer (pH 7.4, 37°C). The passive tension was fixed at 2 g. After a period of equilibration (1 h), the contractile function was tested twice by the application of 80 mM KCl. Then aortic rings were used to test the contractile response to phenylephrine (10−9–10−5 M), and after a submaximal pre-contraction with phenylephrine, a concentration–response curve to acetylcholine (10−9–10−6 M) was obtained as a measure of endothelial function. Contractile responses were evaluated as a percentage of maximal KCl contraction.
Isolated rat heart infarction study
The isolated rat heart experiment was performed essentially as described earlier (Liepinsh et al., 2006). Hearts were excised and retrogradely perfused via the aorta at a constant pressure with oxygenated Krebs-Henseleit buffer at 37°C. The heart rate, left ventricular end-diastolic pressure and left ventricular developed pressure (LVDP) were continuously recorded. Coronary flow was measured using an ultrasound flow detector (HSE) and the PowerLab8/30 system from ADInstruments. The hearts were perfused for 20 min to stabilize the haemodynamic functions and then coronary flow was occluded for 60 min by constricting threads through a plastic tube. Successful occlusion was confirmed by a coronary flow decrease of about 40%. Reperfusion was achieved by releasing the threads. At the end of the 150-min reperfusion period, the risk zone was delineated with 0.1% methylene blue. The hearts were then sectioned transversely from the apex to the base in five slices 2 mm in thickness and incubated in 1% triphenyltetrazolium chloride in phosphate buffer (pH 7.4, 37°C) for 10 min to stain viable tissue red and necrotic tissue white. Computerized planemetric analysis of Sony A700 photographs was performed using Image-Pro Plus 4.5.1 software to determine the area at risk and area of necrosis expressed as % of the left ventricle. The obtained values were then used to calculate the infarct size as a % of risk area according to the formula: Infarct size = (area of necrosis/area at risk) × 100%.
Statistical methods
Data are presented as means ± SEM. Statistically significant differences in the mean values were tested by anova and Tukey's test, using the GK control (untreated) rats as a reference group. The differences were considered significant when P < 0.05. The data were analysed using Graph Pad Prism 3.0 statistical software (Graph Pad, USA).
Materials
Mildronate (3-(2,2,2-trimethylhydrazinium) propionate dihydrate) was obtained from JSC Grindeks (Riga, Latvia). Methanol and acetonitrile were purchased from Merck (Darmstadt, Germany). Acetone, acetic acid, chloroform, diethyl ether, potassium dihydrogen phosphate, sodium chloride, calcium chloride, potassium chloride, potassium hydrogen orthophosphate, EDTA, L-carnitine and ammonium acetate were purchased from Acros Organics (Geel, Belgium). Phenylmethylsulphonylfluoride, diamide, GBB, pepstatin, leupeptin, aprotinin, Tris, 1-deoxy-1-morpholinofructose, triphenyltetrazolium chloride, methylene blue, phenylephrine, acetylcholine, sodium carbonate and bovine serum albumin were obtained from Sigma (USA).
Results
Plasma L-carnitine, GBB and mildronate concentrations
As seen in Table 1, the L-carnitine concentration in GK rat plasma was 25% lower than that in control (Wistar) rats. Mildronate treatment at doses of 100 and 200 mg·kg−1 for 8 weeks induced a significant decrease in L-carnitine concentration for 85% and 95% respectively (Table 1). The GBB plasma concentration was similar in Wistar and GK control animals, whereas mildronate treatment induced a fourfold increase in GBB concentration. At the end of the experiment, the mildronate concentration in treated GK rat plasma reached similar concentrations for both doses (Table 1).
Table 1.
