

Abstract
Acute lung injury and the acute respiratory distress syndrome are common syndromes with a high mortality rate that affect both medical and surgical patients. Better understanding of the pathophysiology of acute lung injury and the acute respiratory distress syndrome and advances in supportive care and mechanical ventilation have led to improved clinical outcomes since the syndrome was first described in 1967. Although several promising pharmacological therapies, including surfactant, nitric oxide, glucocorticoids and lysofylline, have been studied in patients with acute lung injury and the acute respiratory distress syndrome, none of these pharmacological treatments reduced mortality. This article provides an overview of pharmacological therapies of acute lung injury and the acute respiratory distress syndrome tested in clinical trials and current recommendations for their use as well as a discussion of potential future pharmacological therapies including β2- adrenergic agonist therapy, keratinocyte growth factor, and activated protein C.
Keywords: acute lung injury, acute respiratory distress syndrome, pulmonary edema, pharmacologic therapies
The objective of this article is to review pharmacological therapeutic strategies for acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Following a brief overview of definitions, epidemiology, pathophysiology, and major nonpharmacological therapies of ALI/ARDS, we will focus on pharmacological therapies. Given the large number of therapeutic agents tested or implicated for therapy of ALI/ARDS, we attempted to organize this article according to the level of available evidence. The pharmacological therapeutic strategies are divided into 3 sections. The first 2 sections, pulmonary targeted (inhaled) and systemic pharmacological therapy, review all therapeutic agents for which human, phase I, II, or III clinical trials have been conducted. The third section focuses on potential therapies for which there are plausible pathophysiological rationale and preliminary experimental data but no clinical trials. All phase II and III clinical trials are summarized in a table that includes level [1,2] and grade [2,3] of evidence and resulting current recommendations.
We searched the MEDLINE database (since 1967), the Cochrane Library, and bibliographies of retrieved articles. The literature search was performed by both authors. In the first 2 sections of this article, we include all therapies for which clinical trials in patients with ALI/ARDS have been conducted and all important studies relevant to these therapies. In the third section, potential therapies, we discuss the treatments that are currently the most promising.
Definitions and Epidemiology
The acute respiratory distress syndrome is a clinical syndrome of acute inflammatory lung injury resulting from a variety of etiologies. The syndrome was first described by Ashbaugh and colleagues [4] in 1967 in a series of 12 patients, and the first set of criteria for diagnosis (acute respiratory distress, cyanosis refractory to oxygen therapy, decreased lung compliance, and diffuse alveolar infiltrates on the chest radiograph) was published in 1971 [5]. The definition was modified in 1988 by Murray and colleagues [6] using a 4-point lung injury score (LIS), predisposing condition, and presence or absence of nonpulmonary organ dysfunction in the assessment. Even though this expanded definition of ARDS allowed for more systematic identification of patients with ARDS, it still lacked specific criteria to exclude a diagnosis of cardiogenic pulmonary edema and was not predictive of outcome. In 1994, a new definition was recommended by the American–European Consensus Conference (AECC) on ARDS Committee [7]. The AECC criteria include (1) acute onset, (2) bilateral infiltrates on chest radiograph, (3) pulmonary artery wedge pressure ≤ 18 mm Hg or the absence of clinical evidence of left atrial hypertension, and (4) PaO2/FiO2 ratio ≤ 300 (defining ALI) or PaO2/FiO2 ratio ≤ 200 (defining ARDS as a more severe form of ALI).
The incidence of ARDS varies significantly between reports, which may reflect the different definitions of ARDS used in literature since 1971. An early estimate by the National Institutes of Health in 1972 was an incidence of 75/100 000 population per year (incidence of 150 000 cases per year in the United States) [8]. More recent studies showed lower incidences [9–11]; the most recent study, the first to use the 1994 AECC definition, showed an incidence of 17.9/100 000 for ALI and 13.5/100 000 for ARDS [12]. However, a large prospective epidemiologic study was recently completed in Seattle showing an incidence of ALI/ARDS of approximately 200 000 cases per year in the United States [13].
Pathogenesis
The initial (early) phase of ALI/ARDS is characterized by increased permeability of the endothelial and epithelial barriers of the lung, with accumulation of protein-rich and highly cellular edema fluid in the interstitium and alveoli [14]. The edema fluid contains hyaline membranes (which consist of different types of proteins, eg, fibrin, albumin, immunoglobulins, and matrix molecules) and a variety of inflammatory cells, predominantly neutrophils (Fig 1). The inflammatory cells, lung epithelial cells, and fibroblasts produce cytokines that amplify the inflammatory response. Procoagulant pathways are up-regulated and fibrinolysis is depressed, leading to extravascular and intravascular fibrin deposition [15,16]. Intravascular fibrin deposition and thrombi formation lead to vascular obstruction and alterations in the microvasculature. A pathological correlate of the early phase of ALI has been termed diffuse alveolar damage and consists of hyaline membranes plus at least 1 of the following: alveolar type I or endothelial cell necrosis, edema, interstitial fibrosis, or prominent alveolar cell type II proliferation [17–19].
Fig 1.
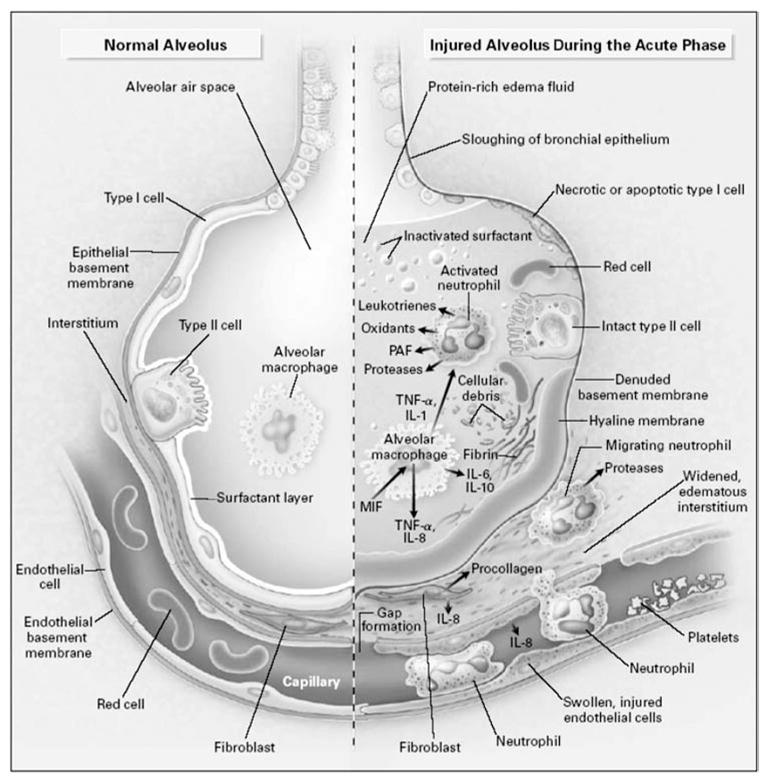
The normal alveolus (left-hand side) and the injured alveolus in the acute phase of acute lung injury and the acute respiratory distress syndrome. In the acute phase of the syndrome (right-hand side), there is sloughing of both the bronchial and alveolar epithelial cell, with the formation of protein-rich hyaline membranes on the denuded basement membrane. Neutrophils are shown adhering to the injured capillary endothelium and marginating through the interstitium into the air space, which is filled with protein-rich edema fluid. In the air space, an alveolar macrophage is secreting cytokines, interleukin (IL)-1, IL-6, IL-8, IL-10, and tumor necrosis factor (TNF)-α, which act locally to stimulate chemotaxis and activate neutrophils. IL-1 can also stimulate the production of extracellular matrix by fibroblasts. Neutrophils can release oxidants, proteases, leukotrienes, and other proinflammatory molecules, such as platelet-activating factor (PAF). A number of anti-inflammatory mediators are also present in the alveolar milieu, including IL-1-receptor antagonist, soluble TNF receptor, autoantibodies against IL-8, and cytokines such as IL-10 and IL-11 (not shown). The influx of protein-rich edema fluid into the alveolus has led to the inactivation of surfactant. MIF denotes macrophage inhibitory factor. Reprinted with the permission of the publisher [146]. Copyright © 2000 Massachusetts Medical Society. All rights reserved.
Some patients recover from ALI/ARDS during the first week of the disease, some die during that period of time, but some progress into a subacute (late) phase of ALI/ARDS that develops 5 to 7 days after onset. During the subacute phase of ALI/ARDS, the alveolar space becomes filled with mesenchymal cells, their products, and new blood vessels [20] (Fig 2). Pathologically, there is evidence of interstitial and alveolar fibrosis with proliferation of type II cells and destruction of portions of microcirculation in the lungs [20,21]. Finally, in some patients respiratory failure persists beyond 14 days, and this chronic phase I is characterized by extensive pulmonary fibrosis with obliteration of normal alveolar architecture and progressive development of emphysematous regions in the lung [20,21].
Fig 2.
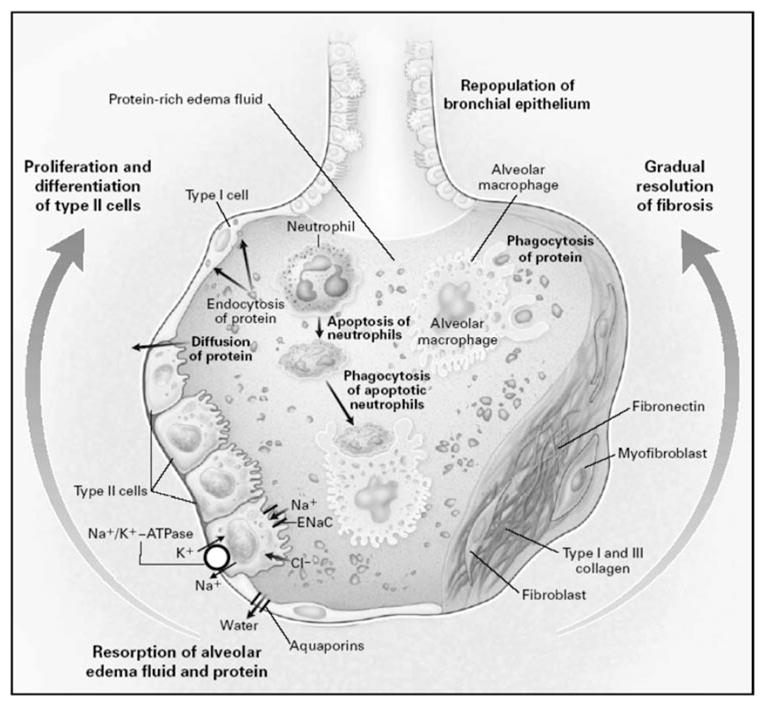
Mechanisms important in the resolution of acute lung injury and acute respiratory distress syndrome. On the left side of the alveolus, the alveolar epithelium is being repopulated by the proliferation and differentiation of alveolar type II cells. Resorption of alveolar edema fluid is shown at the base of the alveolus, with sodium and chloride being transported through the apical membrane of type II cells. Sodium is taken up by the epithelial sodium channel and through the apical membrane of type II cells by the sodium pump (Na+/K+-ATPase). The relevant pathways for chloride transport are unclear. Water is shown moving through water channels, the aquaporins, located primarily on type I cells. Some water may also cross by a paracellular route. Soluble protein is probably cleared primarily by paracellular diffusion and secondarily by endocytosis by alveolar epithelial cells. Macrophages remove insoluble protein and apoptotic neutrophils by phagocytosis. On the right side of the alveolus, the gradual remodeling and resolution of intra-alveolar and interstitial granulation tissue and fibrosis are shown. Reprinted with the permission of the publisher [146]. Copyright © 2000 Massachusetts Medical Society. All rights reserved.
Nonpharmacologic Therapy of ALI/ARDS
This section briefly reviews nonpharmacologic treatment of ALI/ARDS that are generally classified as supportive therapies.
Mechanical Ventilation
Optimal mechanical ventilation strategy in patients with ALI/ARDS has been one of the most controversial issues in management of ALI/ARDS since the syndrome was first described. Over the past 30 years some of the questions have been answered and some need further investigation.
Lower Tidal Volume (Lung-Protective) Versus Traditional Tidal Volume (Better Oxygenation) Ventilation Strategy
Early observational studies suggested a benefit of a low-tidal volume strategy [22,23]. However, early phase III clinical trials had conflicting results. Two small studies, one by Brochard et al [24] (116 patients) and one by Stewart et al [25] (120 patients) in 1998 showed no benefit, whereas a small study by Amato et al [26] (53 patients) the same year showed a decrease in 28-day mortality rate in the low–tidal volume group. This uncertainty was settled by a large, multiple-center, randomized controlled trial of 861 patients with ALI/ARDS conducted by the National Heart Lung and Blood Institute (NHLBI) ARDS network [27]. Patients were randomized to 12 mL/kg tidal volume versus 6 mL/kg tidal volume with a plateau pressure restriction (<30 cm H2O). The study showed a statistically significant relative reduction in mortality rate of 22% (P = .007) in the low–tidal volume group. Although low–tidal volume lung-protective strategy is the only intervention to decrease the mortality rate of ALI/ARDS and therefore should be the standard of care in the management of ALI/ARDS, it is only slowly being adopted in clinical practice. A recent study by Weinert et al [28] suggested that 2 years after the publication of the NHLBI ARDS network data, the mean prescribed tidal volume in academic medical centers was still 10 mL/kg. However, we anticipate that a lung-protective ventilatory strategy as used in the NHLBI ARDS network trials will be increasingly used.
High Positive End-Expiratory Pressure (PEEP) Versus Low PEEP Ventilation Strategy
With a lower level of PEEP, a greater fraction of the lung is nonaerated secondary to atelectasis/edema and shear forces generated by repeated opening and closing of alveoli, and excessive mechanical forces in the aerated part of the lung may exacerbate ventilator-induced lung injury. A higher level of PEEP (“open lung strategy”) may minimize this type of injury and improve oxygenation, allowing the use of a lower FiO2. Therefore, the NHLBI ARDS network has conducted a multiple-center, randomized controlled trial of 549 patients with ALI/ARDS using lung-protective ventilation strategy with a low tidal volume in all patients who were randomized to either higher or lower level of PEEP. The difference in the PEEP levels (8.3 ± 3.2 vs 13.2 ± 3.5 cm H2O) between the groups was statistically significant, but there was no statistical difference in survival [29]. Thus, a moderate level of PEEP seems to be optimal for the benefit of a lung-protective ventilatory strategy, although other clinical trials are testing the value of higher PEEP and recruitment maneuvers.
Other Strategies
Extracorporeal membrane oxygenation has not been shown to have benefit in prospective trials of patients with ALI/ARDS. Other strategies, such as high-frequency oscillatory ventilation, prone positioning, and recruitment maneuvers, will need further study, although recent data on prone positioning have not been encouraging [30–32]. Discussing these strategies is beyond the scope of this article.
Hemodynamic Management and Oxygen Delivery
One of the most controversial issues in the management of ALI/ARDS is optimal fluid management. There are 2 major competing concepts of fluid management in patients with ALI/ARDS.
A lower vascular filling pressure/volume status prioritizes the lungs (reduced pulmonary edema, improved compliance, better gas exchange). Early observational studies in humans showed that patients with ARDS who had negative fluid balance on day 14 [33], who gained less than 1 L of fluid after 36 hours [34], or who had a reduction in pulmonary capillary wedge pressure of at least 25% had improved survival [35]. In 2 single-center randomized controlled trials of restrictive fluid management, patients with pulmonary edema were randomized to fluid management guided by pulmonary wedge pressure measurements compared with extravascular lung water measurement using double-indication technique. The patients in the latter group had significantly lower lung water at 24 hours, as well as fewer ventilator days, but there was no survival benefit [34,36]. These studies also included patients with cardiogenic pulmonary edema, who are expected to benefit from aggressive diuresis.
