

Abstract
Background/Aims
We have previously shown that phagocytosis of apoptotic bodies (AB) by hepatic stellate cells (HSC) is profibrogenic. As HSC survival is central to the progression of liver fibrosis, our goal was to investigate if phagocytosis induces HSC survival.
Methods
Apoptosis of phagocytosing HSC was studied in the presence of known apoptotic agents. The JAK/STAT and PI3K/Akt dependent pathways, NF-κB activation and expression of the anti-apoptotic proteins Mcl-1 and A1 were evaluated. Apoptosis was assessed after blocking A1 by an siRNA approach.
Results
Phagocytosing HSC were resistant to FasL/cycloheximide or TRAIL-induced apoptosis. Inhibition of the JAK/STAT or PI3K-mediated pathways induced apoptosis of HSC. Phagocytosis induced JAK1/STAT3 phosphorylation, and this was prevented by inhibiting JAK. Translocation of STAT3 to the nucleus was also blocked by JAK inhibition. Mcl-1 expression was upregulated in a JAK-dependent manner. PI3K-dependent phosphorylation of Akt depended on NADPH oxidase activity and superoxide production. NF-κB activation and subsequent upregulation of A1 was observed, and A1 inhibition induced apoptosis of HSC.
Conclusion
Phagocytosis of AB promotes HSC survival by two pathways, of which the A1 dependent is more significant. This represents a new mechanism by which engulfment of AB contributes to the propagation of liver fibrosis.
Keywords: Apoptosis, phagocytosis, NADPH oxidase, oxidative radicals, liver fibrogenesis
Introduction
Hepatic stellate cells (HSC) play a pivotal role in liver fibrogenesis (1). Upon induction by fibrogenic stimuli, HSC undergo activation in which they transdifferentiate from a quiescent phenotype to myofibroblasts with production of type I collagen α1(1). Survival of HSC is a hallmark of liver fibrosis, and the induction of HSC apoptosis was shown to induce recovery from fibrosis (2-4). Activated human HSC are resistant to many proapoptotic stimuli, such as serum deprivation, Fas-ligand (FasL), and the main survival signal is the overexpression of Bcl-2 family members (5, 6). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) however, was shown to induce apoptosis of activated HSC via upregulation of TRAIL-R2/DR5 expression (4). Accumulating evidence suggests that reactive oxidative species (ROS) are not only pathological but in many instances they act as second messengers in cell survival pathways (7, 8). The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) are activated during oxidative stress (7, 9-11). Upon phosphorylation, STATs translocate to the nucleus and bind DNA (12). STAT3 phosphorylation is strongly linked to cell survival due to the induction of Bcl-xL (13) and myeloid cell leukemia-1 protein (Mcl-1) (14). We have recently shown that phagocytosis of apoptotic bodies (AB) by HSC activates nicotinamide adenine dinucleotide phosphate reduced (NADPH) oxidase, with resulting upregulation of procollagen α1(I) (15). Superoxide and hydrogen peroxide (H2O2) are known to induce Akt-dependent survival in different systems (16, 17). Because phosphoinositide-3 kinase (PI3K) and Akt are known to regulate nuclear factor κB (NF-κB) activity, it is tempting to speculate that NADPH oxidase would activate NF-κB-dependent survival signals. NF-κB activity is central to the expression of anti-apoptotic proteins, including the Bcl-2 family protein A1 in HSC (4).
Here we report that phagocytosis of AB induces different survival pathways in HSC leading to the propagation of myofibroblasts in the liver. Following the engulfment of AB, an NF-κB-dependent upregulation of the anti apoptotic protein A1 and a STAT3-mediated induction of Mcl-1 are demonstrated.
Materials and methods
Cell culture
Immortalized human HSC line, LX-2 (kindly provided by SL Friedman MD, Mt-Sinai Medical School, NY) and primary rat HSC were used. Rat HSC isolation was performed according to Geerts et al. (18), and the purity was always >95% as assessed by vitamin A fluorescence. Primary HSC were maintained in Medium 199 (Sigma-Aldrich, St. Louis, MO) with 20% FBS. LX-2 cells were cultured in DMEM (Invitrogen) with 5% FBS. Before the experiments the medium was changed to serum-free.
Generation of apoptotic bodies
To generate carboxytetramethyl rhodamine succinimidyl ester-(TAMRA, Invitrogen, Carlsbad, CA) labeled AB, HepG2 cells were incubated with TAMRA (10μM), then exposed to UV irradiation (100 mJ/cm2, 142 seconds) as described previously (19). Labeled AB were added to primary HSC or LX-2 cells. Phagocytosis of AB was detected by fluorescence and phase microscopy (15, 19).
Apoptosis experiments
Primary HSC were incubated with AB for 24 hours then incubated further in the presence or absence of FasL (5ng/ml, EMD Chemicals, San Diego, CA), and/or cycloheximide (10μg/ml, Sigma-Aldrich) for 18 hours, JAK inhibitor, AG490 (50μM, 1 hour, EMD Chemicals.), a selective PI3K inhibitor, LY294002 (600 nM, 1 hour, EMD Chemicals). For the experiments involving TRAIL, primary HSC were culture-activated for 7 days then exposed to 500 ng/ml recombinant TRAIL (R&D Systems, Minneapolis, MN) overnight in the presence or absence of AB. Active caspase-3 was detected by an in situ labeling method using an antibody to active caspase-3 (FITC/PE detection kit, Cell Technology, Mountain View, CA). Nuclei were labeled with DAPI (Vector Laboratories, Burlingame, CA). Apoptosis was assessed by the characteristic nuclear apoptotic changes (DAPI) and by the presence of active caspase-3. Biochemical assays to assess caspase activity were not done as HSC with engulfed AB would have given false positive signal. 500 cells in 3 different fields in each of 3 different experiments were counted.