Effects of mildronate treatment for 8 weeks on some biochemical and diabetes markers in blood plasma
| Wistar | GK control | GK+M100 | GK+M200 | |
|---|---|---|---|---|
| Fed glucose (mM) | 6.0 ± 0.17* | 12.7 ± 1.2 | 10.8 ± 0.68 | 9.5 ± 0.63* |
| Fructosamine (mM) | 0.41 ± 0.05* | 0.68 ± 0.08 | 0.49 ± 0.06* | 0.45 ± 0.06* |
| Insulin (ng·mL−1) | 4.9 ± 0.3* | 2.1 ± 0.3 | 1.7 ± 0.2 | 2.1 ± 0.3 |
| HDL cholesterol (mg·mL−1) | 26.9 ± 0.98 | 29.1 ± 1.5 | 32.9 ± 1.3* | 32.7 ± 0.98* |
| LDL cholesterol (mg·mL−1) | 35.4 ± 1.5* | 39.5 ± 1.3 | 41.5 ± 2.2 | 42.9 ± 2.7 |
| FFA (mM) | 0.61 ± 0.07 | 0.54 ± 0.09 | 0.53 ± 0.08 | 0.58 ± 0.07 |
| Carnitine (µM) | 46.8 ± 5.1* | 30.5 ± 1.7 | 4.5 ± 1.0* | 1.7 ± 0.2* |
| GBB (µM) | 1.7 ± 0.2 | 1.4 ± 0.1 | 6.4 ± 0.3* | 5.5 ± 0.3* |
| Mildronate (µM) | – | – | 41.9 ± 7.2 | 45.3 ± 1.0 |
Treatment for 8 weeks with mildronate (M, 100 or 200 mg·kg−1) induced changes in fed glucose, insulin, FFA, LDL-cholesterol, HDL-cholesterol and fructosamine concentrations in rat plasma. Each value represents the mean ± SEM, n= 8–10 in each group.
GK, Goto-Kakizaki; FFA, free fatty acid; LDL, low-density lipoprotein; HDL, high-density lipoprotein; GBB, γ-butyrobetaine.
Significantly different from GK control group (Tukey's test P < 0.05).
Blood glucose, fructosamine and insulin concentrations
At the beginning of the experiment (rats 12 weeks of age), the average glucose concentration in fasted control GK rat blood plasma was higher than that in Wistar rats (Figure 1). After 4 weeks, the glucose concentration in control GK rats remained unchanged, but after 8 weeks it decreased slightly. In Wistar rats, after 4 and 8 weeks, the fasted plasma glucose levels remained low. As seen in Figure 1, mildronate treatment lowered the fasted blood glucose level in GK rats. Thus, after 4 weeks of treatment, a statistically significant blood glucose lowering effect was observed at a dose of 200 mg·kg−1 (Figure 1). As expected, at the end of the experiment, the blood glucose concentration in fed GK control rats was significantly higher compared with Wistar rats (Table 1). Furthermore, rats treated with mildronate at doses of 100 and 200 mg·kg−1 exhibited dose-dependent decreases in the fed-stage blood glucose.
Figure 1.
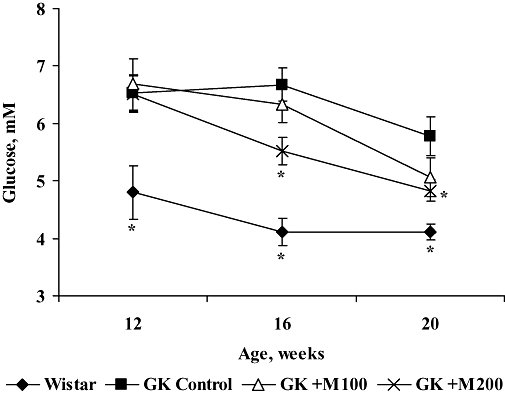
Effect of 4 and 8 weeks of treatment with mildronate (100 and 200 mg·kg−1) on blood glucose concentrations in fasted Goto-Kakizaki (GK) rats. Values shown are means ± SEM from at least eight animals. *Significantly different from GK control group (Tukey's test P < 0.05).
As seen in Table 1, the fructosamine concentration in control GK rats was significantly higher than that in Wistar rats. In turn, the 8-week mildronate treatment, at either dose, significantly reduced the fructosamine level in GK rat blood to values close to those observed in Wistar rats (Table 1). In contrast, at the end of experiment, the insulin level in control GK rats was less than half that in Wistar rats, and mildronate treatment did not induce any significant changes (Table 1).