A higher vascular filling pressure/volume status prioritizes cardiac output, organ perfusion (improved kidney function, etc), and oxygen delivery. The hypothesis that supranormal levels of oxygen delivery achieved by vigorous volume and red blood cell repletion and inotropic support would improve outcome has been addressed in several randomized controlled trials. In the early studies in critically ill patients and patients with septic shock, this strategy either showed no benefit [37,38] or actually conferred increased mortality rate [39]. However, a recent study by Rivers et al [40] of early goal-directed therapy in patients with severe sepsis and septic shock showed a mortality benefit in the intervention group.
The question of optimal fluid management and hemodynamic monitoring is being addressed by a large randomized trial conducted by the NHLBI ARDS network. The study enrolled 1000 patients randomized to either restricted or liberal fluid management guided by central venous or pulmonary artery catheter measurements. The results will be published in the near future.
Pulmonary Targeted Pharmacological Therapy
This section focuses on therapies that can be administered directly to the lung.
Surfactant Therapy
Surfactant, a lipid–protein complex secreted by type II cells in the lungs, lowers alveolar surface tension at the air–fluid interface of the alveoli and is critical to maintaining lung inflation at low transpulmonary pressures. The composition of natural surfactant is 80% phospholipids, 8% neutral lipids, and 12% proteins and it is quite constant in all mammals. Three unique surfactant-associated proteins (surfactant protein, SP) have been identified: SP-A, SP-B, and SP-C [41]. SP-B and SP-C proteins are small, lipophilic proteins that facilitate formation of surfactant monolayer. SP-A, a larger protein, has more complex function involving also regulation of secretory and reuptake pathways and may also have a role in lung host defenses [42].
Surfactant replacement therapy has been shown to be beneficial in respiratory distress syndrome of premature infants, a disorder caused by deficient production of surfactant by the immature lung [43–47]. In ALI/ARDS, the surfactant abnormalities are more complex. Apart from decreased surfactant production, other factors contribute to poor surfactant function: alterations in the surfactant phospholipids composition, loss of surfactant protein SP-B and SP-A content, and inhibition of surfactant function by leaked plasma proteins, oxygen radicals, and proteases [48–50]. The resulting increase in alveolar surface tension leads to atelectasis and decreased lung compliance [51] and may also increase edema formation [52].
Several studies in experimental ARDS models have documented improved pulmonary function following endobronchial surfactant administration [51]. Early case reports [53] demonstrated that administration of synthetic, porcine, or bovine surfactant into the lungs of patients with severe ARDS resulted in a sustained improvement in oxygenation. These case reports were followed by several phase I and II trials [54–56] that showed improvement in pulmonary function and a trend toward a reduction in mortality. In contrast to the encouraging results of these preliminary studies, a phase III, randomized, controlled trial of aerosolized, protein-free, artificial surfactant in 725 patients with sepsis-induced ARDS [57] failed to show benefit. Disappointing results of this trial were attributed to several issues: the type and dose of the surfactant and inadequate delivery to the alveoli. The protein-free artificial surfactant (Exosurf) is thought to be less effective than natural surfactants. Also, the dose might have been inadequate especially given the fact that only about 5% of the dose administered in this trial reached the alveoli. Therefore, further studies focused on the use of protein-containing surfactants with direct intrabronchial surfactant delivery. The surfactants used in these studies were natural bovine surfactants (Survanta, Alveofact) and in later studies artificial apoprotein-containing preparations (Surfaxin, Venticute) that could be more easily produced in larger quantities than the natural surfactants. Again, preliminary phase I and II studies were promising [58–62]. However, in the most recent study, a phase III, multiple-center, randomized, controlled trial of recombinant surfactant protein C–based surfactant in 448 patients [63] given intratracheally, the intervention did not change the duration of mechanical ventilation or survival. It did demonstrate transient improvement in oxygenation for the period of the therapy (24 hours) which was not apparent at 48 hours, suggesting that maybe therapy more than 24 hours was needed to confer mortality benefit.
The failure of the phase III trials again might be explained by the nature of the surfactant preparation: less efficacious recombinant surfactant, inadequate dose, preferential deposition in healthy lung units, inactivation in damaged alveoli, timing, and delivery method [64]. It is also possible that surfactant therapy, even if technically optimal, might simply not be effective in improving survival rate, because patients with ALI/ARDS usually die of sepsis and multiple organ failure. On the other hand, a recent NHLBI ARDS network low–tidal volume trial proved that changes in ventilator management alone can alter outcome [27]. Moreover, 1 recent pediatric phase III, randomized, controlled trial of intratracheally instilled Calfactant (modified natural lung surfactant containing apoprotein B) in 153 children (mean age 7 ± 6.4) showed a significant mortality benefit (19% in surfactant group vs 36% in placebo group, P= .03) [65], although there was no difference in the primary end point, ventilator-free days.
In conclusion, there is currently level 1 (grade A for all types of surfactant pooled; Tables 1 and 2) evidence that surfactant does not improve mortality rate and ventilator-free days in adults with ALI/ARDS. However, given the recent positive pediatric trial, future clinical trials to evaluate different surfactant preparations and regimens in adult and pediatric patients with ALI/ARDS may need to be carried out.
Table 1.
Summary of Clinical Studies of Surfactant in Acute Lung Injury (ALI)/Acute Respiratory Distress Syndrome (ARDS)
| Intervention | Author & Year | Study Design | Study Population & Number of Patients | Results |
|---|---|---|---|---|
| Exosurf with DPPC 40.5 mg/mL or 81 mg/mL or placebo Continuously aerosolized for up to 5 d. | Reines et al 1992 [54] | Prospective, randomized, controlled trial | Patients with sepsis-induced ARDS, N = 49 | 14-d mortality (all surfactant doses vs control): RR 0.61 (95% CI 0.3–1.24) |
| Porcine surfactant; single dose; 4 g in 50 mL Delivered via bronchoscope to each lobar bronchi | Spragg et al 1994 [55] | Case series | Patients with ARDS, N = 6 | Modest, transient improvement in gas exchange; no change in lung compliance |
| Exosurf with DPPC 13.5 mg/mL Continuously aerosolized, 175 mL every 4 h × 12 h/d or 175 mL × 24 h/d or placebo for up to 5 d | Weg et al 1994 [56] | Multiple-center, prospective, randomized, double-blind, placebo- controlled trial phase II study | Patients with ALI and sepsis <18 h, N = 51 | No difference in mortality at 30 d (RR 0.81; 95% CI 0.42–1.57) Dose-dependent trend in reduction of mortality (47% in placebo, 41% in 12 h/d group and 35% in 24 h/d group) |
| Exosurf with DPPC 13.5 mg/mL or placebo Continuously aerosolized 240 mL daily for up to 5 d | Anzueto et al 1996 [57] | Multiple-center, prospective, randomized, double-blind, placebo- controlled trial, phase III study | Patients with sepsis-induced ARDS, N = 725 | Mortality at 30 d in both groups was 40% (RR 1.01; 95% CI 0.84–1.20) No difference in length of mechanical ventilation, did not improve oxygenation |
| Survanta 50 mg/kg × 8 doses or 100 mg/kg × 4 doses or 100 mg/kg × 8 doses or standard therapy Delivered intratracheally | Gregory et al 1997 [62] | Multiple-center, prospective, randomized trial, not blinded, phase II study | Patients with ARDS <48 h, N = 51 | Mortality in the Survanta 100 mg/kg × 4 doses 18.8% vs control group 43.8% (P = .075) |
| Surfaxin: 3 dosing regimens Administered via bronchoscope to each of the 19 segments | Wiswell et al 1999 [60] | Case series, phase I study | Patients with ARDS, N = 12 | Decrease FiO2 (0.8–052) and PEEP (10.3-7.6) at 96 h |
| Alveofact 300 mg/kg × 1 treatment; delivered via bronchoscope into segmental bronchi within 45 min | Walmrath et al 1996 [58], 2002 [59] | Case series | Patients with severe ARDS and septic shock APAPCHE II 33.2 ± 1.3, N = 10, N = 12 | Improved PaO2/FiO2 (109–210) within 12 h Overall mortality rate 44.4% lower than calculated risk of death given the APACHE II (74 ± 3.5%) |
| Venticute 1 mL/kg up to 4 doses/24 h or 0.5 mL/kg up to 4 doses/24 h or standard therapy | Spragg et al 2003 [61] | Multiple-center, randomized, controlled trial, phase I/II study | Patients with ARDS <48 h, N = 40 | No significant difference in 28-d mortality rate or ventilator-free days |
| Venticute 1 mL/kg up to 4 doses/24 h or standard therapy; intratracheal instillation | Spragg et al 2004 [63] | Multiple-center, randomized, double-blind controlled trial, phase III | Patients with ARDS, N = 448 | Mortality rate at 28 d 36% in surfactant group vs 32% in placebo (P = .54); no difference |
| (a) North American study | 2 independent trials, published as pooled data | ARDS <48 h, N = 221 | Improvement of gas exchange during the 24-h period in surfactant group | |
| (b) European/African study | ARDS <72 h, N = 227 |
DPPC – dipalmitoylphosphatidylcholine; RR – relative risk; CI – confidence interval; PEEP – positive end-expiratory pressure; APACHE –Acute Physiology and Chronic Health Evaluation.
Surfactant preparations used in the clinical trials:
Exosurf: artificial surfactant (lipid complex only, no protein)
Survanta: modified natural bovine surfactant (lipid-protein complex, contains apoprotein)
Alveofact: natural bovine surfactant (lipid-protein complex)
Surfaxin: KL4-surfactant; artificial preparation containing a synthetic 21-amino-acid peptide (KL4-peptide)
Venticute: recombinant surfactant protein C-based surfactant; containing 1 mg recombinant surfactant protein C and 50 mg phospholipid per milliliter
Table 2.
Summary of Pharmacologic Therapies With the Level of Evidence and Recommendations
| Intervention | End Points | Highest Level of Evidencea | Recommendation | Grade of Recommendationb |
|---|---|---|---|---|
| Surfactant: pooled data | Mortality | I | No | A |
| Ventilator-free days | ||||
| Surfactant: protein-free surfactants | Mortality | I | No | B |
| Ventilator-free days | ||||
| Surfactant: protein-containing surfactants | Mortality | I | No | B |
| Ventilator-free days | ||||
| Nitric oxide | Mortality | I | No | A |
| Oxygenation | Yesc | |||
| Inhaled prostacyclins | Oxygenation | V | Yesc | F |
| Vasodilators: IV PGE 1 | Mortality | II | No | C |
| Ventilator-free days | ||||
| Vasodilators: liposomal IV PGE | Mortality | I | No | B |
| Ventilator-free days | ||||
| High-dose methylprednisolone for prevention of ARDS in patients at risk | Incidence of ARDS | I | No | B |
| High-dose methylprednisolone for early therapy of ARDS | Mortality | II | No | D |
| Severity of lung injury | ||||
| High-dose methylprednisolone for subacute (late) phase of ARDS | Mortality | I | No | B |
| Ventilator-free days | ||||
| Ketoconazole | Mortality | I | No | B |
| Ventilator-free days | ||||
| Lisofyline | Mortality | I | No | B |
| Ventilator-free days | ||||
| Pentoxifylline | Safety | V | No | F |
| N-Acetylcysteine | Mortality | II | No | C |
| Ventilator-free days | ||||
| Procysteine | Mortality | II | No | D |
| Ventilator-free days |
IV – intravenously; PGE 1 – prostaglandin E1; ARDS – acute respiratory distress syndrome.
Evidence level:
I: Large randomized controlled trial
II: Small randomized controlled trial
III: Nonrandomized, concurrent controls
IV: Nonrandomized, historical controls
V: Case series reports
Level of evidence definition adapted from Sackett [1]; large randomized trial defined as study of ≥200 patients.
Grade of recommendation:
Grade A: Supported by at least 2 level I investigations
Grade B: Supported by at least 1 level I investigations
Grade C: Supported by at least 2 level II investigations
Grade D: Supported by at least 1 level II investigations
Grade E: Supported by at least 1 level III investigations
Grade F: Supported by level IV or level V investigation
Grade of recommendation adapted from the Pulmonary Artery Catheter Consensus Conference [3].
Severe ARDS with refractory hypoxemia only.
Inhaled Vasodilators
ALI/ARDS is characterized by increased permeability edema, inflammatory and procoagulant changes in the lung with loss of hypoxic vasoconstriction in the nonventilated lung regions, and impaired vasodilation in the aerated regions. Maldistribution of blood flow in the injured lung results in increased intrapulmonary right-to-left shunting, pulmonary hypertension, and severe hypoxemia [66]. Increased pulmonary artery pressures in patients with ALI/ARDS were well documented in earlier studies using right heart catheterization, and in several studies pulmonary artery hypertension in patients with ALI/ARDS was associated with poor prognosis [67–69]. Therapeutic interventions that selectively vasodilate pulmonary vasculature in the ventilated regions of the lungs result in a decrease in pulmonary artery pressure, a decrease in right-to-left shunting, and an increase in PaO2/FiO2. Two selective pulmonary vasodilators have been investigated in the therapy of ALI/ARDS: inhaled nitric oxide and inhaled prostacyclins.
Nitric Oxide
Nitric oxide (NO) is a potent endogenous vasodilator that can be exogenously delivered to the pulmonary vasculature by inhalation. NO is rapidly inactivated on contact with hemoglobin [70] at the site of administration or generation and thus causes selective vasodilation of pulmonary vasculature of the aerated lung units without causing systemic effects [71,72]. These physiological properties of NO seemed ideal for potential use of NO as a therapeutic agent for ALI/ARDS. Initial studies of inhaled NO in animal models of ALI/ARDS demonstrated encouraging results and were followed by human studies. A pilot observational study by Rossaint et al [73] in 9 ARDS patients compared inhaled NO at 18 ppm with intravenous prostacyclin (PGI2 at 4 ng/kg). The mean decrease in PA pressure was 37 to 30 mm Hg for NO and 36 to 30 mm Hg for PGI2. Whereas PGI2 caused significant changes in systemic arterial pressure and cardiac output, NO had no systemic effects. NO inhalation also caused a significant improvement in oxygenation and a decrease in intrapulmonary shunt fraction (36% to 31%). Whether these favorable physiological effects would translate into important clinical outcomes has been the focus of several subsequent studies. Two small randomized, controlled, unblinded, single-center studies of inhaled NO versus conventional therapy in ARDS patients (40 and 30 patients) [74,75] showed a transient improvement in oxygenation and a decrease in the shunt fraction, but no mortality benefit. These studies were followed by larger randomized controlled trials. Dellinger et al [76] conducted a randomized controlled, double-blind, multiple-center study of different doses of inhaled NO (1.25–80 ppm; N = 120 patients) vs placebo (n = 57) in patients meeting the AECC criteria for ARDS. The improvements in hemodynamics and oxygenation in the intervention group were modest and transient (during the first 24 hours only), and there was no mortality benefit or difference in ventilator-free days compared with placebo. On post hoc analysis, the 5 ppm inhaled NO group had lower 28-day mortality rate (24% vs 30%), but this difference was not statistically significant on multiple comparisons analysis. Concurrently, a European multiple-center study of inhaled NO in ALI showed similar results [77]. Of 286 patients enrolled, 180 NO responders were randomized to NO at the lowest effective dose (1–40 ppm) or placebo. The primary outcome, the reversal of ALI, was not increased in the NO group, and although the frequency of severe respiratory failure was reduced in the NO group, NO did not alter mortality rate (44% in NO group vs 40% placebo). The most recent study, by Taylor et al [78], examined the efficacy of inhaled NO at a dose of 5 ppm in patients with moderately severe ALI not attributable to sepsis and without nonpulmonary organ dysfunction. This study was the largest to date, enrolling 385 patients. The results were consistent with the previous trials—despite the short-term improvement in oxygenation, NO therapy had no impact on the duration of ventilatory support or mortality.