Cytochrome c reduction assay
Primary HSC were plated and exposed to AB. The supernatant was collected 2 hours after exposure to AB. Cytochrome c (160 uM) (Sigma-Aldrich, St. Louis, MO) was added in the presence (100 U/ml) or absence of superoxide dismutase (SOD) (Sigma-Aldrich, St. Louis, MO), and incubated for 20 minutes at 37°C. The values of superoxide-dependent reduction of cytochrome c were obtained by subtracting the absorbance readings at 550 nm of the samples without SOD from those with SOD. The final result was reported as fold changes above the control values (20).
Western blot analysis
To detect JAK/STAT and Akt phosphorylation, LX-2 cells were incubated in serum free medium for 16 hours then exposed to AB with or without inhibitors, AG490 (50 μM), LY294002 (20 μM), or apocynin (100 μM). Cells were then collected at different time points. The protein concentration was determined with the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA), and 20 μg of protein was separated by SDS-PAGE. The blots were incubated with the appropriate antibodies for 16 hours at 4°C: anti-phospho-JAK1 polyclonal antibody (1:500, EMD-Calbiochem), anti-phospho-STAT3 monoclonal antibody (1:250, Cell Signaling Technology, Danvers, MA), anti-JAK1 and anti-STAT3 polyclonal antibodies (1:300, Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho Akt and anti-Akt polyclonal antibodies (1:1000, Cell Signaling Technology), and anti-GAPDH polyclonal antibody (1:3000, Trevigen, Gaithersburg, MD). The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and the blots were developed by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL). To detect Mcl-1 and A1 expression, primary HSC were exposed to AB in the presence or absence of AG490 (50 μM), LY294002 (600 nM), apocynin (100 μM) or CAPE (1ng/ml). Western blot analysis was performed as above using Mcl-1 antibody (1:1000, Millipore Billerica, MA), A1 polyclonal antibody (1:300, Biovision, Mountain View, CA). The data were normalized to the expression of β-actin (1:1000, Santa Cruz Biotechnology).
cDNA Transfection
pCMV6-XL6-SOCS3 (Origene, Rockville, Md), and the control vector were transfected into LX-2 cells (transfection efficiency 60-70%), using the Lipofectamine reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s recommendation.
RNA Interference
siRNA to A1 (Ambion Austin, TX), and negative control labeled (Alexa Fluor 488) or unlabeled siRNA (Qiagen, Valencia, CA) were transfected into primary HSC with 30nM siRNA in RiboJuice™ (Novagen, Madison, WI), according to the manufacturer’s instructions. Scrambled siRNA in the same concentration was used as control. The cells were exposed to AB in the presence or absence of FasL and cycloheximide, as above and apoptosis was assessed.
STAT3 nuclear translocation assay
Primary HSC were incubated with TAMRA-labeled AB in the presence or absence of AG490 (50μM) for 3 hours, then fixed with 1% paraformaldehyde. After washes, the cells were incubated in methanol at -20° for 2 minutes then washed, incubated in 10% FBS/PBS for 1 hour at 37°C, and stained with the anti-STAT3 antibody (1:200, Santa Cruz Biotechnology). An FITC-conjugated secondary antibody (1:2000, Invitrogen) was applied, and the nuclei were stained with DAPI. The results were analyzed by fluorescence microscopy.
Electrophoretic mobility shift assay (EMSA)
LX-2 cells were serum-starved and incubated with AB for 2 hours then nuclear extracts were prepared using a cytosolic/nuclear extraction kit (Pierce Biotechnology). The NF-κB nuclear translocation was determined using Biotin 3’ prime DNA labeling kit and lightshift chemiluminescent EMSA kit (Pierce Biotechnology). The NF-κB consensus oligonucleotide (5’-AGTTGAGGGGACTTTCCCAGGC-3’) was end-labeled with biotin then the nuclear extract was mixed with the biotin-labeled NF-κB oligonucleotide. The lysate was subjected to electrophoresis then transferred to a nylon membrane. The membrane was exposed to the chemiluminescent substrate, and the resulting chemiluminescence was detected by autoradiography.
Statistical Analysis
All data represent at least three experiments and expressed as the mean ± STD. Differences between groups were compared using one-way analysis of Variance (ANOVA) associated with the Dunnett’s test. Statistical significance was assumed when p<0.05.
Results
Primary HSC engulfing apoptotic bodies are protected against apoptotic stimuli
To determine if phagocytosis is protective against known proapoptotic stimuli, primary HSC were exposed to AB in the presence or absence of cycloheximide and Fas ligand or TRAIL. The treatment including cycloheximide, blocking protein synthesis is known to induce apoptosis of primary HSC while FasL alone is less effective (21). Apoptosis was detected by active caspase 3/DAPI labeling. Cyloheximide and Fas ligand treatment induced apoptosis of HSC, while in phagocytosing HSC this was reduced by 31% compared to cells not exposed to AB (Figure 1A). As TRAIL induces apoptosis of activated HSC, primary HSC were culture-activated and exposed to TRAIL in the presence or absence of AB. TRAIL induced a significant apoptosis of HSC while phagocytosis of AB decreased this by 46%. These data suggest that phagocytosis of AB has a protective, anti-apoptotic effect on HSC.