To evaluate the effect of mildronate on impaired glucose tolerance in GK rats, the glucose tolerance test was performed on the day after the final drug treatment. When 2 g·kg−1 of glucose was used as the oral load in control GK and Wistar rats after overnight fasting, plasma glucose levels at all time points in GK control rats were significantly higher than those in Wistar control rats (data not shown). Mildronate treatment did not reduce plasma glucose and insulin levels at the measured time points in the oral glucose tolerance test.
Plasma lipid and β-hydroxybutyrate levels
To characterize the plasma lipid levels, the concentrations of FFA, HDL-cholesterol and LDL-cholesterol were determined. The lipid profile of Wistar and GK rats is shown in Table 1. As seen in the Table, mildronate treatment significantly increased the HDL-cholesterol concentration in GK rats, but did not influence the other parameters.
In the beginning of the experiment, the β-hydroxybutyrate concentration in Wistar and GK rats was similar (Figure 2). In Wistar rats, the β-hydroxybutyrate concentration did not change during the course of the experiment. However, in control GK rats, it was significantly lower than that in Wistar rats after 4 and 8 weeks. Compared with control GK rats, mildronate treatment at a dose of 200 mg·kg−1 significantly decreased the β-hydroxybutyrate concentration in the group after 4 and 8 weeks of treatment.
Figure 2.
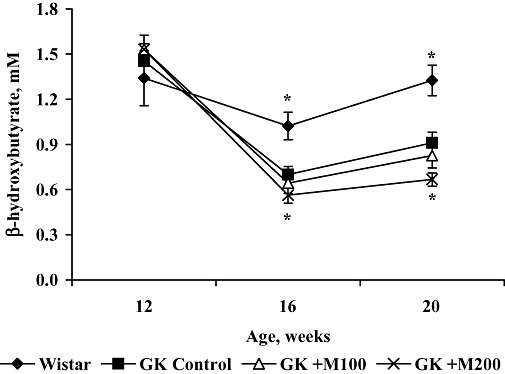
Effect of 4 and 8 weeks of treatment with mildronate (100 and 200 mg·kg−1) on blood-hydroxybutyrate concentrations in fasted Goto-Kakizaki (GK) rats. Values shown are means ± SEM from at least eight animals. *Significantly different from GK control group (Tukey's test P < 0.05).
Peripheral pain perception
To measure the changes in GK rat peripheral thermal pain perception, the tail-flick test was performed at the end of the 8-week treatment. As seen in Figure 3, control GK rats exhibited a 40% longer tail-flick latency (12.6 s) than control Wistar rats (7.2 s). This effect was significantly ameliorated (by 75%) by the administration of mildronate at a dose of 200 mg·kg−1. Mildronate treatment at dose of 100 mg·kg−1 did not induce significant changes in tail-flick latency time in GK rats.
Figure 3.
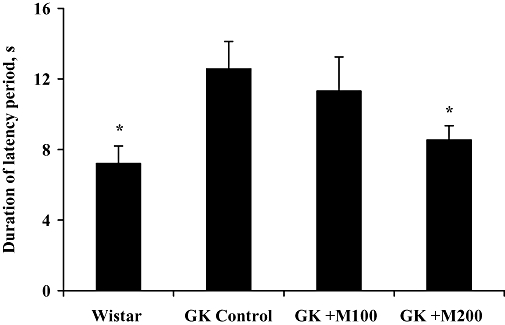
Effect of 8 weeks of treatment with mildronate (100 and 200 mg·kg−1) on the reaction time in the tail-flick test. Each value represents the mean ± SEM of at least eight animals. *Significantly different from Goto-Kakizaki (GK) control group (Tukey's test P < 0.05).
Rat weight and haemodynamic parameters in vivo
As seen in Table 2, the heart rate, systolic blood pressure was similar across the groups at the end of the experiment. The in vivo systolic blood pressure was almost the same, but the heart rate of GK rats was higher than that in Wistar rats; however, the results were not statistically different. Additionally, mildronate treatment did not affect the haemodynamic parameters (Table 2).
Table 2.