In summary, current evidence (level I, grade A; Table 2) suggests that NO has no impact on important clinical outcomes and should not be recommended as standard therapy of ARDS. Because NO does transiently improve oxygenation, it can be useful as a rescue therapy for severe ARDS with refractory hypoxemia.
Inhaled Prostacyclins
Prostaglandins are endogenously produced derivatives of arachidonic acid and were shown to cause vasodilation, inhibit platelet aggregation, and have anti-inflammatory properties via inhibition of macrophage and neutrophil activation. Inhaled prostacyclins cause selective pulmonary vasodilation, thus improving ventilation/perfusion ratio mismatch, oxygenation, and decrease in pulmonary artery pressure and pulmonary vascular resistance. Inhaled prostacyclins may also decrease pulmonary artery pressure through their antithrombotic properties preventing obstruction of the pulmonary microcirculation. The currently available inhaled prostacyclins are prostaglandin I2 (PGI2; generic name epoprostenol, brand name Flolan), which in its intravenous form is approved by the Food and Drug Administration (FDA) for the therapy of primary pulmonary hypertension, and prostaglandin E2 (PGE2). Both PGI2 and PGE2 have an in vivo half-life of 3 to 10 minutes. Iloprost (brand name Ventavis), a stable analog of PGI2 with a longer half-life, is not available in the United States.
Walmrath et al [79] were the first to demonstrate the selective vasodilatory effects of aerosolized PGI2 on pulmonary vasculature at doses of 17 to 50 ng/kg/min in 3 patients with severe ARDS. Several subsequent observational studies compared the efficacy of aerosolized PGI2 with inhaled NO in both adult [80–82] and pediatric [83] patients with ARDS. In these trials, which included relatively small numbers of patients (5–16), inhaled PGI2 had a comparable effect to NO on pulmonary vasculature and oxygenation with minimal systemic effects. However, the selectivity of prostacyclin is dependent on the dose. With larger doses, there is a spillover to systemic circulation and nonaerated lung units despite the relatively short half-life. Because the amount of the drug delivered to the alveolar space varies with different nebulizers used, it is difficult to dose PGI2 as precisely as NO. The problems with dosing of PGI2 and the discouraging results of large randomized trials of NO are probably the reason there are no randomized trials of prostacyclins evaluating clinical outcomes. At this time, there is no evidence that inhaled prostacyclins improve survival in patients with ALI/ARDS (level V, grade F, Table 2). Inhaled prostacyclins can be used as an alternative to very expensive inhaled NO in the treatment of refractory hypoxemia in patients with severe ARDS.
Systemic Pharmacologic Therapy
Vasodilators
Prostaglandin E1 is a naturally occurring prostaglandin that has vasodilatory properties and can modulate neutrophil function and inhibit mediator release [84]. When PGE1 is delivered intravenously, its vasodilatory effects are nonselective and cause systemic vasodilation and hypotension and increase intrapulmonary shunting [85,86]. The potential benefit of systemic administration of prostaglandins was thought to be primarily through its anti-inflammatory effect by inhibition of neutrophil activation. PGE1 was also reported to increase oxygen extraction capabilities in the setting of reduced oxygen delivery [87,88]. In animal models of ALI, PGE1 reduced neutrophil aggregation and pulmonary capillary leak [89,90]. The initial clinical trial of PGE1 in 41 patients with ARDS, conducted by Holcroft et al [91] in 1986, showed a trend toward a survival benefit. In contrast, larger studies in the late 1980s and early 1990s showed no mortality benefit [92,93] and no significant effect on functional activity of polymorphonuclear leukocytes [94]. One explanation for lack of its efficacy was that PGE1 was not able to target neutrophils sufficiently at nontoxic doses. With the introduction of liposomal PGE1 in the mid-1990s, which provides a means of more selective delivery of PGE1 to neutrophils [95], PGE1 for therapy of ARDS was revisited. In a pilot study by Abraham et al [96] in 25 patients with ARDS, liposomal PGE1 caused statistically significant improvement in the oxygenation, lung compliance, and ventilator dependency. Unfortunately, these encouraging results were not confirmed in 2 recent large randomized trials of liposomal PGE1—a North American trial by Abraham et al [97], which enrolled 350 patients, and a European study [98] with 102 patients. Neither of these trials showed any difference in clinically important outcomes. Intravenous prostaglandins therefore cannot be recommended for treatment of patients with ALI/ARDS (level of evidence II, grade C, Table 2). Table 3 summarizes major clinical trials of vasodilators in ARDS.
Table 3.
Summary of Clinical Studies of Vasodilators in Acute Lung Injury (ALI)/Acute Respiratory Distress Syndrome (ARDS)
| Intervention | Author & Year | Study Design | Study Population & Number of Patients | Results |
|---|---|---|---|---|
| Continuous IV PGE 1, dose titrated to 30 ng/kg/min for 48–72 h or placebo | Shoemaker et al 1986 [207] | Prospective, randomized, blinded, placebo- controlled trial, phase I/II | Severely ill adult surgical patients with ARDS, N = 9 | Decrease in PA pressures, PVR, and SVR and increase in oxygenation, oxygen delivery, and cardiac output |
| Continuous IV PGE 1, maximum dose 30 ng/kg/min for 7 d or placebo | Holcroft et al 1986 [91] | Prospective, randomized, double-blind, placebo controlled trial, phase II | Adult surgical patients with ARDS, N = 41 | Trend toward survival benefit; 30-d mortality in PGE 1 group vs placebo 29% vs 65%, respectively (P = .08) |
| Continuous IV PGE 1, maximum dose 30 ng/kg/min for 7 d or placebo | Bone et al 1989 [93] Slotman et al 1992 [92] | Prospective, randomized, double-blind, placebo- controlled, multiple-center trial, phase II | Adult patients with ARDS attributable to trauma, surgery, or sepsis, N = 100; N = 147 | No survival benefit; 30-d mortality in PGE 1 group vs placebo was 30/50 vs 24/50, respectively; RR 1.15 (95% CI 0.89–1.55) |
| Continuous IV PGE 1, dose titrated to 30 ng/kg/min for 7 d or placebo | Rossignon et al 1990 [94] | Prospective, randomized, double-blind study, phase II | Adult patients with ARDS, N = 23 | No significant effect on the function activity of polymorphonuclear leukocytes |
| Liposomal IV PGE 1, dose titrated to 3.6 μg/kg every 6 h for 7 d or placebo | Abraham et al 1996 [96] | Prospective, randomized, double-blind, placebo- controlled, multiple-center trial, phase II | Adult patients with ARDS, N = 25 | Statistically significant improvement in oxygenation (day 3), lung compliance, and ventilator dependency (day 8) in PGE 1 group. Mortality difference in PGE 1 group vs placebo (6% vs 25%, P = .23) not significant |
| Liposomal IV PGE 1, dose titrated to 3.6 μg/kg every 6 h for 7 d or placebo | Abraham et al 1999 [97] | Prospective, randomized, double-blind, placebo- controlled, multiple-center trial, phase III | Adult patients with ARDS per AECC criteria, N = 350 | No difference in mortality or duration of mechanical ventilation. Mortality rate in PGE 1 group vs placebo 32% vs 29% (P = .55) and median number of days to off mechanical ventilation 16.9 vs 19.6 (P = .94) |
| Liposomal IV PGE 1, dose titrated to 1.8 μg/kg every 6 h for 7 d or placebo | Vincent et al 2001 [98] | Prospective, randomized, double-blind, placebo- controlled, multiple-center trial, phase III | Adult patients with ARDS per AECC criteria, N = 102 | No difference in mortality or duration of mechanical ventilation; 28-d mortality rate in PGE1 group 30% vs 28% for placebo (P = .78). Ventilator-free days 16 d in PGE1 group vs. 16.6 with placebo (P = .94) |
IV – intravenously; PGE 1 – prostaglandin E1; PA – pulmonary artery; PVR – pulmonary vascular resistance; SVR - systemic vascular resistance; RR – relative risk; CI – confidence interval; AECC – American European Consensus Conference.
Adverse events reported: hypotension, nonfatal arrythmias, diarrhea, and flushing.
Anti-inflammatory Agents
Glucocorticoids
Glucocorticoids can reduce inflammation and fibrosis through inhibition of several cytokines including interleukin (IL)-1, IL-3, IL-5, IL-6, IL-8, tumor necrosis factor (TNF)-α, and granulocyte macrophage–colony stimulating factor [99,100]. Glucocorticoids exert inhibitory effect on cytokine transcription and some cytokine receptors and inhibit activation of transcription factors activator protein-1 and nuclear factor-κB, known to be involved in the up-regulation of a number of gene products that play a central role in inflammatory response [101–103]. Glucocorticoids are also thought to have a direct effect on the expression of adhesion molecules such as intercellular adhesion molecule-1 and E-selectin [104,105] and to suppress the synthesis of phospholipase A2, cyclooxygenase, and inducible NO synthase [106], decreasing production of prostanoids, platelet activating factor, and NO. In addition to their action through various cytokines, glucocorticoids may also increase type I collagen degradation and in an experimental model of lung injury glucocorticoids prevented excessive collagen deposition [107]. Given their anti-inflammatory and antifibrotic properties, glucocorticosteroids seemed to be the ideal candidates for therapy of the early, exudative as well as late, proliferative phase of ALI/ARDS. Table 4 summarizes all clinical trials of corticosteroids in ARDS.
Table 4.
Summary of Clinical Studies of Glucocorticoids in Acute Lung Injury (ALI)/Acute Respiratory Distress Syndrome (ARDS)
| Intervention | Author & Year | Study Design | Study Population & Number of Patients | Results |
|---|---|---|---|---|
| Methylprednisolone (30 mg/kg IV every 6 h × 48 h) or placebo | Weigelt et al 1985 [108] | Prospective, randomized, placebo- controlled, double-blind study, phase III | Acutely ill, mechanically ventilated patients with 350 PaO2/FiO2 ≤ (considered high risk for ARDS), N = 81 | Increased incidence of ARDS in steroid group (64% vs 33%; P = .008). Increase in mortality rate in the steroid group 46% vs 31% was not statistically significant (P = .177)a |
| Methylprednisolone 30 mg/kg IV or dexamethasone 6 mg/kg (×1 or 2 doses) or nothing | Sprung et al 1984 [109] | Prospective, randomized study, phase III | Patients with septic shock, N = 59 | No difference in incidence of ARDS (23% vs 13% vs 12.5%, respectively.). No difference in overall mortality rate and reversal of septic shocka |
| Methylprednisolone (30 mg/kg IV every 6 h × 24 h) or placebo | Bone et al 1987 [110] | Prospective, randomized, placebo- controlled, double-blind study, phase III | Patients with sepsis, N = 304 | Trend toward increased incidence of ARDS in steroid group (32% vs 25%). Increased mortality in steroid group (61% vs 22%, P = .004) |
| Methylprednisolone (30 mg/kg IV every 6 h × 24 h) or placebo | Luce et al 1988 [111] | Prospective, randomized, placebo- controlled, double-blind study, phase III | Patients with septic shock, N = 75 | No difference in incidence of ARDS (34% vs 37%, P not significant). No difference in overall mortality rate: 22/38 in steroid group vs 20/37 in placebo group |
| Methylprednisolone (30 mg/kg IV every 6 h × 24 h) or placebo | Bernard et al 1987 [112] | Prospective, randomized, placebo- controlled, double-blind study, phase III | Patients with ARDS, N = 99 | No difference in mortality rate: 60% in the steroid group vs 63% in placebo, P = .74a |
| Methylprednisolone IV 125 mg every 6 h (taper on improvement of PaO2/FiO2), course of 21 d | Asbaugh & Maier 1985 [113] | Case series | Patients with persistent ARDS and idiopathic pulmonary fibrosis on open lung biopsy, N = 10 | Mortality rate 20% (2/10 patients); mostly young patients with trauma (mean age 37) |
| Methylprednisolone IV 125–250 mg × 72–96 h then taper (course >21 d) | Hooper & Kearl 1990 [114], 1996 [115] | Case series | Patients with established ARDS (>72 h), positive 67Ga scan at 48 h, N = 10, N = 26 (including 10 patients above) | Mortality rate 19% (5/26 patients) |
| Methylprednisolone IV, initial bolus 200 mg, then 2–3 mg/kg/d, taper after extubation or after 14 d | Meduri et al 1991 [116], 1994 [117] | Case series | Patients with ARDS with progressive respiratory failure with worsening LIS and no infection, N = 8, N = 25 (including previous 8) | Overall mortality rate 24% (6/25 patients) |
| Methylprednisolone IV 2 mg/kg/d × 32 d | Meduri et al 1998 [118] | Prospective, randomized, placebo- controlled, double-blind study, phase III | Patients with severe ARDS who failed to improve LIS by day 7. Patients with infection excluded, N = 24 | Overall mortality rate 12% (2/16) in steroid group vs placebo 63% (5/9); P = .03. Infection rate similar in both groups |
| Methylprednisolone IV, initial loading dose 2 mg/kg followed by 0.5 mg/kg every 6 h × 14 d and 0.5 mg/kg every 12 h × 7 d | ARDS network, unpublished data | Prospective, randomized, multiple-center, placebo- controlled study, phase III | Patients with persistent ARDS (>7 d) and no evidence of infection, N = 180 | No difference in mortality. |
IV – intravenously; LIS – lung injury score.
Higher incidence of infections in the steroid group.
No increase in infectious complications, but increased incidence of myopathy in the steroid group (9 vs 0 for placebo) and higher incidence of reintubations.
Glucocorticoids—Early-Phase ARDS
Clinical trials conducted in the middle to late 1980s demonstrated that patients with early ARDS or at risk for ARDS do not benefit from a short course of high-dose corticosteroids.
Prevention of ARDS
Several clinical trials conducted in the middle to late 1980s in patients at high risk for ARDS (patients with sepsis and high-risk mechanically ventilated patients) showed no benefit of short-course, high-dose corticosteroids to prevent development of ARDS. Weigelt et al [108] in 1985 performed a double-blind, placebo-controlled, randomized trial of methylprednisolone (MPSS) therapy (30 mg/kg intravenously every 6 hours × 48 hours) in 81 acutely ill, mechanically ventilated patients at high risk for ARDS. There were 25/39 (64%) in the MPSS group and 14/42 (33%) in the placebo group who developed ARDS (P = .008), and early infectious complications occurred in 77% of the MPSS-treated patients compared with 43% in the placebo group. There was no difference in mortality rate (46% in the steroid group vs 31% in the placebo group, P = .177). In 1984, Sprung and coworkers [109] conducted a prospective, randomized trial of 59 patients with septic shock randomly assigned to 1 or 2 doses of MPSS 30 mg/kg (21 patients), dexamethasone 6 mg/kg (22 patients), and a control group (16 patients). Apart from primary end points (no difference in reversal of shock and mortality rate), there was also no difference between the 3 groups in developing ARDS as a complication of sepsis (5 patients in the MPSS group, 3 patients in the dexamethasone group, and 2 patients in the control group; differences not statistically significant). In 1987, Bone et al [110] conducted a large, prospective, randomized, double-blind, placebo-controlled study to determine if early therapy with MPSS would decrease the incidence of ARDS in patients with sepsis. There were 304 patients randomized to MPSS (30 mg/kg intravenously every 6 hours × 24 hours) or placebo. There was a trend toward an increased incidence of ARDS in the MPSS group, 50/152 (32%), versus the placebo group, 38/152 (25%). Also, the 14-day mortality rate in patients with ARDS in the MPSS group, 26/50 (61%), was higher compared with placebo, 8/38 (22%), a difference that was statistically significant (P = .004). Similarly, Luce and coworkers [111] in 1988 carried out a prospective, randomized, double-blind study to evaluate the efficacy of high-dose MPSS to prevent ARDS in patients with septic shock. Patients who met the criteria for septic shock received either MPSS (30 mg/kg every 6 hours × 24 hours) or placebo. Of patients receiving MPSS, 13/38 developed ARDS compared with 14/37 patients in the placebo group, a difference that was not statistically significant. There was also no difference in overall mortality rate (58% in the steroid group vs 54% in the placebo group), and in contrast to the study by Bone et al, there was no difference in mortality rate in patients who developed ARDS (9/13 in the steroid group and 8/14 in the placebo group). In both trials by Bone and Luce, there was faster resolution of ARDS in the placebo group (P < .05)
Therapy of Early ARDS
The only randomized, double-blind placebo controlled trial of early corticosteroid therapy for ARDS is a study by Bernard et al [112] conducted in 1987 with 99 patients from 7 centers with the diagnosis of ARDS. The etiology of ARDS was infectious in half of the patients (44% sepsis and 5% pneumonia). The patients were randomized to high-dose MPSS (30 mg/kg intravenously every 6 hours) or placebo × 24 hours. The mortality rate at 45 days was 30/50 (60%) in the MPSS group and 31/49 (63%) in the placebo group (P = .74). Infectious complications were similar in the 2 groups.