Figure 1. Phagocytosis of Apoptotic Bodies Induces HSC Survival.

(A) Exposure to AB did not change the apoptotic rate of primary HSC (3.7% ± 0. 9) compared to control (2.9% ± 0.2). FasL (FSL) combined with cycloheximide (CHX) induced a 15.8% (± 1.1) increase in HSC apoptosis. This was decreased to 11% (±1.0) in phagocytosing HSC N+5,*p<0.001, **p<0.05. (B) TRAIL-induced apoptosis of primary, culture activated HSC (50.6%±6.1) was significantly decreased in phagocytosing HSC (27.6%± 1.8). Apoptosis was based on the detection of active caspase-3/DAPI staining. Data are normalized to control, set as 1. N=4, **p<0.05
Inhibition of JAK1/STAT3 and PI3K induces apoptosis of phagocytosing HSC
As ROS production has been linked to the activation of the JAK/STAT and PI3K-dependent survival pathways (7, 9, 22, 23), next we studied these pathways in phagocytosing HSC. Primary HSC 2 days after isolation were exposed to TAMRA-labeled AB in the presence or absence of the JAK inhibitor AG490 (50 μM) or the PI3K inhibitor LY294002 (600 nM), or both to assess if these pathways play a role. Immunohistochemistry detecting the active caspase 3 subunit and DAPI staining were performed, and the rate of apoptosis quantified. Biochemical assessment of apoptosis was not done as the lysis of the cells with intracellular AB would have resulted in a false-positive signal. In parallel, propidium iodide (PI) staining was performed as well, to detect cellular necrosis. In both the control group and the AB treated group, the rate of apoptosis was low (Figure 2A a, b) however, it increased significantly in the AG490 plus AB-treated group (Figure 2A c). Combining the inhibitors of both pathways, we detected further increase in apoptosis of HSC, as shown by the number of active caspase 3 positive cells with the characteristic nuclear condensation/fragmentation (Figure 2A d, overlay image). As illustrated in Figure 2B, AG490 and LY294002 treatment with these conditions induced apoptosis which was slightly decreased by phagocytosis. Combining JAK and PI3K inhibition, the rate of apoptosis rose further (78.3% ±3.1), and there was some reduction in phagocytosing cells however, the apoptotic rate still remained significant. The percentage of PI positive cells was lower than 0.5% among all groups (data not shown). These data indicate that in phagocytosing HSC cell survival mediated by a JAK/STAT and PI3K-dependent pathway.
Figure 2. Blocking the JAK/STAT and/or PI3K pathways Induces Apoptosis of Phagocytosing HSC.

(A) DAPI (blue) nuclear staining and immunofluorescence to detect active caspase-3 (green) were performed on rat primary HSC treated for 24 hours with TAMRA-labeled AB (red) in the presence or absence of AG490 (AG) and/or PI3K inhibitor LY294002 (LY). DMSO treated cells served as control. a) control HSC were only DAPI-positive. b) In HSC exposed to AB, no active caspase 3 signal is detectable. c) AG490-treated phagocytosing HSC are positive for active caspase 3, indicating higher apoptotic rate. d) In phagocytosing HSC exposed to both AG490 and LY294002 the apoptosis was further induced. Arrows point to AB where active caspase-3 (green), DAPI (blue), and TAMRA (red) co-localize. Arrowhead: caspase-3 positive HSC. Bar: 10μm. (B) AB exposure did not change the apoptotic rate (4.0% ±0.9) compared to control (2.9% ± 0.8). AG490 induced 26.9% (±1.4) of apoptosis and LY294002 induced 40.8% (±1.0) of apoptosis in HSC which was slightly decreased by phagocytosis (not statistically significant). Combined inhibition of both pathways induced 78.3% (±3.1) apoptosis, and this somewhat decreased by phagocytosis of AB however, the apoptotic rate remained significant (57.4% ±2.8). N=4, *p<0.001.
Engulfment of apoptotic bodies induces activation of JAK1 and STAT3, and STAT3 translocation to the nucleus
Because inhibition of JAK activation after AB engulfment induced apoptosis, we next investigated whether the JAK1/STAT3 pathways are activated in phagocytosing HSC. Western blot analysis showed that phagocytosis of AB induced both JAK1 and STAT3 phosphorylation in a time-dependent manner (Figure 3A). As chemical inhibition of JAK1 phosphorylation may not be specific, to confirm these data, LX-2 cells were transfected with a SOCS3 expression plasmid prior to exposing them to AB. JAK1 and STAT3 phosphorylation were inhibited in the SOCS3-transfected cells (data not shown). As a positive control for JAK1 and STAT3 phosphorylation, we used leptin, a known cytokine to induce these pathways in HSC (24, 25) (Figure 3B). To assess if the phosphorylated STAT3 translocates to the nucleus, immunohistochemistry on primary, phagocytosing HSC in the presence or absence of the JAK inhibitor, AG490 was performed. STAT3 translocated to the nucleus in AB-exposed cells (Figure 3C b, c), and this was blocked by AG490 treatment (Figure 3C d), confirming that engulfment of AB indeed induces STAT3 activation and nuclear translocation. Interestingly, JAK1 and STAT3 phosphorylation could also be partially reduced by inhibiting PI3K activity, suggestive of a crosstalk between the two signaling pathways (Figure 3D).
Figure 3. Phagocytosis of AB Induces JAK/STAT Phosphorylation and STAT3 Nuclear Translocation.