Rat weight and haemodynamic parameters in Wistar and Goto-Kakizaki (GK) rats
| Wistar | GK control | GK+M100 | GK+M200 | |
|---|---|---|---|---|
| Rat weight (g) | 376 ± 25 | 372 ± 8 | 378 ± 8 | 371 ± 42 |
| Systolic blood pressure (mmHg) | 117 ± 4 | 121 ± 2 | 114 ± 3 | 123 ± 4 |
| Heart rate (bpm) | 369 ± 9 | 417 ± 8 | 391 ± 23 | 393 ± 11 |
Effects of mildronate treatment on Wistar and GK rat weight, systolic arterial blood pressure and heart rate. Values are expressed as mean ± SEM, n= 8–10 in each group.
Contractile responsiveness of rat-isolated aorta
To measure the contractile responsiveness of vascular tissue, the vasoconstrictive action of phenylephrine was tested in aortic rings. Aortic rings from diabetic GK control rats showed significantly higher sensitivity to phenylephrine, compared with the aortic rings from the Wistar rats. Accordingly, at phenylephrine concentrations of 0.01, 0.1 and 1 µM the effect was higher by 13%, 36% and 29% (P < 0.05) respectively. In contrast, mildronate treatment at dose of 200 mg·kg−1 for 8 weeks significantly prevented this hypersensitivity to phenylephrine, compared with that in GK controls, the effect of phenylephrine at concentrations of 0.01 and 0.1 µM being lower, at 7% and 20% respectively. The endothelium-mediated vascular relaxation of noradrenaline pre-contracted mesenteric arterial rings in response to acetylcholine was also tested. The arterial ring response to acetylcholine was similar in Wistar and control GK rats, and long-term mildronate treatment did not induce any changes (data not shown).
Effects in rat-isolated heart infarction model
In isovolumetrically contracting hearts, there were no differences in control Wistar and GK rat heart rates, peak left ventricular pressure, LV +dp/dt or LV −dp/dt. In contrast, coronary flow in GK rat hearts was significantly lower than that in Wistar rat hearts (Table 3). After 8 weeks of mildronate treatment at a dose of 200 mg·kg−1, a significant improvement in coronary flow in GK rat hearts was observed (Table 3).
Table 3.
Cardiovascular parameters in isolated hearts from Wistar and Goto-Kakizaki (GK) rats
| Wistar | GK control | GK+M100 | GK+M200 | |
|---|---|---|---|---|
| Heart index | 2.2 ± 0.3 | 2.2 ± 0.2 | 2.3 ± 0.4 | 2.4 ± 0.2 |
| HR (bpm) | 237 ± 7 | 213 ± 8 | 208 ± 17 | 256 ± 11 |
| LVP (mmHg) | 44 ± 5 | 52 ± 6 | 47 ± 5 | 47 ± 4 |
| CF (mL·min−1) | 7.1 ± 0.2* | 5.8 ± 0.3 | 6.3 ± 0.8 | 6.8 ± 0.2* |
| +dp/dt (mmHg·s−1) | 1365 ± 209 | 1602 ± 209 | 1490 ± 96 | 1456 ± 188 |
| −dp/dt (mmHg·s−1) | −945 ± 93 | −1032 ± 110 | −1015 ± 45 | −937 ± 86 |
Effects of mildronate treatment on heart index (heart weight/body weight), heart rate (HR), left ventricular pressure (LVP), coronary flow (CF) and maximal and minimal dp/dt in isolated hearts from Wistar and GK rats (untreated (GK control) and treated). Values are expressed as mean ± SEM, n= 8–10 in each group.
Significantly different from GK control group (Tukey's test P < 0.05).
The anti-infarction effect of mildronate was investigated in an isolated heart infarction model, using hearts from GK rats. During occlusion of left coronary artery, the coronary flow in all experimental groups was decreased for 40% (from 11 to 7 mL·min−1). Moreover, a drop of LVDP, from about 50 to 25 mmHg, was observed. The heart rate during the occlusion period did not change significantly. During reperfusion, coronary flow, LVDP, ±dp/dt values were restored to about 80% of control values. There were no significant differences between Wistar, GK control and mildronate-treated groups.
Values for the infarction area at risk were similar in hearts of all experimental groups, and the area at risk in all groups was about 55% of the area of the left ventricle (data not shown). The infarct size in control GK and Wistar rat hearts was similar. As seen in Figure 4, mildronate treatment at doses of 100 or 200 mg·kg−1 decreased the infarct size by 30% (P= 0.03) and 25% (P= 0.07) of the infarct size in the GK control group respectively.