Glucocorticoids—Late-Phase ARDS
Observational Studies
The failure of the randomized trials to show efficacy of glucocorticosteroids to prevent or treat the early phase of ARDS prompted interest in using glucocorticoids to treat the late, fibrosing-alveolitis phase of the disease. The first report, by Ashbaugh and Maier [113] in 1985, described 10 patients with ARDS who did not respond to conventional treatment and had fibrotic process on open lung biopsy in the absence of infection (biopsy performed on average on day 11.5). Patients received MPSS 125 mg intravenously every 6 hours; the dose was tapered on response over the course of 3 to 6 weeks. Mortality rate was 2 of 10 patients (20%, the 2 patients died of sepsis), which was much lower than the 60% to 80% mortality rate for ARDS usually reported at the time. Encouraging results of this study stimulated a series of case reports in the early 1990s. Hooper and Kearl [114,115] published 2 case series of 10 and 26 patients (1990 and 1996, respectively) with established ARDS (more than 72 hours). These authors did not perform open lung biopsy, but in 18 of 26 patients the investigators performed 67Ga scan to document active inflammatory process in the lungs. Patients with documented infection (positive blood or wound cultures, any fungal infection) were excluded. Patients received MPSS 125 to 250 mg intravenously every 6 hours as an initial dose, which was tapered after 72 to 96 hours based on clinical response. The overall mortality rate in this series was 5 of 26 (19%), confirming the observation from the previous report. Similarly, Meduri and coworkers [116,117] published 2 case series of 8 and 25 patients (including the previous 8) with ARDS with progressive respiratory failure and severe fibroproliferation (open lung biopsy in 13 patients with fibrosis). Patients with active infection were excluded. Glucocorticoid therapy with MPSS was started on day 15 ± 7.5 after the diagnosis of ARDS. The overall mortality rate was 24% (6 of 25).
Randomized Controlled Trials
Meduri et al followed their case series with a very small, prospective, randomized, placebo-controlled trial of corticosteroids for persistent ARDS in 24 patients [118]. Patients who had failed to improve their lung injury score by day 7 and had no evidence of active infection were enrolled; 16 patients were randomized to MPSS (2 mg/kg/d times 32 days) and 8 to placebo. Overall mortality rate in the study was 12% (2 of 16) in the MPSS group vs placebo 63% (5 of 9, P = .03). The infection rate was similar in both groups. Per protocol patients who failed to improve their lung injury score by day 10 of the study drug were blindly crossed over to the other arm. Because all patients in the MPSS group improved, there was no crossover from MPSS to placebo. In the placebo arm, 2 patients improved and 2 died within the first 10 days, and 4 were crossed over to MPSS after 10 days of placebo. Of those 4 patients, 3 died and 1 survived. To resolve this issue, the NHLBI ARDS network undertook a much larger multiple-center randomized controlled trial to determine efficacy of glucocorticoids in patients with persistent ARDS. In this study, 180 patients with ARDS of at least 7 days duration were assigned to receive either MPSS or placebo. Based on a presentation of the results of the Late Steroid Rescue Study at the American Thoracic Society meeting in May 2005 in San Diego, the primary end point, 60-day mortality, was not different between the placebo and the methylprednisolone-treated groups. Overall, the results of this study do not support the routine use of MPSS for persistent ARDS (unpublished data, 2005).
In conclusion, current evidence suggests that glucocorticoids should not be used in prevention (evidence level I, grade B) or therapy of either early (evidence level II, grade D) or late (evidence level I, grade B) phases of ALI/ARDS (Table 2).
Ketoconazole
Ketoconazole is an antifungal agent but also a potent inhibitor of leukotriene and thromboxane A2 synthesis. Thromboxane A2 acts as a potent vasoconstrictor and aggregator of platelets and neutrophils [119] and has been implicated as an important mediator in septic shock and development of ALI/ARDS. Ketoconazole has also been shown to inhibit secretion of proinflammatory cytokines by alveolar macrophages [120]. Two preliminary studies [121,122] have shown that ketoconazole may prevent development of ARDS in high-risk surgical patients. Slotman et al [122] conducted a randomized, double-blind, placebo-controlled study of ketoconazole at 200 mg orally daily (for 21 days or until discharge) in 71 surgical intensive care unit patients. The incidence of ARDS was lower in the ketoconazole group (6% vs 31% in the placebo group; P < .01) as was median surgical intensive care unit stay (7 vs 15.5 days, P < .05). Yu and Tomasa [121] in 1993 performed a randomized, double-blind, placebo controlled trial of ketoconazole 400 mg or placebo in 54 consecutive surgical intensive care unit patients with sepsis. There was significant reduction in the incidence of ARDS in the ketoconazole group (15% vs 64%, P = .002). However, a recent large, multiple-center, randomized trial conducted by NHLBI ARDS network [123] of 234 patients with ARDS randomized to ketoconazole 400 mg/d enterally up to 21 days or placebo showed no mortality benefit (35% in the treatment group vs 34% in placebo group, P = not significant). There was also no difference in ventilator-free days (10 vs 9 days, respectively). At this time, ketoconazole should not be used for therapy of ALI/ARDS (evidence level I, grade B; Table 2). Ketoconazole might be more useful as prophylaxis in patients at risk for ARDS, but this needs further study.
Lisofylline and Pentoxifylline
Pentoxifylline, a phosphodiesterase inhibitor, and its derivative lisofylline(1-[5R-hydroxyhexyl]-3, 7-dimethyl-xanthine) exert their anti-inflammatory properties through different mechanisms.
Pentoxifylline has been shown to inhibit neutrophil chemotaxis and activation and to inhibit the release of TNF-α by macrophages in response to endotoxin in animal models of sepsis and ALI/ARDS [124–126]. In animal models of ALI, pretreatment with pentoxifylline decreased endothelial permeability and pulmonary edema [126,127]. However, there is limited experience in humans to make definite recommendations for clinical use. A small, uncontrolled, phase I pilot study of high-dose pentoxifylline in 6 ARDS patients showed no improvement in either gas exchange or hemodynamic parameters [128].
Lisofylline, like pentoxifylline, has been shown to diminish proinflammatory cytokine expression (TNF-α, IL-1, and IL-6) [129,130] and to inhibit neutrophil accumulation and edema formation [131]. Unlike pentoxifylline, lysofylline partly exerts its anti-inflammatory properties by inhibiting the release of oxidized free fatty acids from cell membranes under oxidative stress. Activation of phospholipid-signaling pathways involving the acyl chains oleate and linoleate that leads to an increase in oxidized free fatty acids may initiate and amplify the inflammatory response. In patients with sepsis, circulating levels of free fatty acids increase several-fold, and this increase is suppressible by lisofylline [132]. In critically ill patients, increases in unsaturated serum acyl chain ratios were demonstrated to predict development of ARDS [132]. In animal studies of sepsis and ARDS, treatment with lisofylline showed promising results. In murine model of sepsis, lisofylline significantly improved survival even if administered 4 hours after a lethal dose of endotoxin [133], and in pigs, lisofylline 1 hour after induction of sepsis reduced the severity of ALI [129]. Given the encouraging preliminary data, the NHLBI ARDS network conducted a phase III, randomized, double-blind, placebo-controlled clinical trial of lisofylline in 235 patients meeting the AECC criteria for ALI [134]. Patients were randomized to lisofylline 3 mg/kg intravenously every 6 hours for 20 days or until 48 hours of unassisted breathing or placebo. The study was stopped at an interim analysis by the Data Safety Monitoring Board for futility. The mortality rate at 28 days was 31.9% in the lisofylline group vs 24.7% in the placebo group (P = .215). There was no difference in ventilator-free days or resolution of organ failure.
Antioxidant Therapy
N-acetylcysteine and Procystein
There is convincing experimental evidence that reactive oxygen species produced by activated neutrophils, macrophages, and stimulated pulmonary endothelial cells play a major role in mediating injury to the lung endothelium in sepsis or hyperoxic lung injury [135]. Three major antioxidants, superoxide dismutase, catalase, and glutathione, neutralize free radicals and prevent their injurious effects. Patients with ALI/ARDS have been shown to have both a decrease in total glutathione and a relative increase in oxidized glutathione [136,137], and ALI/ARDS patients who do not survive sustain much greater levels of oxidative molecular damage [138]. N-acetylcysteine (NAC) and procysteine effectively replete intracellular glutathione via their metabolism to the glutathione precursor cysteine [139]. In some animal models of ALI/ARDS, antioxidant therapy has been shown to have benefit. NAC significantly improved all monitored pathophysiological parameters in an endotoxin model of ARDS in sheep [140]. NAC administration 2.5 hours after intratracheal instillation of IL-1 in rats decreased lung leak, neutrophil count in bronchoalveolar lavage, and defects in lung histology. Initial phase II clinical studies showed equivocal results. A small, randomized trial by Jepsen et al [141] in 1992 of intravenous NAC (150 mg/kg bolus followed by 20 mg/kg/h infusion for 7 days) vs placebo in 66 patients with ARDS showed no mortality benefit at day 60 (53% in the NAC group vs 50% in placebo), and there was also no significant difference in pulmonary physiological parameters. In 1994, Suter et al [142] evaluated 61 patients presenting with mild to moderate ALI (200 mm Hg < PaO2/FiO2 < 300 mm Hg) and predisposing factors for ARDS who received either NAC 40 mg/kg/d or placebo intravenously for 3 days. NAC treatment improved systemic oxygenation and reduced the need for ventilatory support in this study population. Development of ARDS and mortality were not significantly reduced. Given the encouraging results of this study, the same group of investigators assessed the effects of intravenous NAC in a randomized controlled trial of 42 patients with established ARDS (PaO2/FiO2 <200 mm Hg) [143]. Patients were randomized to NAC 190 mg/kg/d or placebo infusion over 72 hours. In this study, NAC therapy neither improved systemic oxygenation nor reduced the need for ventilatory support. Mortality rate was 32% for the NAC group and 25% for the placebo group (P = not significant). Bernard et al [144] conducted another phase II study of NAC (70 mg/kg) versus procysteine (63 mg/kg) versus placebo intravenously every 8 hours for 10 days in 44 patients with ARDS. The study failed to show a mortality benefit (mortality rate at day 30 was 36%, 35%, and 40%, respectively), but patients treated with either antioxidant had a trend toward fewer ALI days and less development of new organ failure. Unfortunately, despite these encouraging trends in phase II trials, a recent large, phase III, double-blind, placebo-controlled trial of procysteine in patients with ALI/ARDS was stopped prematurely because of concern regarding mortality in the intervention group [unpublished data, 145,146]. There is currently no evidence (NAC, level II, grade C; procysteine, level II, grade D; see table 2) to support use of antioxidants in therapy of ALI/ARDS.
Albumin
Albumin, the most abundant plasma protein, is an important natural antioxidant in plasma. An exposed thiol group (–SH) on the surface of albumin molecules which has the ability to be reversibly oxidized [147], is thought to be responsible for antioxidant properties of albumin. As albumin plasma levels in critical illness decline, plasma levels of thiols decrease. Quinlan et al [148] reported in 1994 in an observational study of 8 patients with ARDS that survivors (3 patients) had higher total plasma thiol values, which increased as the lung injury resolved. The same authors [149] reported in 1998 that administration of albumin to patients with sepsis leads to a sustained increase in plasma thiols and therefore might have a beneficial effect in reducing oxidative stress. In 2004, Quinlan et al [150] conducted a small prospective study of the influence of albumin on antioxidant status in 20 ARDS patients randomized to albumin and placebo (normal saline). The study showed that albumin administration favorably influences plasma thiol-dependent antioxidant status as well as levels of protein oxidative damage. This study was not powered to assess important clinical outcomes, but it is interesting in view of the results of the recently published SAFE trial, a large trial of albumin versus normal saline for fluid resuscitation in the intensive care unit [151]. This prospective, randomized, double-blind controlled trial of 6997 patients showed no overall difference in mortality rate between the albumin and normal saline groups. Based on a prespecified subgroup analysis, the patients with severe sepsis (1218 patients) who were given albumin showed a trend toward lower mortality rate, relative risk 0.87 (95% confidence interval [CI] 0.74–1.02). A subgroup analysis of patients with ARDS in the group that received albumin showed a relative risk of death 0.93 (95% CI 0.61–1.41) but there were only 127 patients with ARDS enrolled in the study. The relative risk of death of patients with no ARDS was 1.00 (95% CI 0.91–1.09). These trends toward improved outcome in a subgroup of patients with sepsis and ARDS who received albumin will need further study. Most recently, Martin et al [152] evaluated the effect of albumin administered with furosemide therapy in a small, randomized, placebo-controlled trial of 40 hypoproteinemic patients (total protein <6.0 g/dL) with ALI/ARDS. The addition of albumin to furosemide compared with placebo in this patient population significantly improved oxygenation, with greater net negative fluid balance and better maintenance of hemodynamic stability. This needs further evaluation.
Potential New Therapies
Anticoagulant Therapies
Activation of coagulation pathways and suppression of fibrinolysis play an important role in the pathogenesis of ALI/ARDS, and extravascular and intravascular fibrin deposition [15,16] is one of the hallmarks of diffuse alveolar damage, a pathological correlate of early ALI/ARDS. There is evidence both from radiological studies [153] and pathology [154] that abnormalities of pulmonary blood flow secondary to pulmonary vascular obstruction and injury to the lung microcirculation are prominent features of ALI/ARDS. These changes in pulmonary vasculature can at least in part explain the increased pulmonary vascular resistance and elevated dead space fraction early in the course of ALI. Interestingly, elevated early dead space fraction has been shown to be an independent predictor of mortality in patients with ARDS [155]. The coagulation abnormalities present in ALI/ARDS do not differ significantly from those observed in sepsis. Several anticoagulant therapies, including tissue factor pathway inhibitor (TFPI), factor VIIai (competitive inhibitor of tissue factor), antithrombin (AT) III, activated protein C (APC), heparin, and thrombomodulin, have been implicated as potential therapeutic interventions in patients with severe sepsis and ALI/ARDS.