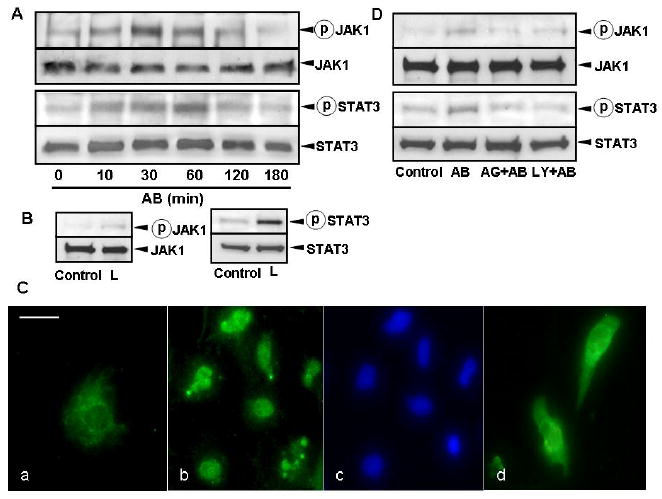
(A) LX-2 cells were exposed to AB, and the cell lysate was collected for immunoblotting using antibodies to phosho-JAK1, total JAK1, phospho-STAT3, and total STAT3. The assay showed that AB reduced JAK1 and STAT3 phosphorylation in a time-dependent manner. (B) As a positive control, phosphorylation of JAK1 and STAT3 were induced in LX-2 cells by leptin (75ng/ml, 10 min). (C) Immunocytochemistry using anti-STAT3 antibody was performed on rat primary HSC. (a) In control cells (not exposed to AB), STAT3 is cytoplasmic. (b, c) Incubation with AB induced STAT3 nuclear translocation in HSC, detected by the colocalization of the STAT3 and DAPI signal (d) AG490 treatment in AB-exposed HSC inhibited STAT3 translocation. STAT3: green, DAPI: blue. Bar: 20μm. (D) JAK1 and STAT3 phosphorylation could also partially be inhibited by inhibiting PI3K activity with preincubation of HSC by LY294002, suggestive of a crosstalk between the two signaling pathways.
Mcl-1 expression is upregulated in phagocytosing HSC
Since STAT3 is known to be at the center of cell survival pathways (14, 26, 27), and Mcl-1 expression is regulated by STAT3 at a transcriptional level (28, 29), next we tested if phagocytosis of AB upregulates Mcl-1 in HSC. Western blot analysis was performed on cytosolic extracts of primary HSC. After engulfment of AB, Mcl-1 was upregulated, and this was inhibited by prior incubation with AG490. Densitometry data show that phagocytosis of AB induces 1.83-fold upregulation of Mcl-1, while this is inhibited by blocking JAK activity (Figure 4). IL-6 served as positive control to study induction of Mcl-1.
Figure 4. Phagocytosis of AB Induces Mcl-1 Expression in HSC.
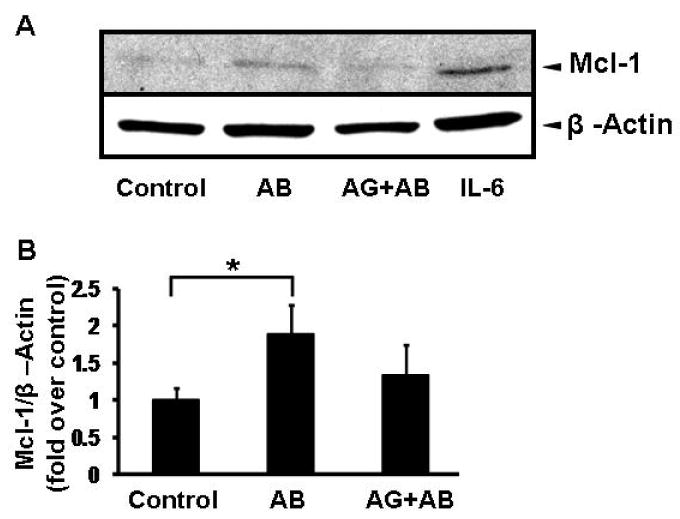
Primary rat HSC were treated with AB for 24 hours in the presence or absence of AG490. Western blot analysis was performed on the cytosolic extracts to detect changes in Mcl-1 expression. Mcl-1 expression increased in AB treated cells; this was inhibited by preincubation of cells with the JAK inhibitor, AG490. IL-6 treatment was used as a known inducer of Mcl-1 expression. Densitometry showed that phagocytosis of AB induced a 1.8-fold increase (±0.3) in expression, normalized to β-actin and presented as fold over control. N=4, *p<0.05.
Phagocytosis of AB induces PI3K-dependent phosphorylation of Akt
Our previous data showed that inhibition of PI3K with LY294002 induced apoptosis in phagocytosing HSC therefore survival of HSC may also involve the activation of an Akt-dependent pathway. As PI3K activation following phagocytosis is known to occur in concert with the production of reactive oxidative species (ROS) (16), first we performed a cytochrome c assay to demonstrate ROS release (Figure 5A). Next, HSC were exposed to AB in the presence or absence of LY294002, and AG490, to see if there is a crosstalk between the two signaling pathways. Treatment of the cells with the NADPH oxidase inhibitor apocynin was done as well, to test if Akt phosphorylation is dependent on NADPH oxidase activity and ROS production. Engulfment of AB induced phosphorylation of Akt in a time-dependent manner, and this was inhibited by the PI3K inhibitor LY294002 and apocynin (Figure 5B). Inhibition of JAK had less effect on Akt phosphorylation. Taken together, phagocytosis of AB induces an Akt-dependent survival pathway, which depends on ROS production by NADPH oxidase and PI3K activation.