Figure 4.
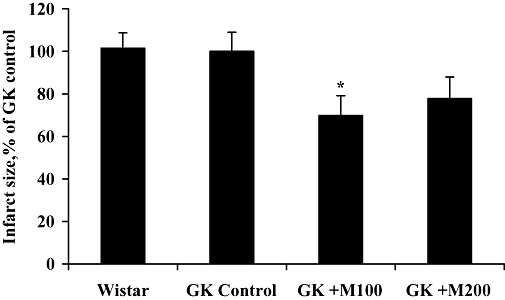
Effect of 8 weeks of treatment with mildronate (100 and 200 mg·kg−1) on infarct size in the rat-isolated heart infarction experiment. Each value represents the mean ± SEM of at least eight animals. *Significantly different from Goto-Kakizaki (GK) control group (Tukey's test P < 0.05).
Discussion
In the present study, the effects of mildronate on L-carnitine and blood glucose concentrations and diabetes-related complications were examined in an experimental model of type 2 diabetes for the first time. The main finding of our study was that the decreased availability of L-carnitine was accompanied by dose-dependent beneficial effects on fed and fasted blood glucose concentrations in diabetic GK rats. Furthermore, a protective effect of long-term mildronate treatment was observed in cardiovascular and pain sensitivity assays.
Our study demonstrated that long-term treatment with mildronate dose- and time-dependently reduced blood glucose in both the fasted and fed state without increasing insulin levels (Figure 1, Table 1). In addition, the concentration of fructosamine or glycated serum protein was reduced to the same level that was observed in non-diabetic Wistar rats after treatment with mildronate (Table 1), which indicates the effectiveness of the treatment of the diabetes. Our results are in line with previous studies that showed the effects of mildronate on insulin-stimulated glucose metabolism in non-diabetic mice (Liepinsh et al., 2008). Thus far, the primary mechanism of pharmacological action of mildronate has been discussed mainly on the grounds of decreased L-carnitine availability and the partial inhibition of fatty acid oxidation (Spaniol et al., 2001; Dambrova et al., 2002; Liepinsh et al., 2006). However, more recent data provide direct evidence that mildronate also acts as a metabolic modulator that shifts the energy metabolism substrate preference to glucose in the heart (Liepinsh et al., 2008). In the present study, we measured the concentration of β-hydroxybutyrate, the decreased concentration of which could also be considered a marker of overall fatty acid oxidation (van Knegsel et al., 2005). The concentration of β-hydroxybutyrate in control GK rats was significantly lower than that in Wistar rats, and it was further decreased after prolonged treatment with mildronate at a dose of 200 mg·kg−1 (Figure 2). Thus, the beneficial effects of mildronate treatment in GK rats could be explained by both the partial inhibition of fatty acid oxidation and an increase in glucose metabolism.
In the present study, both doses of mildronate significantly reduced L-carnitine concentrations in the plasma of diabetic GK rats. After 8 weeks of treatment, a 6–18 fold decrease of L-carnitine plasma concentrations was observed (Table 1). In spite of previous reports (Zaugg et al., 2003; Broderick, 2006) suggesting severe complications in the case of L-carnitine depletion, we did not observe any problems with motor function (Rota-rod test, data not shown) or signs of cardiomyopathy in GK rats after treatment with higher doses of mildronate (Tables 2 and 3). On the contrary, a significant cardioprotective effect of mildronate was observed. As seen in Figure 4, mildronate treatment at doses of 100 and 200 mg·kg−1 decreased the infarct size by 30% (P= 0.03) and 25% (P= 0.07) respectively, while it did not affect the workload. If we compare the effects of mildronate in diabetic GK and non-diabetic Wistar rats reported before (Liepinsh et al., 2006), a comparable cardioprotective effect is observed. Interestingly, mildronate at a dose of 200 mg·kg−1 induced significantly better effects on blood glucose concentrations and diabetic complications, but its protective effect on myocardial infarction in GK rats was less pronounced than at 100 mg·kg−1.