Tissue factor activates the extrinsic coagulation pathway binding to coagulation factor V, which in turn activates factor X. TFPI and factor VIIai exert their anticoagulant effect through suppression of the activity of the tissue factor/VIIa/Xa complex. TFPI and factor VIIai have been shown to improve survival in animal models of sepsis [156] and reduce alveolar inflammation and fibrin deposition in animal models of ALI/ARDS [157,158]. Recombinant TFPI has been evaluated in a phase II, randomized, placebo-controlled, single-blind study in patients with severe sepsis. There was a 20% relative reduction in mortality rate and improvement in lung dysfunction scores in patients treated with TFPI [159]. A subsequent phase III, randomized, double-blind, placebo-controlled, multiple-center clinical trial [160] of 1754 patients with severe sepsis failed to a show mortality benefit and there was an increase in the risk of bleeding in the intervention group. There have been no randomized controlled trials in patients with ALI/ARDS.
Antithrombin III inhibits activated coagulation factors including IXa, Xa, and thrombin. Antithrombin III therapy has been evaluated in animal models of ALI/ARDS and was shown to improve several physiological and pathological indices of lung injury. Plasma levels of AT III decrease in experimental and clinical sepsis and correlate with higher mortality rate [161]. Several small randomized clinical trials [162–164] together with a review including meta-analysis of AT III therapy in patients with severe sepsis and septic shock suggested improved survival. Unfortunately, in a large, randomized, multiple-center, placebo-controlled trial of 2314 patients with severe sepsis, there was no reduction in mortality and there was a higher incidence of bleeding events in patients receiving concomitant heparin prophylaxis [165]. There have been no randomized controlled trials in patients with ALI/ARDS.
Heparin is a potent activator of AT III and is widely used in therapy and prevention of venous and arterial thrombosis. The results of preclinical studies to demonstrate the effectiveness of heparin to block fibrin deposition in the lung and improve physiological and pathological indices of lung function in experimental models of ALI/ARDS have been equivocal [166–169]. There have been no randomized clinical trials of heparin in patients with ALI/ARDS or sepsis. Davidson et al [170] conducted a retrospective subgroup analysis of pooled data from 2 large clinical trials evaluating other anticoagulants (protein C [171] and AT III [165]) in the therapy of severe sepsis. They concluded that among the 1995 pooled placebo recipients from the 2 trials, the odds of survival for the patients who received heparin, compared with those who did not, was 1.45 (95% CI 1.18–1.78, P < .001). These results need to be interpreted with caution because of the possible selection and allocation bias of post hoc crossover studies [172].
Activated protein C is an endogenous protein that promotes fibrinolysis and inhibits thrombosis and inflammation. Exogenous recombinant activated protein C (drotrecogin alfa) was recently tested for treatment of patients with severe sepsis in a large, phase III, randomized controlled trial and was shown to significantly reduce mortality rate from 30% to 24% [171]. This large trial, which was the first clinical study of pharmacological therapy in severe sepsis apart from antimicrobials to demonstrate a favorable impact on clinical outcomes, underscores the importance of both inflammatory and procoagulant pathways in the pathogenesis of severe sepsis. Unfortunately, even though 75% of patients in this study were mechanically ventilated at enrollment, there is no information from this study on the effect of APC in patients with coexisting ALI. Interestingly, a recent clinical study by Ware et al [173] demonstrated that plasma levels of protein C were lower in 45 patients with ARDS than in normal controls and that in patients with ARDS, lower levels of protein C correlated with a worse clinical outcome. Lower protein C levels in pulmonary edema fluid in these patients also conferred a higher mortality rate. In addition, plasma and pulmonary edema fluid levels of plasminogen activator inhibitor (PAI)-1 [174], which in excess can impair normal fibrinolysis, were significantly higher in patients with ARDS compared with patients with hydrostatic pulmonary edema. Most recently, van der Poll et al [175] demonstrated that in healthy humans with endotoxin-induced activation of coagulation (after endotoxin administration to the lung), APC suppressed coagulation and promoted fibrinolysis. Activated protein C decreased the levels of thrombin–antithrombin complexes, soluble tissue factor, and PAI-1 [175]. Overall, the pathophysiological rationale from observational and experimental studies of ARDS and the results of APC in severe sepsis clinical trial are encouraging to test APC for treatment of ARDS. There is currently a phase II study of APC in ALI underway at the University of California San Francisco.
β2-Adrenergic Agonist
There is convincing clinical evidence that impaired alveolar fluid clearance in patients with ALI/ARDS is associated with higher mortality rate [14,176]. In addition to epithelial and endothelial injury, the impaired alveolar fluid clearance may also be caused by down-regulation of specific ion and fluid transporters in the alveolar epithelium [177]. Restoration of the alveolar fluid transport capacity of the lung to a normal level may hasten the resolution of acute lung injury [178]. β2-adrenergic agonists have been shown to accelerate the airspace fluid clearance by increasing transepithelial sodium and chloride transport [177]. Several experimental animal and ex vivo human lung studies demonstrated that β2-agonists accelerate the rate of alveolar epithelial fluid clearance in normal lung models [178–183] as well as moderate hyperoxic or ventilator-associated lung injury models [184–187]. Apart from up-regulation of transepithelial ion transport, β2-agonists may reduce lung edema by reducing pulmonary endothelial permeability [188–191] and also may have an anti-inflammatory effect by decreasing release of proinflammatory cytokines [192]. There is some evidence from clinical studies that supports the results from animal studies. Wright et al in 1994 [193] examined the effect of standard aerosolized β2-agonist in 8 patients with ARDS in a randomized, placebo-controlled, crossover trial. This study found that β2-agonist therapy significantly reduced peak airway pressure, airflow resistance, and plateau pressure, suggesting an improvement in total respiratory system compliance. Also, a recent randomized, double-blind, placebo-controlled study of pretreatment with aerosolized β2-agonist (salmeterol) for the prevention of high-altitude pulmonary edema (HAPE) in 37 HAPE-susceptible subjects showed that salmeterol administration was associated with a decrease in the incidence of HAPE (33% in the salmeterol group vs 74% in the placebo group, P = .02) [194]. Most recently, a randomized controlled trial of intravenous salbutamol in 40 patients with ARDS showed that salbutamol-treated patients had a significant reduction in extravascular lung water and a trend toward improved survival (Perkins and McAuley, unpublished data, 2005). These studies provide substantial support for a large, randomized, multiple-center study powered to assess important clinical outcomes.
Keratinocyte Growth Factor
Keratinocyte growth factor (KGF), a member of the fibroblast growth factor family, is expressed predominantly by mesenchymal cells and has epithelial specificity—its receptor KGFR is expressed only on epithelial cells. KGF plays an important role in lung morphogenesis and epithelial differentiation in the developing lung, and there is a growing body of evidence that KGF also plays a role in inflammation and repair of injured lung [195]. Endogenous KGF production has been shown to be up-regulated in several animal models of lung injury [196,197]. The protective effect of exogenous KGF in an animal model of acute lung injury was first reported by Panos in 1995 [198]. Intratracheal pretreatment with KGF has since been shown to have a protective effect in a variety of lung injury models, including direct epithelial injury [199–201], direct endothelial injury [202,203], and T-cell–mediated injury [204]. The protection against acute lung injury in experimental models is explained by several mechanisms. KGF reduces apoptosis and cell death of alveolar epithelium, protects alveolar epithelium against mechanically induced lung injury, increases surfactant protein and phospholipid production, causes epithelial type II cell proliferation that hastens restoration of alveolar epithelium, and up-regulates alveolar epithelial fluid transport [195]. The major limitation is that in all of these studies, pretreatment with KGF was necessary for the protective effect. This is the major reason that there have been no clinical trials to evaluate the role of KGF in human acute lung injury. However, it should be pointed out that the clinical course of ARDS given mechanical ventilation and other supportive therapies, unlike animal models of acute lung injury, is often prolonged and therefore might provide enough time for KGF to exert its beneficial effects. Moreover, KGF is currently being evaluated for therapy in experimental and clinical studies of conditions other than acute lung injury. Recently, Chen et al [205] reported the successful prevention of late radiation-induced pulmonary toxicity in rats by a single dose of KGF given 10 minutes after the last of 5 doses of fractionated unilateral thoracic irradiation. In December 2004, human recombinant KGF (Palifermin) was FDA approved for prevention and treatment of mucositis in patients with cancer treated with high-dose chemotherapy, based on several phase II and III studies of patients with colorectal cancer and after autologous stem-cell transplant for hematological malignancies. KGF reduced the incidence of moderate to severe mucositis and was well tolerated [206]. The potential of KGF therapy for ALI needs further clinical study.
Summary
There has been significant progress in understanding the pathophysiology of ALI/ARDS as well as in therapy of ALI/ARDS since the syndrome was first described in 1967. Mortality rate, ranging from 60% to 80% in the early years, has declined to about 35% to 40% [13]. This reduction in mortality rate over the past 5 decades is attributable to important advances in supportive measures, particularly a better ventilation strategy. The low–tidal volume lung-protective ventilation strategy is the only specific intervention for therapy of ALI tested by randomized trial that has been shown to translate into mortality benefit. Future refinements in mechanical ventilation may improve outcomes further.
Despite promising preliminary studies, none of the pharmacological interventions tested in large clinical trials have been of significant clinical benefit. Table 2 summarizes all therapeutic interventions discussed in this article, with the level of evidence and current recommendations for clinical use. There is no pharmacological intervention that can be currently recommended for use in standard therapy of ALI. Nitric oxide or inhaled prostacyclins might be used in severe ARDS with refractory hypoxemia to improve oxygenation, but this intervention does not confer a mortality benefit.
The reasons for the lack of success of clinical trials of pharmaceutical agents in the therapy of ALI are multifactorial. Animal studies can never accurately reflect true clinical situations, especially in a condition as complex as ALI. The heterogeneity of patients with ALI also makes it more difficult to find a universal pharmacological therapy. It is possible that future studies might have to target more specific populations of ALI patients according to specific mechanisms of the clinical disorder (eg, direct vs indirect ALI). Also, with the mortality rate as low as 25% in recent clinical trials [29], the patient selection for clinical studies might need to focus on patients with more severe forms of lung injury or the end points may need to include both mortality and other end points such as the duration of mechanical ventilation or even longer term functional outcomes.
There are some promising potential therapies. The recent success of APC in the therapy of severe sepsis is encouraging, and APC is currently being tested in patients with ALI in a phase II clinical trial. Other potential therapies, β2-agonists, KGF, and albumin, need to be evaluated in clinical trials. There is also an ongoing phase II trial of granulocyte-macrophage colony stimulating factor for ALI/ARDS, a treatment that may inhibit epithelial cell apoptosis. Some of the therapies that have already been tested in clinical trials might need to be revisited under modified conditions (eg, surfactant). Finally, advances in basic research continue to drive clinical research [208]. There are some emerging areas of basic research that may eventually apply to ALI, including stem cell research or even gene therapy.
Acknowledgments
This manuscript was supported in part by an NIH SCCOR grant in Acute Lung Injury P50HL 74005 and two other individual NIH grants (HL 51856 and HL 51854).
References
- 1.Sackett DL. Rules of evidence and clinical recommendations on the use of antithrombotic agents. Chest. 1986;89 (2suppl):2S–3S. [PubMed] [Google Scholar]
- 2.Kopp R, Kuhlen R, Max M, et al. Evidence-based medicine in the therapy of the acute respiratory distress syndrome. Intensive Care Med. 2002;28:244–255. doi: 10.1007/s00134-002-1232-7. [DOI] [PubMed] [Google Scholar]
- 3.Pulmonary Artery Catheter Consensus conference: consensus statement. Crit Care Med. 1997;25:910–925. doi: 10.1097/00003246-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Ashbaugh DG, Bigelow DB, Petty TL, et al. Acute respiratory distress in adults. Lancet. 1967;2:319–323. doi: 10.1016/s0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- 5.Petty TL, Ashbaugh DG. The adult respiratory distress syndrome. Clinical features, factors influencing prognosis and principles of management. Chest. 1971;60:233–239. doi: 10.1378/chest.60.3.233. [DOI] [PubMed] [Google Scholar]
- 6.Murray JF, Matthay MA, Luce JM, et al. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:720–723. doi: 10.1164/ajrccm/138.3.720. [DOI] [PubMed] [Google Scholar]
- 7.Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 8.National Heart and Lung Institute. Task force on problems, research approaches, needs: the lung program. Washington DC: Department of Health, Education and Welfare; 1972. pp. 165–180. [Google Scholar]