Figure 5. Phagocytosis of AB Induces ROS Production and Akt Phosphorylation.

(A) LX-2 cells were incubated with AB for 6 hours and cytochrome c assay was used to evaluate ROS production. Phagocytosis of AB induced 2.2±0.4-fold of superoxide activity compared to control. N=4, *p<0.05. (B) LX-2 cells were exposed to AB with or without apocynin (NADPH oxidase inhibitor), AG490 or LY294002, and the cell lysates were collected for immunoblotting using antibodies to phosho-Akt and total Akt. AB transiently induced Akt phosphorylation, which was blocked by apocynin and LY294002 respectively, but not AG490.
Phagocytosis of AB induces NF-κB activation
NF-κB is central to regulating apoptosis and cell survival in HSC (30, 31), and phagocytosis was shown to activate NF-κB in macrophages (32). To study if NF-κB activation occurs in HSC after phagocytosis, and to explore the signaling pathways leading to its activation; HSC were exposed to AB after preincubation with the NF-κB inhibitor CAPE, JAK inhibitor AG490, the PI3K inhibitor LY294002, or the NADPH oxidase inhibitor apocynin. Nuclear extracts were obtained and an EMSA was performed. Phagocytosis of AB strongly induced NF-κB activation, and this was inhibited by the NF-κB inhibitor CAPE (Figure 6). Inhibiting the PI3K also reduced NF-κB activation significantly, while the reduction by inhibiting the JAK-mediated pathway or the NADPH oxidase was more modest. This is consistent with our previous data where inhibition of the PI3K activation induced apoptosis of phagocytosing HSC. This suggests that NF-κB activation is an integral part of the phagocytosis-induced HSC survival, and that this depends on PI3K activity.
Figure 6. Phagocytosis of AB Induces NF-κB activation.
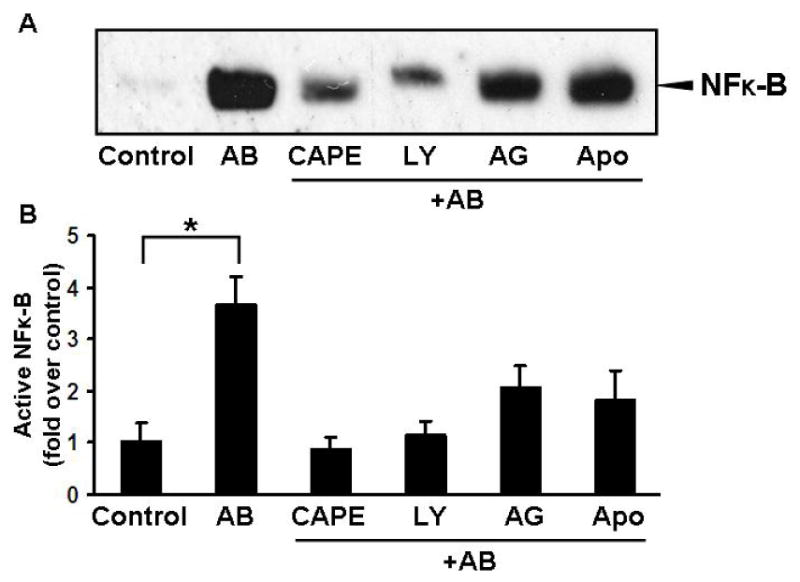
LX-2 cells were incubated in a serum free medium then exposed to AB with or without prior preincubation with CAPE (1ng/ml, 14 hours), LY294002 (600 nM, 1 hour), AG490 (50μM, 1 hour), or apocynin (100μM, 1 hour). Engulfment of the AB by LX-2 cells induced a significant activation of NF-κB (3.65±0.55-fold), and this was inhibited by CAPE, PI3K inhibition, and to a lesser extent by JAK and NADPH oxidase inhibition. Densitometry shows data from 3 experiments. *p<0.005
Phagocytosis of AB induces upregulation of the anti-apoptotic protein A1 and inhibition of A1 expression induces apoptosis of HSC
One of the main targets of NF-κB is the Bcl-2 family member A1, and it is known to play a major role in HSC survival (4). Thus, the next question is whether A1 is upregulated following phagocytosis. We performed western blot analysis on the cytosols of phagocytosing HSC cultured in the presence or absence of AG 490 and LY294002. We found that A1 indeed was upregulated following engulfment of AB in a PI3K-dependent manner (Figure 7A, B). To study if inhibition of A1 induces apoptosis of phagocytosing HSC, the cells were transfected with either scrambled siRNA or siRNA to A1. A1 expression was confirmed by western blot analysis (Figure 7C). At baseline (HSC exposed to AB) apoptosis increased following A1 inhibition suggesting that A1 is an important anti apoptotic protein in HSC (Figue 7D). After treating phagocytosing HSC with FasL and cycloheximide, A1 inhibition caused a significant increase in apoptosis even in phagocytosing cells. These data indicate that A1 expression is one of the major survival pathways induced in HSC following phagocytosis.
Figure 7. A1 expression is induced in phagocytosing hepatic stellate cells.