The effect of long-term treatment with mildronate on the functional manifestation of diabetes complications was also assessed. Several studies have demonstrated that impairment of endothelium-dependent relaxation is associated with diabetes (Azuma et al., 2006; Rosen and Wiernsperger, 2006). Thus, endothelium-dependent vasodilatation is impaired in diabetic rats, and there is also some evidence that aortic rings from diabetic rats are more sensitive to the vasoconstrictive action of phenylephrine (Rosen and Wiernsperger, 2006). In our study, the contractile responsiveness to phenylephrine was significantly enhanced in the aortas of GK rats, and this hypersensitivity was normalized by mildronate at a dose of 200 mg·kg−1. Instead, differences in the response to acetylcholine between Wistar and GK rats were not observed, and mildronate treatment did not induce any changes. The functional impairment of coronary arteries characterized by significantly lower coronary flow was observed in isolated hearts from GK rats, in comparison with those from Wistar rats, but after mildronate treatment at a dose of 200 mg·kg−1, the coronary flow significantly increased (Table 3). These findings, together with increased HDL levels, may indicate that long-term treatment with mildronate prevents endothelial dysfunction in this experimental model of type 2 diabetes.
In previous reports, a peripheral neuropathy characterized by abnormality in the thermal pain threshold of diabetic GK rats was associated with the development of diabetes (Suzuki et al., 1990; Ueta et al., 2005). The loss of thermal response is dependent on the severity and duration of hyperglycaemia, and is also a characteristic diabetic complication in patients (Ueta et al., 2005). Long-term mildronate administration both controlled hyperglycaemia and prevented the diabetic neuropathy (Figure 3). It is very unlikely that mildronate directly caused neuronal sensitization, as there was no effect on the tail-flick response after a single administration of mildronate (data not shown). Hence, our results indicate that mildronate improves blood glucose control, and might also be beneficial for the inhibition of neuropathy in type 2 diabetes.
The beneficial effects of L-carnitine supplementation in animal models of diabetes have been demonstrated (Malone et al., 1999; Malone et al., 2006; Sena et al., 2008); however, the effects of L-carnitine and acetyl L-carnitine in healthy subjects or in type 2 diabetic patients are controversial (Mingrone, 2004; Gonzalez-Ortiz et al., 2008). The suggested mechanisms of pharmacological action of L-carnitine are increased pyruvate dehydrogenase complex activity, a regulation of acetyl- and acyl-cellular trafficking, and changes in the ascetyl CoA/CoA ratio and expression of key enzymes in glycolysis and gluconeogenesis (Uziel et al., 1988; Mingrone, 2004). We have recently shown that decreased L-carnitine concentrations induced by mildronate treatment brings about an increase in glucose transporter 4, insulin receptor, hexokinase II and pyruvate dehydrogenase complex component protein expression in heart tissue (Liepinsh et al., 2008). Thus, taking into account the results of the present study, we could conclude that both mildronate-induced decrease in L-carnitine availability and pharmacological supplementation with L-carnitine can stimulate glucose metabolism, and that both of these treatments could be effective in diabetes treatment. Altogether, we conclude that energy metabolism could be regulated differentially by the availability of L-carnitine under certain physiological or pathological conditions and the overall metabolic state.
In conclusion, using GK rats as an animal model of type 2 diabetes, we demonstrated that oral treatment with mildronate improved blood glucose levels, protected diabetic rat heart against ischaemia-reperfusion injury and prevented diabetes-related endothelial dysfunction and loss of pain sensitivity. The overall beneficial effects of mildronate on type 2 diabetes may be related to the metabolic actions of the drug mediating a decrease in L-carnitine availability.
Acknowledgments
We thank JSC Grindeks for financial support. Supported by European Social Foundation (ESF), Latvian State Research Program ‘New medicines and biocorrection tools: design, transport forms and mechanisms of action’.