- 9.Villar J, Slutsky AS. The incidence of the adult respiratory distress syndrome. Am Rev Respir Dis. 1989;140:814–816. doi: 10.1164/ajrccm/140.3.814. [DOI] [PubMed] [Google Scholar]
- 10.Webster NR, Cohen AT, Nunn JF. Adult respiratory distress syndrome—how many cases in the UK? Anaesthesia. 1988;43:923–926. doi: 10.1111/j.1365-2044.1988.tb05652.x. [DOI] [PubMed] [Google Scholar]
- 11.Thomsen GE, Morris AH. Incidence of the adult respiratory distress syndrome in the state of Utah. Am J Respir Crit Care Med. 1995;152:965–971. doi: 10.1164/ajrccm.152.3.7663811. [DOI] [PubMed] [Google Scholar]
- 12.Luhr OR, Antonsen K, Karlsson M, et al. Incidence and mortality after acute respiratory failure and acute respiratory distress syndrome in Sweden, Denmark, and Iceland. The ARF Study Group. Am J Respir Crit Care Med. 1999;159:1849–1861. doi: 10.1164/ajrccm.159.6.9808136. [DOI] [PubMed] [Google Scholar]
- 13.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 14.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- 15.Chapman HA, Jr, Bertozzi P, Reilly JJ., Jr Role of enzymes mediating thrombosis and thrombolysis in lung disease. Chest. 1988;93:1256–1263. doi: 10.1378/chest.93.6.1256. [DOI] [PubMed] [Google Scholar]
- 16.Idell S, Peters J, James KK, et al. Local abnormalities of coagulation and fibrinolytic pathways that promote alveolar fibrin deposition in the lungs of baboons with diffuse alveolar damage. J Clin Invest. 1989;84:181–193. doi: 10.1172/JCI114139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esteban A, Fernandez-Segoviano P, Frutos-Vivar F, et al. Comparison of clinical criteria for the acute respiratory distress syndrome with autopsy findings. Ann Intern Med. 2004;141:440–445. doi: 10.7326/0003-4819-141-6-200409210-00009. [DOI] [PubMed] [Google Scholar]
- 18.Katzenstein AL, Bloor CM, Leibow AA. Diffuse alveolar damage—the role of oxygen, shock, and related factors. A review. Am J Pathol. 1976;85:209–228. [PMC free article] [PubMed] [Google Scholar]
- 19.Tomashefski JF., Jr Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 2000;21:435–466. doi: 10.1016/s0272-5231(05)70158-1. [DOI] [PubMed] [Google Scholar]
- 20.Fukuda Y, Ishizaki M, Masuda Y, et al. The role of intraalveolar fibrosis in the process of pulmonary structural remodeling in patients with diffuse alveolar damage. Am J Pathol. 1987;126:171–182. [PMC free article] [PubMed] [Google Scholar]
- 21.Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis. 1977;116:589–615. doi: 10.1164/arrd.1977.116.4.589. [DOI] [PubMed] [Google Scholar]
- 22.Hickling KG, Walsh J, Henderson S, et al. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: a prospective study. Crit Care Med. 1994;22:1568–1578. doi: 10.1097/00003246-199422100-00011. [DOI] [PubMed] [Google Scholar]
- 23.Hickling KG, Henderson SJ, Jackson R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med. 1990;16:372–377. doi: 10.1007/BF01735174. [DOI] [PubMed] [Google Scholar]
- 24.Brochard L, Roudot-Thoraval F, Roupie E, et al. Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. The Multicenter Trial Group on Tidal Volume reduction in ARDS. Am J Respir Crit Care Med. 1998;158:1831–1838. doi: 10.1164/ajrccm.158.6.9801044. [DOI] [PubMed] [Google Scholar]
- 25.Stewart TE, Meade MO, Cook DJ, et al. Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N Engl J Med. 1998;338:355–361. doi: 10.1056/NEJM199802053380603. [DOI] [PubMed] [Google Scholar]
- 26.Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338:347–354. doi: 10.1056/NEJM199802053380602. [DOI] [PubMed] [Google Scholar]
- 27.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 28.Weinert CR, Gross CR, Marinelli WA. Impact of randomized trial results on acute lung injury ventilator therapy in teaching hospitals. Am J Respir Crit Care Med. 2003;167:1304–1309. doi: 10.1164/rccm.200205-478OC. [DOI] [PubMed] [Google Scholar]
- 29.Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 30.Guerin C, Gaillard S, Lemasson S, et al. Effects of systematic prone positioning in hypoxemic acute respiratory failure: a randomized controlled trial. JAMA. 2004;292:2379–2387. doi: 10.1001/jama.292.19.2379. [DOI] [PubMed] [Google Scholar]
- 31.Curley MA, Thompson JE, Arnold JH. The effects of early and repeated prone positioning in pediatric patients with acute lung injury. Chest. 2000;118:156–163. doi: 10.1378/chest.118.1.156. [DOI] [PubMed] [Google Scholar]
- 32.Curley MA, Hibbard PL, Fineman LD, et al. Effect of prone positioning on clinical outcomes in children with acute lung injury: a randomized controlled trial. JAMA. 2005;294:229–237. doi: 10.1001/jama.294.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simmons RS, Berdine GG, Seidenfeld JJ, et al. Fluid balance and the adult respiratory distress syndrome. Am Rev Respir Dis. 1987;135:924–929. doi: 10.1164/arrd.1987.135.4.924. [DOI] [PubMed] [Google Scholar]
- 34.Eisenberg PR, Hansbrough JR, Anderson D, et al. A prospective study of lung water measurements during patient management in an intensive care unit. Am Rev Respir Dis. 1987;136:662–668. doi: 10.1164/ajrccm/136.3.662. [DOI] [PubMed] [Google Scholar]
- 35.Humphrey H, Hall J, Sznajder I, et al. Improved survival in ARDS patients associated with a reduction in pulmonary capillary wedge pressure. Chest. 1990;97:1176–1180. doi: 10.1378/chest.97.5.1176. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell JP, Schuller D, Calandrino FS, et al. Improved outcome based on fluid management in critically ill patients requiring pulmonary artery catheterization. Am Rev Respir Dis. 1992;45:990–998. doi: 10.1164/ajrccm/145.5.990. [DOI] [PubMed] [Google Scholar]
- 37.Yu M, Levy MM, Smith P, et al. Effect of maximizing oxygen delivery on morbidity and mortality rates in critically ill patients: a prospective, randomized, controlled study. Crit Care Med. 1993;21:830–838. doi: 10.1097/00003246-199306000-00009. [DOI] [PubMed] [Google Scholar]
- 38.Tuchschmidt J, Fried J, Astiz M, et al. Elevation of cardiac output and oxygen delivery improves outcome in septic shock. Chest. 1992;102:216–220. doi: 10.1378/chest.102.1.216. [DOI] [PubMed] [Google Scholar]
- 39.Hayes MA, Timmins AC, Yau EH, et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients. N Engl J Med. 1994;330:1717–1722. doi: 10.1056/NEJM199406163302404. [DOI] [PubMed] [Google Scholar]
- 40.Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 41.Weaver TE, Whitsett JA. Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem J. 1991;273:249–264. doi: 10.1042/bj2730249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Possmayer F. The role of surfactant-associated proteins. Am Rev Respir Dis. 1990;142:749–752. doi: 10.1164/ajrccm/142.4.749. [DOI] [PubMed] [Google Scholar]
- 43.Hoekstra RE, Jackson JC, Myers TF, et al. Improved neonatal survival following multiple doses of bovine surfactant in very premature neonates at risk for respiratory distress syndrome. Pediatrics. 1991;88:10–18. [PubMed] [Google Scholar]
- 44.Long W, Corbet A, Cotton R, et al. A controlled trial of synthetic surfactant in infants weighing 1250 g or more with respiratory distress syndrome. The American Exosurf Neonatal Study Group I, and the Canadian Exosurf Neonatal Study Group. N Engl J Med. 1991;325:1696–1703. doi: 10.1056/NEJM199112123252404. [DOI] [PubMed] [Google Scholar]
- 45.Liechty EA, Donovan E, Purohit D, et al. Reduction of neonatal mortality after multiple doses of bovine surfactant in low birth weight neonates with respiratory distress syndrome. Pediatrics. 1991;88:19–28. [PubMed] [Google Scholar]
- 46.Corbet AJ, Long WA, Murphy DJ, et al. Reduced mortality in small premature infants treated at birth with a single dose of synthetic surfactant. J Paediatr Child Health. 1991;27:245–249. doi: 10.1111/j.1440-1754.1991.tb00401.x. [DOI] [PubMed] [Google Scholar]
- 47.Corbet A, Bucciarelli R, Goldman S, et al. Decreased mortality rate among small premature infants treated at birth with a single dose of synthetic surfactant: a multicenter controlled trial. American Exosurf Pediatric Study Group 1. J Pediatr. 1991;118:277–284. doi: 10.1016/s0022-3476(05)80502-5. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt R, Meier U, Yabut-Perez M, et al. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am J Respir Crit Care Med. 2001;163:95–100. doi: 10.1164/ajrccm.163.1.9903029. [DOI] [PubMed] [Google Scholar]
- 49.Pison U, Seeger W, Buchhorn R, et al. Surfactant abnormalities in patients with respiratory failure after multiple trauma. Am Rev Respir Dis. 1989;140:1033–1039. doi: 10.1164/ajrccm/140.4.1033. [DOI] [PubMed] [Google Scholar]
- 50.Hallman M, Spragg R, Harrell JH, et al. Evidence of lung surfactant abnormality in respiratory failure. Study of bronchoalveolar lavage phospholipids, surface activity, phospholipase activity, and plasma myoinositol. J Clin Invest. 1982;70:673–683. doi: 10.1172/JCI110662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewis JF, Jobe AH. Surfactant and the adult respiratory distress syndrome. Am Rev Respir Dis. 1993;147:218–233. doi: 10.1164/ajrccm/147.1.218. [DOI] [PubMed] [Google Scholar]
- 52.Albert RK, Lakshminarayan S, Hildebrandt J, et al. Increased surface tension favors pulmonary edema formation in anesthetized dogs’ lungs. J Clin Invest. 1979;63:1015–1018. doi: 10.1172/JCI109369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richman PS, Spragg RG, Robertson B, et al. The adult respiratory distress syndrome: first trials with surfactant replacement. Eur Respir J Suppl. 1989;3:109s–111s. [PubMed] [Google Scholar]
- 54.Reines HD, Silverman H, Hurst J, et al. Effects of two concentrations of nebulized surfactant (Exosurf) in sepsis-induced adult respiratory distress syndrome (ARDS) Crit Care Med. 1992;20:S61. [Google Scholar]
- 55.Spragg RG, Gilliard N, Richman P, et al. Acute effects of a single dose of porcine surfactant on patients with the adult respiratory distress syndrome. Chest. 1994;105:195–202. doi: 10.1378/chest.105.1.195. [DOI] [PubMed] [Google Scholar]
- 56.Weg JG, Balk RA, Tharratt RS, et al. Safety and potential efficacy of an aerosolized surfactant in human sepsis-induced adult respiratory distress syndrome. JAMA. 1994;272:1433–1438. [PubMed] [Google Scholar]
- 57.Anzueto A, Baughman RP, Guntupalli KK, et al. Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group. N Engl J Med. 1996;334:1417–1421. doi: 10.1056/NEJM199605303342201. [DOI] [PubMed] [Google Scholar]
- 58.Walmrath D, Gunther A, Ghofrani HA, et al. Bronchoscopic surfactant administration in patients with severe adult respiratory distress syndrome and sepsis. Am J Respir Crit Care Med. 1996;154:57–62. doi: 10.1164/ajrccm.154.1.8680699. [DOI] [PubMed] [Google Scholar]
- 59.Walmrath D, Grimminger F, Pappert D, et al. Bronchoscopic administration of bovine natural surfactant in ARDS and septic shock: impact on gas exchange and haemodynamics. Eur Respir J. 2002;19:805–810. doi: 10.1183/09031936.02.00243402. [DOI] [PubMed] [Google Scholar]
- 60.Wiswell TE, Smith RM, Katz LB, et al. Bronchopulmonary segmental lavage with Surfaxin (KL(4)-surfactant) for acute respiratory distress syndrome. Am J Respir Crit Care Med. 1999;160:1188–1195. doi: 10.1164/ajrccm.160.4.9808118. [DOI] [PubMed] [Google Scholar]
- 61.Spragg RG, Lewis JF, Wurst W, et al. Treatment of acute respiratory distress syndrome with recombinant surfactant protein C surfactant. Am J Respir Crit Care Med. 2003;167:1562–1566. doi: 10.1164/rccm.200207-782OC. [DOI] [PubMed] [Google Scholar]
- 62.Gregory TJ, Steinberg KP, Spragg R, et al. Bovine surfactant therapy for patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1997;155:1309–1315. doi: 10.1164/ajrccm.155.4.9105072. [DOI] [PubMed] [Google Scholar]
- 63.Spragg RG, Lewis JF, Walmrath HD, et al. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med. 2004;351:884–892. doi: 10.1056/NEJMoa033181. [DOI] [PubMed] [Google Scholar]
- 64.Baudouin SV. Exogenous surfactant replacement in ARDS—one day, someday, or never? N Engl J Med. 2004;351:853–855. doi: 10.1056/NEJMp048172. [DOI] [PubMed] [Google Scholar]
- 65.Willson DF, Thomas NJ, Markovitz BP, et al. Effect of exogenous surfactant (calfactant) in pediatric acute lung injury: a randomized controlled trial. JAMA. 2005;293:470–476. doi: 10.1001/jama.293.4.470. [DOI] [PubMed] [Google Scholar]
- 66.Kaisers U, Busch T, Deja M, et al. Selective pulmonary vasodilation in acute respiratory distress syndrome. Crit Care Med. 2003;31:S337–342. doi: 10.1097/01.CCM.0000057913.45273.1A. [DOI] [PubMed] [Google Scholar]
- 67.Jardin F, Gurdjian F, Fouilladieu JL, et al. Pulmonary and systemic haemodynamic disorders in the adult respiratory distress syndrome. Intensive Care Med. 1979;5:127–133. doi: 10.1007/BF01683194. [DOI] [PubMed] [Google Scholar]
- 68.Villar J, Blazquez MA, Lubillo S, et al. Pulmonary hypertension in acute respiratory failure. Crit Care Med. 1989;7:523–526. doi: 10.1097/00003246-198906000-00007. [DOI] [PubMed] [Google Scholar]
- 69.Squara P, Dhainaut JF, Artigas A, et al. Hemodynamic profile in severe ARDS: results of the European Collaborative ARDS Study. Intensive Care Med. 1998;24:1018–1028. doi: 10.1007/s001340050710. [DOI] [PubMed] [Google Scholar]
- 70.Rimar S, Gillis CN. Selective pulmonary vasodilation by inhaled nitric oxide is due to hemoglobin inactivation. Circulation. 1993;88:2884–2887. doi: 10.1161/01.cir.88.6.2884. [DOI] [PubMed] [Google Scholar]
- 71.Pepke-Zaba J, Higenbottam TW, Dinh-Xuan AT, et al. Inhaled nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertension. Lancet. 1991;338:1173–1174. doi: 10.1016/0140-6736(91)92033-x. [DOI] [PubMed] [Google Scholar]
- 72.Frostell C, Fratacci MD, Wain JC, et al. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation. 1991;83:2038–2047. doi: 10.1161/01.cir.83.6.2038. [DOI] [PubMed] [Google Scholar]
- 73.Rossaint R, Falke KJ, Lopez F, et al. Inhaled nitric oxide for the adult respiratory distress syndrome. N Engl J Med. 1993;328:399–405. doi: 10.1056/NEJM199302113280605. [DOI] [PubMed] [Google Scholar]
- 74.Michael JR, Barton RG, Saffle JR, et al. Inhaled nitric oxide versus conventional therapy: effect on oxygenation in ARDS. Am J Respir Crit Care Med. 1998;157:1372–1380. doi: 10.1164/ajrccm.157.5.96-10089. [DOI] [PubMed] [Google Scholar]
- 75.Troncy E, Collet JP, Shapiro S, et al. Inhaled nitric oxide in acute respiratory distress syndrome: a pilot randomized controlled study. Am J Respir Crit Care Med. 1998;157:1483–1488. doi: 10.1164/ajrccm.157.5.9707090. [DOI] [PubMed] [Google Scholar]
- 76.Dellinger RP, Zimmerman JL, Taylor RW, et al. Effects of inhaled nitric oxide in patients with acute respiratory distress syndrome: results of a randomized phase II trial. Inhaled Nitric Oxide in ARDS Study Group. Crit Care Med. 1998;26:15–23. doi: 10.1097/00003246-199801000-00011. [DOI] [PubMed] [Google Scholar]