(A) LX-2 cells were exposed to AB in the presence or absence of LY294002, or AG490. Western blot analysis was done to study the anti apoptotic protein, A1 expression. (B) The quantitative densitometry analysis shows that phagocytosis of AB upregulated A1 expression (1.7±0.2-fold), and this was inhibited by blocking the PI3K activity. Inhibiting the JAK pathway however, had only a modest effect on A1 expression N=4, *p<0.05. To study the anti-apoptotic effect of A1, primary rat HSC were transfected with A1 siRNA (30 nM) and exposed to AB. (C) Western blot analysis shows the inhibition of A1 expression by the siRNA. (D) Primary HSC were transfected with either scrambled (Scr) or A1 siRNA (A1si), exposed to AB, in the presence or absence of FasL and cycloheximide. Caspase-3/DAPI staining was performed to assess apoptosis. In the Scr+AB group the apoptosis was (14.1%±0.8). This was increased by FasL and CHX (29.6%±1.2). A1 siRNA transfection induced higher apoptosis rate at baseline (only AB-exposed cells) and after FasL/CHX treatment (42.1%±0.7). Mean±SED, N=4, *p<0.001, **p<0.005.
Discussion
HSC survival during liver injury is an important factor in propagation of liver fibrosis. The exact mechanisms responsible for maintaining an active HSC population however, are not well understood. Our study describes a novel mechanism linking chronic liver injury with resulting hepatocyte apoptosis to phagocytosis of AB and the induction of survival signals in HSC.
The presence of phagocytic cells in an adequate number at the site of tissue injury is essential to maintain homeostasis, first, by the clearance of apoptotic debris and/or pathogens, second, by producing anti-inflammatory cytokines such as TGF-β. Survival of professional macrophages phagocytosing AB was previously demonstrated (32, 33), playing a role in the resolution of inflammation. Increased production of TGF-β by phagocytosing HSC was shown by us (15). As clearance of AB by HSC induces their activation and consequent production of ECM components, it is imperative that we determine the fate of phagocytosing HSC. In our study we have shown that in phagocytosing HSC there are different anti-apoptotic pathways induced, including a JAK1/STAT3-dependent pathway and a NADPH oxidase-dependent PI3K/Akt/NF-κB induction pathway. ROS in given circumstances have been described as pro-apoptotic (34, 35) and also anti-apoptotic (7, 8). The reason for this controversy is that the effects of ROS largely depend on the redox conditions of the cell, concentration and availability of antioxidants, the source, amount and time course of ROS production (36). ROS can act as second messengers in given circumstances inducing anti-apoptotic pathways in different cell-types (8, 37). The signaling pathways that govern these responses are just beginning to emerge and include the activation of the PI3K/Akt-mediated pathway and NF-κB (37). PI3K is an important regulatory element of the NADPH oxidase complex and plays a role in the regulation of superoxide production in neutrophils (38). Phagocytosis-induced activation of the PI3K leading to Akt phosphorylation was demonstrated in several studies (33, 38, 39). Akt is a crucial survival signaling factor in several cell types including HSC (17). In HSC, leptin protects cells from cycloheximide or TRAIL induced apoptosis via phosphorylation of Akt (40, 41). ROS exposure is also known to induce Akt-dependent survival pathways (22, 37, 42). In the studies by Wang et al. oxidative stress-induced activation of the PI3K/Akt pathway was required for cell survival and inhibition of the PI3K induced apoptosis in multiple cell types (43). Consistent with these, we found that PI3K-dependent Akt phosphorylation follows phagocytosis of AB, and inhibition of this pathway induces apoptosis of HSC. Inhibiting the NADPH oxidase with apocynin reduced phosphorylation of Akt, suggesting that NADPH oxidase activation is preceding Akt phosphorylation. As NF-κB is a potent pro-survival transcription factor in activated HSC (30, 44-46), and its inhibition could cause apoptosis of activated HSC (4), next we tested if phagocytosis of AB induces NF-κB activation. Phagocytosis-induced significant activation of NF-κB, this was PI3K/Akt-dependent, and partially inhibited by apocynin in HSC, suggesting that NADPH oxidase-derived ROS play a role in its activation. To further elucidate the possible target of NF-κB, we found that A1, an anti-apoptotic Bcl-2 family member was upregulated upon phagocytosis, and this was inhibited by the PI3K inhibitor. NF-κB-mediated A1 upregulation was shown to be required for survival of activated macrophages following phagocytosis (47), and also crucial in the survival of activated HSC (4). To assess if indeed A1 expression is a dominant mechanism of HSC survival following phagocytosis, A1 expression was inhibited by an siRNA approach. This has resulted in a significant increase in apoptosis of phagocytosing cells suggesting that A1 is a key anti-apoptotic protein.
We have also described the activation of a JAK/STAT mediated survival pathway in phagocytosing HSC. STAT3 is considered to be central to multiple anti apoptotic pathways (26, 48). Abrogation of STAT3 activation by an antisense strategy increased tumor cell apoptosis and decreased Bcl-xL expression in head and neck tumors (49), while in a different study, expressing the truncated, inhibitory form of STAT3 induced cell death in melanoma (50). In our studies we found that following phagocytosis, STAT3 became phosphorylated and translocated to the nucleus, and these events were inhibited by blocking JAK1 activity. One of the targets of STAT3 is Mcl-1 which belongs to the Bcl-2 family of anti-apoptotic proteins, and contributes to the control of mitochondrial integrity, which is critical for maintaining cell viability (51). We found that Mcl-1 was upregulated following phagocytosis of AB in primary HSC and that this was inhibited by blocking JAK1. This upregulation, albeit less significant as the activation of the NF-κB/A1 axis may also play a role in cell survival following phagocytosis. Other potential targets of STAT3 such as survivin (52, 53) may also induce HSC survival. Survivin was shown to be upregulated in different models of liver fibrosis (54). As to how JAK1 activation occurs following phagocytosis, is not yet elucidated. JAK activation may occur via phagocytosis-induced engagement of integrins (55), or alternatively, it could be the result of crosstalk between the PI3K/JAK pathways (56). Indeed, we found evidence in our studies that inhibition of PI3K may affect JAK1 and STAT3 phosphorylation. This crosstalk has been demonstrated in other experimental models especially involving ROS-mediated signaling pathways (57).