Glossary
Abbreviations:
- FFA
free fatty acid
- GBB
γ-butyrobetaine
- GK
Goto-Kakizaki
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- LV dp/dt
first time derivative of left ventricular pressure
References
- Azuma K, Toyofuku Y, Iesaki T, Otsuka A, Tanaka A, Mita T, et al. Acarbose, an alpha-glucosidase inhibitor, improves endothelial dysfunction in Goto-Kakizaki rats exhibiting repetitive blood glucose fluctuation. Biochem Biophys Res Commun. 2006;345:688–693. doi: 10.1016/j.bbrc.2006.04.090. [DOI] [PubMed] [Google Scholar]
- Bisbis S, Bailbe D, Tormo MA, Picarel-Blanchot F, Derouet M, Simon J, et al. Insulin resistance in the GK rat: decreased receptor number but normal kinase activity in liver. Am J Physiol. 1993;265:E807–E813. doi: 10.1152/ajpendo.1993.265.5.E807. [DOI] [PubMed] [Google Scholar]
- Broderick TL. Hypocarnitinaemia induced by sodium pivalate in the rat is associated with left ventricular dysfunction and impaired energy metabolism. Drugs R D. 2006;7:153–161. doi: 10.2165/00126839-200607030-00002. [DOI] [PubMed] [Google Scholar]
- Broderick TL, Quinney HA, Lopaschuk GD. Carnitine stimulation of glucose oxidation in the fatty acid perfused isolated working rat heart. J Biol Chem. 1992;267:3758–3763. [PubMed] [Google Scholar]
- Choy CK, Rodgers JE, Nappi JM, Haines ST. Type 2 diabetes mellitus and heart failure. Pharmacotherapy. 2008;28:170–192. doi: 10.1592/phco.28.2.170. [DOI] [PubMed] [Google Scholar]
- Dambrova M, Cirule H, Svalbe B, Zvejniece L, Pugovichs O, Zorenko T, et al. Effect of inhibiting carnitine biosynthesis on male rat sexual performance. Physiol Behav. 2008;95:341–347. doi: 10.1016/j.physbeh.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Dambrova M, Liepinsh E, Kalvinsh I. Mildronate: cardioprotective action through carnitine-lowering effect. Trends Cardiovasc Med. 2002;12:275–279. doi: 10.1016/s1050-1738(02)00175-5. [DOI] [PubMed] [Google Scholar]
- Foster DW. The role of the carnitine system in human metabolism. Ann N Y Acad Sci. 2004;1033:1–16. doi: 10.1196/annals.1320.001. [DOI] [PubMed] [Google Scholar]
- Gauguier D, Froguel P, Parent V, Bernard C, Bihoreau MT, Portha B, et al. Chromosomal mapping of genetic loci associated with non-insulin dependent diabetes in the GK rat. Nat Genet. 1996;12:38–43. doi: 10.1038/ng0196-38. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Ortiz M, Hernandez-Gonzalez SO, Hernandez-Salazar E, Martinez-Abundis E. Effect of oral L-carnitine administration on insulin sensitivity and lipid profile in type 2 diabetes mellitus patients. Ann Nutr Metab. 2008;52:335–338. doi: 10.1159/000151488. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Kirimoto T, Asaka N, Nakano M, Tajima K, Miyake H, et al. Beneficial effects of MET-88, a gamma-butyrobetaine hydroxylase inhibitor in rats with heart failure following myocardial infarction. Eur J Pharmacol. 2000;395:217–224. doi: 10.1016/s0014-2999(00)00098-4. [DOI] [PubMed] [Google Scholar]
- Kuwajima M, Fujihara H, Sei H, Umehara A, Sei M, Tsuda TT, et al. Reduced carnitine level causes death from hypoglycemia: possible involvement of suppression of hypothalamic orexin expression during weaning period. Endocr J. 2007;54:911–925. doi: 10.1507/endocrj.k07-044. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Vilskersts R, Loca D, Kirjanova O, Pugovichs O, Kalvinsh I, et al. Mildronate, an inhibitor of carnitine biosynthesis, induces an increase in gamma-butyrobetaine contents and cardioprotection in isolated rat heart infarction. J Cardiovasc Pharmacol. 2006;48:314–319. doi: 10.1097/01.fjc.0000250077.07702.23. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Vilskersts R, Skapare E, Svalbe B, Kuka J, Cirule H, et al. Mildronate decreases carnitine availability and up-regulates glucose uptake and related gene expression in the mouse heart. Life Sci. 2008;83:613–619. doi: 10.1016/j.lfs.