- 77.Lundin S, Mang H, Smithies M, et al. Inhalation of nitric oxide in acute lung injury: results of a European multicentre study. The European Study Group of Inhaled Nitric Oxide. Intensive Care Med. 1999;25:911–919. doi: 10.1007/s001340050982. [DOI] [PubMed] [Google Scholar]
- 78.Taylor RW, Zimmerman JL, Dellinger RP, et al. Low-dose inhaled nitric oxide in patients with acute lung injury: a randomized controlled trial. JAMA. 2004;291:1603–1609. doi: 10.1001/jama.291.13.1603. [DOI] [PubMed] [Google Scholar]
- 79.Walmrath D, Schneider T, Pilch J, et al. Aerosolised prostacyclin in adult respiratory distress syndrome. Lancet. 1993;342:961–962. doi: 10.1016/0140-6736(93)92004-d. [DOI] [PubMed] [Google Scholar]
- 80.Zwissler B, Kemming G, Habler O, et al. Inhaled prostacyclin (PGI2) versus inhaled nitric oxide in adult respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:1671–1677. doi: 10.1164/ajrccm.154.6.8970353. [DOI] [PubMed] [Google Scholar]
- 81.Walmrath D, Schneider T, Schermuly R, et al. Direct comparison of inhaled nitric oxide and aerosolized prostacyclin in acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;153:991–996. doi: 10.1164/ajrccm.153.3.8630585. [DOI] [PubMed] [Google Scholar]
- 82.Van Heerden PV, Blythe D, Webb SA. Inhaled aerosolized prostacyclin and nitric oxide as selective pulmonary vasodilators in ARDS—a pilot study. Anaesth Intensive Care. 1996;24:564–568. doi: 10.1177/0310057X9602400510. [DOI] [PubMed] [Google Scholar]
- 83.Pappert D, Busch T, Gerlach H, et al. Aerosolized prostacyclin versus inhaled nitric oxide in children with severe acute respiratory distress syndrome. Anesthesiology. 1995;82:1507–1511. doi: 10.1097/00000542-199506000-00020. [DOI] [PubMed] [Google Scholar]
- 84.Mikawa K, Akamatsu H, Maekawa N, et al. Inhibitory effect of prostaglandin E1 on human neutrophil function. Prostaglandins Leukot Essent Fatty Acids. 1994;51:287–291. doi: 10.1016/0952-3278(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 85.Putensen C, Hormann C, Kleinsasser A, et al. Cardiopulmonary effects of aerosolized prostaglandin E1 and nitric oxide inhalation in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 1998;157:1743–1747. doi: 10.1164/ajrccm.157.6.9609017. [DOI] [PubMed] [Google Scholar]
- 86.Melot C, Lejeune P, Leeman M, et al. Prostaglandin E1 in the adult respiratory distress syndrome. Benefit for pulmonary hypertension and cost for pulmonary gas exchange. Am Rev Respir Dis. 1989;139:106–110. doi: 10.1164/ajrccm/139.1.106. [DOI] [PubMed] [Google Scholar]
- 87.Zhang H, Benlabed M, Spapen H, et al. Prostaglandin E1 increases oxygen extraction capabilities in experimental sepsis. J Surg Res. 1994;57:470–479. doi: 10.1006/jsre.1994.1172. [DOI] [PubMed] [Google Scholar]
- 88.Groeneveld AB, Vermeij CG, Nauta JJ, et al. Vasodilating prostaglandin E1 decreases critical oxygen delivery by increasing critical oxygen extraction in anesthetized pigs. Circ Shock. 1993;39:253–262. [PubMed] [Google Scholar]
- 89.Grant MM, Burnett CM, Fein AM. Effect of prostaglandin E1 infusion on leukocyte traffic and fibrosis in acute lung injury induced by bleomycin in hamsters. Crit Care Med. 1991;19:211–217. doi: 10.1097/00003246-199102000-00017. [DOI] [PubMed] [Google Scholar]
- 90.Yamanaka K, Saluja AK, Brown GE, et al. Protective effects of prostaglandin E1 on acute lung injury of caerulein-induced acute pancreatitis in rats. Am J Physiol. 1997;272:G23–G30. doi: 10.1152/ajpgi.1997.272.1.G23. [DOI] [PubMed] [Google Scholar]
- 91.Holcroft JW, Vassar MJ, Weber CJ. Prostaglandin E1 and survival in patients with the adult respiratory distress syndrome. A prospective trial. Ann Surg. 1986;203:371–378. doi: 10.1097/00000658-198604000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Slotman GJ, Kerstein MD, Bone RC, et al. The effects of prostaglandin E1 on non-pulmonary organ function during clinical acute respiratory failure. The Prostaglandin E1 Study Group. J Trauma. 1992;32:480–488. discussion 488–489. [PubMed] [Google Scholar]
- 93.Bone RC, Slotman G, Maunder R, et al. Randomized double-blind, multicenter study of prostaglandin E1 in patients with the adult respiratory distress syndrome. Prostaglandin E1 Study Group. Chest. 1989;96:114–119. doi: 10.1378/chest.96.1.114. [DOI] [PubMed] [Google Scholar]
- 94.Rossignon MD, Khayat D, Royer C, et al. Functional and metabolic activity of polymorphonuclear leukocytes from patients with adult respiratory distress syndrome: results of a randomized double-blind placebo-controlled study on the activity of prostaglandin E1. Anesthesiology. 1990;72:276–281. doi: 10.1097/00000542-199002000-00012. [DOI] [PubMed] [Google Scholar]
- 95.Eierman DF, Yagami M, Erme SM, et al. Endogenously opsonized particles divert prostanoid action from lethal to protective in models of experimental endotoxemia. Proc Natl Acad Sci U S A. 1995;92:2815–2819. doi: 10.1073/pnas.92.7.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abraham E, Park YC, Covington P, et al. Liposomal prostaglandin E1 in acute respiratory distress syndrome: a placebo-controlled, randomized, double-blind, multicenter clinical trial. Crit Care Med. 1996;24:10–15. doi: 10.1097/00003246-199601000-00005. [DOI] [PubMed] [Google Scholar]
- 97.Abraham E, Baughman R, Fletcher E, et al. Liposomal prostaglandin E1 (TLC C-53) in acute respiratory distress syndrome: a controlled, randomized, double-blind, multi-center clinical trial. TLC C-53 ARDS Study Group. Crit Care Med. 1999;27:1478–1485. doi: 10.1097/00003246-199908000-00013. [DOI] [PubMed] [Google Scholar]
- 98.Vincent JL, Brase R, Santman F, et al. A multi-centre, double-blind, placebo-controlled study of liposomal prostaglandin E1 (TLC C-53) in patients with acute respiratory distress syndrome. Intensive Care Med. 2001;27:1578–1583. doi: 10.1007/s001340101077. [DOI] [PubMed] [Google Scholar]
- 99.Wiegers GJ, Reul JM. Induction of cytokine receptors by glucocorticoids: functional and pathological significance. Trends Pharmacol Sci. 1998;19:317–321. doi: 10.1016/s0165-6147(98)01229-2. [DOI] [PubMed] [Google Scholar]
- 100.Newton R. Molecular mechanisms of glucocorticoid action: what is important? Thorax. 2000;55:603–613. doi: 10.1136/thorax.55.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saatcioglu F, Claret FX, Karin M. Negative transcriptional regulation by nuclear receptors. Semin Cancer Biol. 1994;5:347–359. [PubMed] [Google Scholar]
- 102.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci U S A. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang-Yen HF, Chambard JC, Sun YL, et al. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 104.van de Stolpe A, Caldenhoven E, Raaijmakers JA, et al. Glucocorticoid-mediated repression of intercellular adhesion molecule-1 expression in human monocytic and bronchial epithelial cell lines. Am J Respir Cell Mol Biol. 1993;8:340–347. doi: 10.1165/ajrcmb/8.3.340. [DOI] [PubMed] [Google Scholar]
- 105.Jantz MA, Sahn SA. Corticosteroids in acute respiratory failure. Am J Respir Crit Care Med. 1999;60:1079–1100. doi: 10.1164/ajrccm.160.4.9901075. [DOI] [PubMed] [Google Scholar]
- 106.Thompson BT. Glucocorticoids and acute lung injury. Crit Care Med. 2003;31:S253–S257. doi: 10.1097/01.CCM.0000057900.19201.55. [DOI] [PubMed] [Google Scholar]
- 107.Dik WA, McAnulty RJ, Versnel MA, et al. Short course dexamethasone treatment following injury inhibits bleomycin induced fibrosis in rats. Thorax. 2003;58:765–771. doi: 10.1136/thorax.58.9.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weigelt JA, Norcross JF, Borman KR, et al. Early steroid therapy for respiratory failure. Arch Surg. 1985;120:536–540. doi: 10.1001/archsurg.1985.01390290018003. [DOI] [PubMed] [Google Scholar]
- 109.Sprung CL, Caralis PV, Marcial EH, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984;311:1137–1143. doi: 10.1056/NEJM198411013111801. [DOI] [PubMed] [Google Scholar]
- 110.Bone RC, Fisher CJ, Jr, Clemmer TP, et al. Early methylprednisolone treatment for septic syndrome and the adult respiratory distress syndrome. Chest. 1987;92:1032–1036. doi: 10.1378/chest.92.6.1032. [DOI] [PubMed] [Google Scholar]
- 111.Luce JM, Montgomery AB, Marks JD, et al. Ineffectiveness of high-dose methylprednisolone in preventing parenchymal lung injury and improving mortality in patients with septic shock. Am Rev Respir Dis. 1988;138:62–68. doi: 10.1164/ajrccm/138.1.62. [DOI] [PubMed] [Google Scholar]
- 112.Bernard GR, Luce JM, Sprung CL, et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N Engl J Med. 1987;317:1565–1570. doi: 10.1056/NEJM198712173172504. [DOI] [PubMed] [Google Scholar]
- 113.Ashbaugh DG, Maier RV. Idiopathic pulmonary fibrosis in adult respiratory distress syndrome. Diagnosis and treatment. Arch Surg. 1985;120:530–535. doi: 10.1001/archsurg.1985.01390290012002. [DOI] [PubMed] [Google Scholar]
- 114.Hooper RG, Kearl RA. Established ARDS treated with a sustained course of adrenocortical steroids. Chest. 1990;97:138–143. doi: 10.1378/chest.97.1.138. [DOI] [PubMed] [Google Scholar]
- 115.Hooper RG, Kearl RA. Established adult respiratory distress syndrome successfully treated with corticosteroids. South Med J. 1996;89:359–364. doi: 10.1097/00007611-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 116.Meduri GU, Belenchia JM, Estes RJ, et al. Fibroproliferative phase of ARDS. Clinical findings and effects of corticosteroids. Chest. 1991;100:943–952. doi: 10.1378/chest.100.4.943. [DOI] [PubMed] [Google Scholar]
- 117.Meduri GU, Chinn AJ, Leeper KV, et al. Corticosteroid rescue treatment of progressive fibroproliferation in late ARDS. Patterns of response and predictors of outcome. Chest. 1994;105:1516–1527. doi: 10.1378/chest.105.5.1516. [DOI] [PubMed] [Google Scholar]
- 118.Meduri GU, Headley AS, Golden E, et al. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1998;280:159–165. doi: 10.1001/jama.280.2.159. [DOI] [PubMed] [Google Scholar]
- 119.Lelcuk S, Huval WV, Valeri CR, et al. Inhibition of ischemia-induced thromboxane synthesis in man. J Trauma. 1984;24:393–396. doi: 10.1097/00005373-198405000-00004. [DOI] [PubMed] [Google Scholar]
- 120.Williams JG, Maier RV. Ketoconazole inhibits alveolar macrophage production of inflammatory mediators involved in acute lung injury (adult respiratory distress syndrome) Surgery. 1992;112:270–277. [PubMed] [Google Scholar]
- 121.Yu M, Tomasa G. A double-blind, prospective, randomized trial of ketoconazole, a thromboxane synthetase inhibitor, in the prophylaxis of the adult respiratory distress syndrome. Crit Care Med. 1993;21:1635–1642. doi: 10.1097/00003246-199311000-00010. [DOI] [PubMed] [Google Scholar]
- 122.Slotman GJ, Burchard KW, D’Arezzo A, et al. Ketoconazole prevents acute respiratory failure in critically ill surgical patients. J Trauma. 1988;28:648–654. doi: 10.1097/00005373-198805000-00015. [DOI] [PubMed] [Google Scholar]
- 123.Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. The ARDS Network. JAMA. 2000;283:1995–2002. doi: 10.1001/jama.283.15.1995. [DOI] [PubMed] [Google Scholar]
- 124.Mandell GL. ARDS, neutrophils, and pentoxifylline. Am Rev Respir Dis. 1988;138:1103–1105. doi: 10.1164/ajrccm/138.5.1103. [DOI] [PubMed] [Google Scholar]
- 125.Welsh CH, Lien D, Worthen GS, et al. Pentoxifylline decreases endotoxin-induced pulmonary neutrophil sequestration and extravascular protein accumulation in the dog. Am Rev Respir Dis. 1988;138:1106–1114. doi: 10.1164/ajrccm/138.5.1106. [DOI] [PubMed] [Google Scholar]
- 126.Seear MD, Hannam VL, Kaapa P, et al. Effect of pentoxifylline on hemodynamics, alveolar fluid reabsorption, and pulmonary edema in a model of acute lung injury. Am Rev Respir Dis. 1990;142:1083–1087. doi: 10.1164/ajrccm/142.5.1083. [DOI] [PubMed] [Google Scholar]
- 127.Kudoh I, Ohtake M, Nishizawa H, et al. The effect of pentoxifylline on acid-induced alveolar epithelial injury. Anesthesiology. 1995;82:531–541. doi: 10.1097/00000542-199502000-00023. [DOI] [PubMed] [Google Scholar]
- 128.Montravers P, Fagon JY, Gilbert C, et al. Pilot study of cardiopulmonary risk from pentoxifylline in adult respiratory distress syndrome. Chest. 1993;103:1017–1022. doi: 10.1378/chest.103.4.1017. [DOI] [PubMed] [Google Scholar]
- 129.Hasegawa N, Oka Y, Nakayama M, et al. The effects of post-treatment with lisofylline, a phosphatidic acid generation inhibitor, on sepsis-induced acute lung injury in pigs. Am J Respir Crit Care Med. 1997;155:928–936. doi: 10.1164/ajrccm.155.3.9117028. [DOI] [PubMed] [Google Scholar]
- 130.Rice GC, Rosen J, Weeks R, et al. CT-1501R selectively inhibits induced inflammatory monokines in human whole blood ex vivo. Shock. 1994;1:254–266. doi: 10.1097/00024382-199404000-00003. [DOI] [PubMed] [Google Scholar]
- 131.Guidot DM, Bursten SL, Rice GC, et al. Modulating phosphatidic acid metabolism decreases oxidative injury in rat lungs. Am J Physiol. 1997;273:L957–L966. doi: 10.1152/ajplung.1997.273.5.L957. [DOI] [PubMed] [Google Scholar]
- 132.Bursten SL, Federighi DA, Parsons P, et al. An increase in serum C18 unsaturated free fatty acids as a predictor of the development of acute respiratory distress syndrome. Crit Care Med. 1996;24:1129–1136. doi: 10.1097/00003246-199607000-00011. [DOI] [PubMed] [Google Scholar]
- 133.Rice GC, Brown PA, Nelson RJ, et al. Protection from endotoxic shock in mice by pharmacologic inhibition of phosphatidic acid. Proc Natl Acad Sci U S A. 1994;91:3857–3861. doi: 10.1073/pnas.91.9.3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Randomized, placebo-controlled trial of lisofylline for early treatment of acute lung injury and acute respiratory distress syndrome. Crit Care Med. 2002;30:1–6. doi: 10.1097/00003246-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 135.Baird BR, Cheronis JC, Sandhaus RA, et al. O2 metabolites and neutrophil elastase synergistically cause edematous injury in isolated rat lungs. J Appl Physiol. 1986;61:2224–2229. doi: 10.1152/jappl.1986.61.6.2224. [DOI] [PubMed] [Google Scholar]
- 136.Pacht ER, Timerman AP, Lykens MG, et al. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100:1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 137.Bunnell E, Pacht ER. Oxidized glutathione is increased in the alveolar fluid of patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1993;148:1174–1178. doi: 10.1164/ajrccm/148.5.1174. [DOI] [PubMed] [Google Scholar]
- 138.Quinlan GJ, Lamb NJ, Tilley R, et al. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am J Respir Crit Care Med. 1997;155:479–484. doi: 10.1164/ajrccm.155.2.9032182. [DOI] [PubMed] [Google Scholar]
- 139.Bernard GR. N-acetylcysteine in experimental and clinical acute lung injury. Am J Med. 1991;91:54S–59S. doi: 10.1016/0002-9343(91)90284-5. [DOI] [PubMed] [Google Scholar]