In summary, these studies have delineated a novel pathway of HSC survival. We have shown that phagocytosis of AB induces different survival signals in HSC, resulting in NF-κB and A1 activation/upregulation, and to a lesser degree Mcl-1 induction. As survival of activated, phagocytosing HSC contributes to the propagation of liver fibrosis, therapeutic strategies, targeting these pathways may be useful in inducing apoptosis of activated HSC, which may result in the reversal of fibrosis.
Figure 8. Phagocytosis of AB by HSC induces different survival pathways.

Engulfment of AB induces a JAK1/STAT3 mediated survival pathway leading to an upregulation of Mcl-1. Phagocytosis also induces Akt phosphorylation via the PI3K, and this contributes to NF-κB activation. As a result, the A1 anti apoptotic protein is upregulated in phagocytosing HSC, in a PI3K-dependent manner.
Acknowledgments
This study was supported by the NIH DK069765 (NJT) and the UC Davis Health System Awards (NJT).
Abbreviations
- HSC
hepatic stellate cells
- TGF-β1
transforming growth factor-β1
- AB
apoptotic bodies
- ASMA
α-smooth muscle actin
- NADPH oxidase
nicotinamide adenine dinucleotide phosphate reduced oxidase
- TAMRA
carboxytetramethyl rhodamine succinimidyl ester
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedman SL. Stellate cells: a moving target in hepatic fibrogenesis. Hepatology. 2004;40:1041–1043. doi: 10.1002/hep.20476. [DOI] [PubMed] [Google Scholar]
- 2.Wright MC, Issa R, Smart DE, Trim N, Murray GI, Primrose JN, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121:685–698. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 3.Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43:335–344. doi: 10.1002/hep.21036. [DOI] [PubMed] [Google Scholar]
- 5.Novo E, Marra F, Zamara E, Valfre di Bonzo L, Monitillo L, Cannito S, et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut. 2006;55:1174–1182. doi: 10.1136/gut.2005.082701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawada N. Human hepatic stellate cells are resistant to apoptosis: implications for human fibrogenic liver disease. Gut. 2006;55:1073–1074. doi: 10.1136/gut.2005.090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu HM, Zhi JL, Cui Y, Tang EH, Sun SN, Feng JQ, et al. Role of the JAK-STAT pathway in protection of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress in PC12 cells. Apoptosis. 2006;11:931–941. doi: 10.1007/s10495-006-6578-9. [DOI] [PubMed] [Google Scholar]
- 8.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 9.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 10.Maziere C, Conte MA, Maziere JC. Activation of JAK2 by the oxidative stress generated with oxidized low-density lipoprotein. Free Radic Biol Med. 2001;31:1334–1340. doi: 10.1016/s0891-5849(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 11.Waris G, Turkson J, Hassanein T, Siddiqui A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J Virol. 2005;79:1569–1580. doi: 10.1128/JVI.79.3.1569-1580.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 13.Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97:4227–4232. doi: 10.1073/pnas.97.8.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107:351–362. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43:435–443. doi: 10.1002/hep.21093. [DOI] [PubMed] [Google Scholar]
- 16.Caro AA, Cederbaum AI. Role of phosphatidylinositol 3-kinase/AKT as a survival pathway against CYP2E1-dependent toxicity. J Pharmacol Exp Ther. 2006;318:360–372. doi: 10.1124/jpet.106.102921. [DOI] [PubMed] [Google Scholar]
- 17.Kim D, Chung J. Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–115. doi: 10.5483/bmbrep.2002.35.1.106. [DOI] [PubMed] [Google Scholar]
- 18.Geerts A, Niki T, Hellemans K, De Craemer D, Van Den Berg K, Lazou JM, et al. Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatology. 1998;27:590–598. doi: 10.1002/hep.510270238. [DOI] [PubMed] [Google Scholar]
- 19.Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 20.Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 21.Gong W, Pecci A, Roth S, Lahme B, Beato M, Gressner AM. Transformation-dependent susceptibility of rat hepatic stellate cells to apoptosis induced by soluble Fas ligand. Hepatology. 1998;28:492–502. doi: 10.1002/hep.510280229. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, et al. The Role of the NADPH Oxidase Complex, p38 MAPK and Akt in Regulating Human Monocyte/Macrophage Survival. Am J Respir Cell Mol Biol. 2006 doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papa S, Bubici C, Zazzeroni F, Pham CG, Kuntzen C, Knabb JR, et al. The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 2006;13:712–729. doi: 10.1038/sj.cdd.4401865. [DOI] [PubMed] [Google Scholar]
- 24.Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao Q, Mak KM, Ren C, Lieber CS. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279:4292–4304. doi: 10.1074/jbc.M308351200. [DOI] [PubMed] [Google Scholar]
- 26.Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–392. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- 27.Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, et al. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001;15:787–793. doi: 10.1038/sj.leu.2402093. [DOI] [PubMed] [Google Scholar]
- 28.Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329–1338. doi: 10.1002/hep.20966. [DOI] [PubMed] [Google Scholar]