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Malone JI, Cuthbertson DD, Malone MA, Schocken DD. Cardio-protective effects of carnitine in streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2006;5:2–7. doi: 10.1186/1475-2840-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone JI, Schocken DD, Morrison AD, Gilbert-Barness E. Diabetic cardiomyopathy and carnitine deficiency. J Diabetes Complications. 1999;13:86–90. doi: 10.1016/s1056-8727(99)00039-2. [DOI] [PubMed] [Google Scholar]
- Mingrone G. Carnitine in type 2 diabetes. Ann N Y Acad Sci. 2004;1033:99–107. doi: 10.1196/annals.1320.009. [DOI] [PubMed] [Google Scholar]
- Rosen P, Wiernsperger NF. Metformin delays the manifestation of diabetes and vascular dysfunction in Goto-Kakizaki rats by reduction of mitochondrial oxidative stress. Diabetes Metab Res Rev. 2006;22:323–330. doi: 10.1002/dmrr.623. [DOI] [PubMed] [Google Scholar]
- Sena CM, Nunes E, Louro T, Proenca T, Fernandes R, Boarder MR, et al. Effects of alpha-lipoic acid on endothelial function in aged diabetic and high-fat fed rats. Br J Pharmacol. 2008;153:894–906. doi: 10.1038/sj.bjp.0707474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti C, Simkhovich BZ, Kalvinsh I, Kloner RA. Mildronate, a novel fatty acid oxidation inhibitor and antianginal agent, reduces myocardial infarct size without affecting hemodynamics. J Cardiovasc Pharmacol. 2006;47:493–499. doi: 10.1097/01.fjc.0000211732.76668.d2. [DOI] [PubMed] [Google Scholar]
- Spaniol M, Brooks H, Auer L, Zimmermann A, Solioz M, Stieger B, et al. Development and characterization of an animal model of carnitine deficiency. Eur J Biochem. 2001;268:1876–1887. [PubMed] [Google Scholar]
- Statsenko ME, Belenkova SV, Sporova OE, Shilina NN. The use of mildronate in combined therapy of postinfarction chronic heart failure in patients with type 2 diabetes mellitus. Klin Med (Mosk) 2007;85:39–42. [PubMed] [Google Scholar]
- Stephens FB, Constantin-Teodosiu D, Greenhaff PL. New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. J Physiol. 2007;581:431–444. doi: 10.1113/jphysiol.2006.125799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Yen-Chung H, Toyota T, Goto Y, Hirata Y, Okada K. The significance of nerve sugar levels for the peripheral nerve impairment of spontaneously diabetic GK (Goto-Kakizaki) rats. Diabetes Res. 1990;14:21–25. [PubMed] [Google Scholar]
- Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: Part I: general concepts. Circulation. 2002;105:1727–1733. doi: 10.1161/01.cir.0000012466.50373.e8. [DOI] [PubMed] [Google Scholar]
- Ueta K, Ishihara T, Matsumoto Y, Oku A, Nawano M, Fujita T, et al. Long-term treatment with the Na+-glucose cotransporter inhibitor T-1095 causes sustained improvement in hyperglycemia and prevents diabetic neuropathy in Goto-Kakizaki Rats. Life Sci. 2005;76:2655–2668. doi: 10.1016/j.lfs.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Uziel G, Garavaglia B, Di DS. Carnitine stimulation of pyruvate dehydrogenase complex (PDHC) in isolated human skeletal muscle mitochondria. Muscle Nerve. 1988;11:720–724. doi: 10.1002/mus.880110708. [DOI] [PubMed] [Google Scholar]
- van Knegsel AT, van den Brand H, Dijkstra J, Tamminga S, Kemp B. Effect of dietary energy source on energy balance, production, metabolic disorders and reproduction in lactating dairy cattle. Reprod Nutr Dev. 2005;45:665–688. doi: 10.1051/rnd:2005059. [DOI] [PubMed] [Google Scholar]
- Zaugg CE, Spaniol M, Kaufmann P, Bellahcene M, Barbosa V, Tolnay M, et al. Myocardial function and energy metabolism in carnitine-deficient rats. Cell Mol Life Sci. 2003;60:767–775. doi: 10.1007/s00018-003-3011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]