- 140.Bernard GR, Lucht WD, Niedermeyer ME, et al. Effect of N-acetylcysteine on the pulmonary response to endotoxin in the awake sheep and upon in vitro granulocyte function. J Clin Invest. 1984;73:1772–1784. doi: 10.1172/JCI111386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jepsen S, Herlevsen P, Knudsen P, et al. Antioxidant treatment with N-acetylcysteine during adult respiratory distress syndrome: a prospective, randomized, placebo-controlled study. Crit Care Med. 1992;20:918–923. doi: 10.1097/00003246-199207000-00004. [DOI] [PubMed] [Google Scholar]
- 142.Suter PM, Domenighetti G, Schaller MD, et al. N-acetylcysteine enhances recovery from acute lung injury in man. A randomized, double-blind, placebo-controlled clinical study. Chest. 1994;105:190–194. doi: 10.1378/chest.105.1.190. [DOI] [PubMed] [Google Scholar]
- 143.Domenighetti G, Suter PM, Schaller MD, et al. Treatment with N-acetylcysteine during acute respiratory distress syndrome: a randomized, double-blind, placebo-controlled clinical study. J Crit Care. 1997;12:177–182. doi: 10.1016/s0883-9441(97)90029-0. [DOI] [PubMed] [Google Scholar]
- 144.Bernard GR, Wheeler AP, Arons MM, et al. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest. 1997;112:164–172. doi: 10.1378/chest.112.1.164. [DOI] [PubMed] [Google Scholar]
- 145.Walsh TS, Lee A. N-acetylcysteine administration in the critically ill. Intensive Care Med. 1999;25:432–434. doi: 10.1007/s001340050876. [DOI] [PubMed] [Google Scholar]
- 146.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 147.Moran LK, Gutteridge JM, Quinlan GJ. Thiols in cellular redox signalling and control. Curr Med Chem. 2001;8:763–772. doi: 10.2174/0929867013372904. [DOI] [PubMed] [Google Scholar]
- 148.Quinlan GJ, Evans TW, Gutteridge JM. Oxidative damage to plasma proteins in adult respiratory distress syndrome. Free Radic Res. 1994;20:289–298. doi: 10.3109/10715769409145628. [DOI] [PubMed] [Google Scholar]
- 149.Quinlan GJ, Margarson MP, Mumby S, et al. Administration of albumin to patients with sepsis syndrome: a possible beneficial role in plasma thiol repletion. Clin Sci (Lond) 1998;95:459–465. [PubMed] [Google Scholar]
- 150.Quinlan GJ, Mumby S, Martin GS, et al. Albumin influences total plasma antioxidant capacity favorably in patients with acute lung injury. Crit Care Med. 2004;32:755–759. doi: 10.1097/01.ccm.0000114574.18641.5d. [DOI] [PubMed] [Google Scholar]
- 151.Finfer S, Norton R, Bellomo R, et al. The SAFE study: saline vs. albumin for fluid resuscitation in the critically ill. Vox Sang. 2004;87(suppl 2):123–131. doi: 10.1111/j.1741-6892.2004.00468.x. [DOI] [PubMed] [Google Scholar]
- 152.Martin GS, Moss M, Wheeler AP, et al. A randomized, controlled trial of furosemide with or without albumin in hypoproteinemic patients with acute lung injury. Crit Care Med. 2005;33:1681–1687. doi: 10.1097/01.ccm.0000171539.47006.02. [DOI] [PubMed] [Google Scholar]
- 153.Greene R, Zapol WM, Snider MT, et al. Early bedside detection of pulmonary vascular occlusion during acute respiratory failure. Am Rev Respir Dis. 1981;124:593–601. doi: 10.1164/arrd.1981.124.5.593. [DOI] [PubMed] [Google Scholar]
- 154.Tomashefski JF, Jr, Davies P, Boggis C, et al. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol. 1983;112:112–126. [PMC free article] [PubMed] [Google Scholar]
- 155.Nuckton TJ, Alonso JA, Kallet RH, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med. 2002;346:1281–1286. doi: 10.1056/NEJMoa012835. [DOI] [PubMed] [Google Scholar]
- 156.Creasey AA, Chang AC, Feigen L, et al. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–2860. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Welty-Wolf KE, Carraway MS, Miller DL, et al. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med. 2001;164:1988–1996. doi: 10.1164/ajrccm.164.10.2105027. [DOI] [PubMed] [Google Scholar]
- 158.Miller DL, Welty-Wolf K, Carraway MS, et al. Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 2002;26:650–658. doi: 10.1165/ajrcmb.26.6.4688. [DOI] [PubMed] [Google Scholar]
- 159.Abraham E, Reinhart K, Svoboda P, et al. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: a multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med. 2001;29:2081–2089. doi: 10.1097/00003246-200111000-00007. [DOI] [PubMed] [Google Scholar]
- 160.Abraham E, Reinhart K, Opal S, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 161.Fourrier F, Chopin C, Goudemand J, et al. Septic shock, multiple organ failure, and disseminated intravascular coagulation. Compared patterns of antithrombin III, protein C, and protein S deficiencies. Chest. 1992;101:816–823. doi: 10.1378/chest.101.3.816. [DOI] [PubMed] [Google Scholar]
- 162.Fourrier F, Chopin C, Huart JJ, et al. Double-blind, placebo-controlled trial of antithrombin III concentrates in septic shock with disseminated intravascular coagulation. Chest. 1993;104:882–888. doi: 10.1378/chest.104.3.882. [DOI] [PubMed] [Google Scholar]
- 163.Baudo F, Caimi TM, de Cataldo F, et al. Antithrombin III (ATIII) replacement therapy in patients with sepsis and/or postsurgical complications: a controlled double-blind, randomized, multicenter study. Intensive Care Med. 1998;24:336–342. doi: 10.1007/s001340050576. [DOI] [PubMed] [Google Scholar]
- 164.Eisele B, Lamy M, Thijs LG, et al. Antithrombin III in patients with severe sepsis. A randomized, placebo-controlled, double-blind multicenter trial plus a meta-analysis on all randomized, placebo-controlled, double-blind trials with antithrombin III in severe sepsis. Intensive Care Med. 1998;4:663–672. doi: 10.1007/s001340050642. [DOI] [PubMed] [Google Scholar]
- 165.Warren BL, Eid A, Singer P, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 166.Tasaki O, Mozingo DW, Dubick MA, et al. Effects of heparin and lisofylline on pulmonary function after smoke inhalation injury in an ovine model. Crit Care Med. 2002;30:637–643. doi: 10.1097/00003246-200203000-00024. [DOI] [PubMed] [Google Scholar]
- 167.Uchiba M, Okajima K, Murakami K, et al. Attenuation of endotoxin-induced pulmonary vascular injury by antithrombin III. Am J Physiol. 1996;270:L921–L930. doi: 10.1152/ajplung.1996.270.6.L921. [DOI] [PubMed] [Google Scholar]
- 168.Abubakar K, Schmidt B, Monkman S, et al. Heparin improves gas exchange during experimental acute lung injury in newborn piglets. Am J Respir Crit Care Med. 1998;158:1620–1625. doi: 10.1164/ajrccm.158.5.9803041. [DOI] [PubMed] [Google Scholar]
- 169.Malik AB, van der Zee H. Time course of pulmonary vascular response to microembolization. J Appl Physiol. 1977;43:51–58. doi: 10.1152/jappl.1977.43.1.51. [DOI] [PubMed] [Google Scholar]
- 170.Davidson BL, Geerts WH, Lensing AW. Low-dose heparin for severe sepsis. N Engl J Med. 2002;347:1036–1037. doi: 10.1056/NEJM200209263471316. [DOI] [PubMed] [Google Scholar]
- 171.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 172.Opal SM. Unintended bias, clinical trial results, and the heparin post hoc crossover fallacy. Crit Care Med. 2004;32:874–875. doi: 10.1097/01.ccm.0000114812.42346.fe. [DOI] [PubMed] [Google Scholar]
- 173.Ware LB, Fang X, Matthay MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L514–L521. doi: 10.1152/ajplung.00442.2002. [DOI] [PubMed] [Google Scholar]
- 174.Prabhakaran P, Ware LB, White KE, et al. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L20–L28. doi: 10.1152/ajplung.00312.2002. [DOI] [PubMed] [Google Scholar]
- 175.van der Poll T, Levi M, Nick JA, et al. Activated protein C inhibits local coagulation after intrapulmonary delivery of endotoxin in humans. Am J Respir Crit Care Med. 2005;171:1125–1128. doi: 10.1164/rccm.200411-1483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 177.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 178.Crandall ED, Matthay MA. Alveolar epithelial transport. Basic science to clinical medicine. Am J Respir Crit Care Med. 2001;163:1021–1029. doi: 10.1164/ajrccm.163.4.2006116. [DOI] [PubMed] [Google Scholar]
- 179.Berthiaume Y, Staub NC, Matthay MA. Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J Clin Invest. 1987;79:335–343. doi: 10.1172/JCI112817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Berthiaume Y, Broaddus VC, Gropper MA, et al. Alveolar liquid and protein clearance from normal dog lungs. J Appl Physiol. 1988;65:585–593. doi: 10.1152/jappl.1988.65.2.585. [DOI] [PubMed] [Google Scholar]
- 181.Sakuma T, Okaniwa G, Nakada T, et al. Alveolar fluid clearance in the resected human lung. Am J Respir Crit Care Med. 1994;150:305–310. doi: 10.1164/ajrccm.150.2.8049807. [DOI] [PubMed] [Google Scholar]
- 182.Sakuma T, Suzuki S, Usuda K, et al. Preservation of alveolar epithelial fluid transport mechanisms in rewarmed human lung after severe hypothermia. J Appl Physiol. 1996;80:1681–1686. doi: 10.1152/jappl.1996.80.5.1681. [DOI] [PubMed] [Google Scholar]
- 183.Sakuma T, Folkesson HG, Suzuki S, et al. Beta-adrenergic agonist stimulated alveolar fluid clearance in ex vivo human and rat lungs. Am J Respir Crit Care Med. 1997;155:506–512. doi: 10.1164/ajrccm.155.2.9032186. [DOI] [PubMed] [Google Scholar]
- 184.Lasnier JM, Wangensteen OD, Schmitz LS, et al. Terbutaline stimulates alveolar fluid resorption in hyperoxic lung injury. J Appl Physiol. 1996;81:1723–1729. doi: 10.1152/jappl.1996.81.4.1723. [DOI] [PubMed] [Google Scholar]
- 185.Saldias FJ, Lecuona E, Comellas AP, et al. Beta-adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am J Respir Crit Care Med. 2000;162:282–287. doi: 10.1164/ajrccm.162.1.9809058. [DOI] [PubMed] [Google Scholar]
- 186.Saldias FJ, Comellas A, Ridge KM, et al. Isoproterenol improves ability of lung to clear edema in rats exposed to hyperoxia. J Appl Physiol. 1999;87:30–35. doi: 10.1152/jappl.1999.87.1.30. [DOI] [PubMed] [Google Scholar]
- 187.Garat C, Meignan M, Matthay MA, et al. Alveolar epithelial fluid clearance mechanisms are intact after moderate hyperoxic lung injury in rats. Chest. 1997;111:1381–1388. doi: 10.1378/chest.111.5.1381. [DOI] [PubMed] [Google Scholar]
- 188.McAuley DF, Frank JA, Fang X, et al. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med. 2004;32:1470–1476. doi: 10.1097/01.ccm.0000129489.34416.0e. [DOI] [PubMed] [Google Scholar]
- 189.Minnear FL, DeMichele MA, Moon DG, et al. Isoproterenol reduces thrombin-induced pulmonary endothelial permeability in vitro. Am J Physiol. 1989;257:H1613–H1623. doi: 10.1152/ajpheart.1989.257.5.H1613. [DOI] [PubMed] [Google Scholar]
- 190.Minnear FL, DeMichele MA, Leonhardt S, et al. Isoproterenol antagonizes endothelial permeability induced by thrombin and thrombin receptor peptide. J Appl Physiol. 1993;75:1171–1179. doi: 10.1152/jappl.1993.75.3.1171. [DOI] [PubMed] [Google Scholar]
- 191.Khimenko PL, Barnard JW, Moore TM, et al. Vascular permeability and epithelial transport effects on lung edema formation in ischemia and reperfusion. J Appl Physiol. 1994;77:1116–1121. doi: 10.1152/jappl.1994.77.3.1116. [DOI] [PubMed] [Google Scholar]
- 192.Zhang H, Kim YK, Govindarajan A, et al. Effect of adrenoreceptors on endotoxin-induced cytokines and lipid peroxidation in lung explants. Am J Respir Crit Care Med. 1999;160:1703–1710. doi: 10.1164/ajrccm.160.5.9903068. [DOI] [PubMed] [Google Scholar]
- 193.Wright PE, Carmichael LC, Bernard GR. Effect of bronchodilators on lung mechanics in the acute respiratory distress syndrome (ARDS) Chest. 1994;106:1517–1523. doi: 10.1378/chest.106.5.1517. [DOI] [PubMed] [Google Scholar]
- 194.Sartori C, Allemann Y, Duplain H, et al. Salmeterol for the prevention of high-altitude pulmonary edema. N Engl J Med. 2002;346:1631–1636. doi: 10.1056/NEJMoa013183. [DOI] [PubMed] [Google Scholar]
- 195.Ware LB, Matthay MA. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol. 2002;282:L924–L940. doi: 10.1152/ajplung.00439.2001. [DOI] [PubMed] [Google Scholar]
- 196.Barton SP, Marks R. Changes in suspensions of human keratinocytes due to trypsin. Arch Dermatol Res. 1981;271:245–257. doi: 10.1007/BF00409453. [DOI] [PubMed] [Google Scholar]
- 197.Adamson IY, Bakowska J. Relationship of keratinocyte growth factor and hepatocyte growth factor levels in rat lung lavage fluid to epithelial cell regeneration after bleomycin. Am J Pathol. 1999;155:949–954. doi: 10.1016/S0002-9440(10)65194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Panos RJ, Bak PM, Simonet WS, et al. Intratracheal instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats. J Clin Invest. 1995;96:2026–2033. doi: 10.1172/JCI118250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yi ES, Salgado M, Williams S, et al. Keratinocyte growth factor decreases pulmonary edema, transforming growth factor-beta and platelet-derived growth factor-BB expression, and alveolar type II cell loss in bleomycin-induced lung injury. Inflammation. 1998;22:315–325. doi: 10.1023/a:1022304317111. [DOI] [PubMed] [Google Scholar]
- 200.Yano T, Deterding RR, Simonet WS, et al. Keratinocyte growth factor reduces lung damage due to acid instillation in rats. Am J Respir Cell Mol Biol. 1996;15:433–442. doi: 10.1165/ajrcmb.15.4.8879176. [DOI] [PubMed] [Google Scholar]
- 201.Sugahara K, Iyama K, Kuroda MJ, et al. Double intratracheal instillation of keratinocyte growth factor prevents bleomycin-induced lung fibrosis in rats. J Pathol. 1998;186:90–98. doi: 10.1002/(SICI)1096-9896(199809)186:1<90::AID-PATH137>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 202.Mason CM, Guery BP, Summer WR, et al. Keratinocyte growth factor attenuates lung leak induced by alpha-naphthylthiourea in rats. Crit Care Med. 1996;24:925–931. doi: 10.1097/00003246-199606000-00009. [DOI] [PubMed] [Google Scholar]
- 203.Guery BP, Mason CM, Dobard EP, et al. Keratinocyte growth factor increases transalveolar sodium reabsorption in normal and injured rat lungs. Am J Respir Crit Care Med. 1997;155:1777–1784. doi: 10.1164/ajrccm.155.5.9154891. [DOI] [PubMed] [Google Scholar]
- 204.Panoskaltsis-Mortari A, Lacey DL, Vallera DA, et al. Keratinocyte growth factor administered before conditioning ameliorates graft-versus-host disease after allogeneic bone marrow transplantation in mice. Blood. 1998;92:3960–3967. [PubMed] [Google Scholar]
- 205.Chen L, Brizel DM, Rabbani ZN, et al. The protective effect of recombinant human keratinocyte growth factor on radiation-induced pulmonary toxicity in rats. Int J Radiat Oncol Biol Phys. 2004;0:1520–1529. doi: 10.1016/j.ijrobp.2004.07.729. [DOI] [PubMed] [Google Scholar]
- 206.Meropol NJ, Somer RA, Gutheil J, et al. Randomized phase I trial of recombinant human keratinocyte growth factor plus chemotherapy: potential role as mucosal protectant. J Clin Oncol. 2003;21:1452–1458. doi: 10.1200/JCO.2003.10.079. [DOI] [PubMed] [Google Scholar]
- 207.Shoemaker WC, Appel PL. Effects of prostaglandin E1 in adult respiratory distress syndrome. Surgery. 1986;99:275–283. [PubMed] [Google Scholar]
- 208.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome; four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]