- 29.Isomoto H, Mott JL, Kobayashi S, Werneburg NW, Bronk SF, Haan S, et al. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132:384–396. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oakley F, Meso M, Iredale JP, Green K, Marek CJ, Zhou X, et al. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128:108–120. doi: 10.1053/j.gastro.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927–939. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- 32.Cui D, Thorp E, Li Y, Wang N, Yvan-Charvet L, Tall AR, et al. Pivotal advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J Leukoc Biol. 2007;82:1040–1050. doi: 10.1189/jlb.0307192. [DOI] [PubMed] [Google Scholar]
- 33.Reddy SM, Hsiao KH, Abernethy VE, Fan H, Longacre A, Lieberthal W, et al. Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J Immunol. 2002;169:702–713. doi: 10.4049/jimmunol.169.2.702. [DOI] [PubMed] [Google Scholar]
- 34.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 35.Pessayre D, Fromenty B, Mansouri A. Mitochondrial injury in steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16:1095–1105. doi: 10.1097/00042737-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Fruehauf JP, Meyskens FL., Jr Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 37.Yang B, Oo TN, Rizzo V. Lipid rafts mediate H2O2 prosurvival effects in cultured endothelial cells. Faseb J. 2006;20:1501–1503. doi: 10.1096/fj.05-5359fje. [DOI] [PubMed] [Google Scholar]
- 38.Hawkins PT, Davidson K, Stephens LR. The role of PI3Ks in the regulation of the neutrophil NADPH oxidase. Biochem Soc Symp. 2007:59–67. doi: 10.1042/BSS0740059. [DOI] [PubMed] [Google Scholar]
- 39.Ruhul Abid M, Spokes KC, Shih SC, Aird WC. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J Biol Chem. 2007 doi: 10.1074/jbc.M702175200. [DOI] [PubMed] [Google Scholar]
- 40.Saxena NK, Titus MA, Ding X, Floyd J, Srinivasan S, Sitaraman SV, et al. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. Faseb J. 2004;18:1612–1614. doi: 10.1096/fj.04-1847fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lafdil F, Chobert MN, Couchie D, Brouillet A, Zafrani ES, Mavier P, Laperche Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology. 2006;44:228–239. doi: 10.1002/hep.21237. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, et al. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am J Respir Cell Mol Biol. 2007;36:68–77. doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- 44.Burow ME, Weldon CB, Melnik LI, Duong BN, Collins-Burow BM, Beckman BS, et al. PI3-K/AKT regulation of NF-kappaB signaling events in suppression of TNF-induced apoptosis. Biochem Biophys Res Commun. 2000;271:342–345. doi: 10.1006/bbrc.2000.2626. [DOI] [PubMed] [Google Scholar]
- 45.Gong L, Li Y, Nedeljkovic-Kurepa A, Sarkar FH. Inactivation of NF-kappaB by genistein is mediated via Akt signaling pathway in breast cancer cells. Oncogene. 2003;22:4702–4709. doi: 10.1038/sj.onc.1206583. [DOI] [PubMed] [Google Scholar]
- 46.Lang A, Schoonhoven R, Tuvia S, Brenner DA, Rippe RA. Nuclear factor kappaB in proliferation, activation, and apoptosis in rat hepatic stellate cells. J Hepatol. 2000;33:49–58. doi: 10.1016/s0168-8278(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 47.Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages require constitutive NF-kappaB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. 2000;20:8855–8865. doi: 10.1128/mcb.20.23.8855-8865.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim DJ, Chan KS, Sano S, Digiovanni J. Signal transducer and activator of transcription 3 (Stat3) in epithelial carcinogenesis. Mol Carcinog. 2007;46:725–731. doi: 10.1002/mc.20342. [DOI] [PubMed] [Google Scholar]
- 49.Lee TL, Yeh J, Friedman J, Yan B, Yang X, Yeh NT, et al. A signal network involving coactivated NF-kappaB and STAT3 and altered p53 modulates BAX/BCL-XL expression and promotes cell survival of head and neck squamous cell carcinomas. Int J Cancer. 2008;122:1987–1998. doi: 10.1002/ijc.23324. [DOI] [PubMed] [Google Scholar]
- 50.Niu G, Heller R, Catlett-Falcone R, Coppola D, Jaroszeski M, Dalton W, et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999;59:5059–5063. [PubMed] [Google Scholar]
- 51.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–271. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 52.Aoki Y, Feldman GM, Tosato G. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003;101:1535–1542. doi: 10.1182/blood-2002-07-2130. [DOI] [PubMed] [Google Scholar]
- 53.Kanda N, Seno H, Konda Y, Marusawa H, Kanai M, Nakajima T, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23:4921–4929. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 54.De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132:1937–1946. doi: 10.1053/j.gastro.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 55.Rossetti G, Collinge M, Bender JR, Molteni R, Pardi R. Integrin-dependent regulation of gene expression in leukocytes. Immunol Rev. 2002;186:189–207. doi: 10.1034/j.1600-065x.2002.18616.x. [DOI] [PubMed] [Google Scholar]
- 56.Weber-Nordt RM, Mertelsmann R, Finke J. The JAK-STAT pathway: signal transduction involved in proliferation, differentiation and transformation. Leuk Lymphoma. 1998;28:459–467. doi: 10.3109/10428199809058353. [DOI] [PubMed] [Google Scholar]
- 57.Sauer H, Neukirchen W, Rahimi G, Grunheck F, Hescheler J, Wartenberg M. Involvement of reactive oxygen species in cardiotrophin-1-induced proliferation of cardiomyocytes differentiated from murine embryonic stem cells. Exp Cell Res. 2004;294:313–324. doi: 10.1016/j.yexcr.2003.10.032. [DOI] [PubMed] [Google Scholar]