

Abstract
Recurrent interstitial deletion of a region of 8p23.1 flanked by the low copy repeats 8p-OR-REPD and 8p-OR-REPP is associated with a spectrum of anomalies that can include congenital heart malformations and congenital diaphragmatic hernia (CDH). Haploinsufficiency of GATA4 is thought to play a critical role in the development of these birth defects. We describe two individuals and a monozygotic twin pair discordant for anterior CDH all of whom have complex congenital heart defects caused by this recurrent interstitial deletion as demonstrated by array comparative genome hybridization. To better define the genotype/phenotype relationships associated with alterations of genes on 8p23.1, we review the spectrum of congenital heart and diaphragmatic defects that have been reported in individuals with isolated GATA4 mutations and interstitial, terminal, and complex chromosomal rearrangements involving the 8p23.1 region. Our findings allow us to clearly define the CDH minimal deleted region on chromosome 8p23.1 and suggest that haploinsufficiency of other genes, in addition to GATA4, may play a role in the severe cardiac and diaphragmatic defects associated with 8p23.1 deletions. These findings also underscore the importance of conducting a careful cytogenetic/molecular analysis of the 8p23.1 region in all prenatal and postnatal cases involving congenital defects of the heart and/or diaphragm.
Keywords: Congenital heart defects, 8p23.1 deletion syndrome, Diaphragmatic hernia, Array comparative genomic hybridization, GATA4, Prenatal diagnosis
INTRODUCTION
Published deletions involving chromosome 8p23.1 range from large terminal deletions that are easily detectable by routine chromosome analysis to small interstitial deletions which are best identified using fluorescence in situ hybridization (FISH) or molecular techniques such as array comparative genomic hybridization (aCGH) [Paez et al., 2008; Reddy 1999; Devriendt et al., 1999]. Recurrent deletions of a region of 8p23.1 flanked by low copy repeats 8p-OR-REPD (distal) and 8p-OR-REPP (proximal) are associated with a spectrum of anomalies including congenital heart malformations, congenital diaphragmatic hernia (CDH), developmental delay, and neuropsychiatric findings [Digilio et al., 1998; Ciccone et al., 2001; Shimokawa et al., 2005; Ciccone et al., 2006; Zuffardi et al., 2006].
This region contains the transcription factor gene GATA4 which is known to play a key role in heart development in humans [Garg et al., 2003; Okubo et al., 2004; Sarkozy et al., 2005]. Disruption of Gata4 in mice has also been shown to cause heart defects and anterior CDH [Molkentin et al., 1997; Kuo et al., 1997; Jay et al., 2007]. Mice homozygous for a Gata4 null allele failed to form a heart tube and died between E7.0 and E9.5 [Kuo et al., 1997; Molkentin et al., 1997]. More recently, Jay et al. reported that a significant fraction of C57BL/6 mice heterozygous for a Gata4 deletion in exon 2—which removed the translation start site and the N-terminal activation domain—died within 1 day of birth, and that developmental defects in these heterozygotes included atrial septal defects (ASD), ventricular septal defects (VSD) and atrioventricular septal defects (AVSD) [Jay et al., 2007].
In this report, we describe two individuals and an identical twin pair discordant for anterior CDH all of whom were diagnosed prenatally with complex congenital heart defects and were subsequently shown to have interstitial 8p23.1 deletions. We review the spectrum of congenital heart and diaphragmatic defects that have been reported in individuals with isolated GATA4 mutations and interstitial, terminal, and complex chromosomal rearrangements involving 8p23.1 and discuss the implications of this data on our understanding of the gene(s) within this region which may play a role in heart and diaphragm development.
CLINICAL REPORTS
Patient 1
Patient 1 is a male born at 39 3/7 weeks gestation to a 26-year-old G2P1→2 mother. Both parents were Hispanic and nonconsanguineous. Prenatal ultrasound revealed pulmonary and tricuspid atresia. No amniocentesis was performed. Birth weight was 2.82 kg (10th centile), length was 48.5 cm (50th centile) and frontooccipital circumference (OFC) was 31.5 cm (25th centile). In the neonatal period he was noted to have a two-vessel umbilical cord, a very large anterior fontanelle, downslanting palpebral fissures, mild micrognathia, a short neck, slender fingers, and proximally implanted thumbs. Head ultrasound revealed normal anatomy with possible thinning of the corpus callosum.
Shortly after birth he was admitted to the neonatal intensive care unit for respiratory distress and tachypnea. Postnatal cardiac evaluation revealed a complex single ventricle anatomy consisting of a double outlet right ventricle, unbalanced complete atrioventricular septal defect (AVSD) with left atrioventricular (AV) valve atresia and left ventricular hypoplasia, pulmonary atresia, and D-transposition of the great vessels. Despite stenting of his ductus ateriosus, he continued to have limited pulmonary outflow. During surgery to create a systemic-to-pulmonary artery (Blalock-Taussig) shunt and to ligate the stented patent ductus arteriosus (PDA), he was found to have bilateral superior vena cavae with absence of a bridging innominate vein.
Approximately 6 weeks after birth, he experienced a cardiopulmonary arrest requiring resuscitation and emergent use of extracorporeal membrane oxygenation (ECMO). While on ECMO he developed seizures. He was weaned, decannulated and maintained for a short period of time on high-dose inotropic support before dying at age one month.
Patient 2
Patient 2 is a female born via cesarean at 35 weeks to a G2P1→3 mother as part of a twin gestation. Prenatal ultrasound showed complex congenital heart disease and intra-uterine growth retardation. No anomalies were identified in the male twin. No amniocentesis was performed.
The family history was significant for postaxial polydactyly in father, paternal half brother and maternal half sister. A paternal half brother had aortic insufficiency but this was felt to be secondary to rheumatic heart disease.
Birth weight was 1320 gm (<5th centile), length was 39 cm (<5th centile) and frontooccipital circumference (OFC) was 27 cm (<5th centile). His weight, length, and OFC were at approximately the 50th centile level for a 29-30 week gestation infant, consistent with symmetric intra-uterine growth retardation. Postaxial polydactyly of the left hand was noted along with prominent heels. No facial dysmorphic features were noted.
Shortly after birth, the patient developed respiratory distress requiring intubation and mechanical ventilation. Cardiac evaluation by ECHO and cardiac angiography revealed a complex cardiovascular malformation consisting of an unbalanced AVSD, left AV valve atresia, hypoplastic left ventricle, severe tricuspid regurgitation, absent intrahepatic inferior vena cava and hemiazygos continuation to a left superior vena cava. Head and abdominal ultrasounds were normal.
Considering the severity of the cardiac condition, life-support was withdrawn with parental consent and the patient died at 88 days of life.
Patient 3
Patient 3 is a male infant born at 34 5/7 weeks via cesarean to a 33-year-old G1P0→2 mother as part of a monozygotic twin gestation (see Patient 4). Prenatal ultrasound at 20-weeks gestation revealed an AVSD with a large inlet ventricular septal defect (VSD) extending to the perimembranous septum, aortic hypoplasia, a dominant pulmonary artery, and mild tricuspid regurgitation. An amniocentesis was performed and aCGH and chromosome analyses revealed a deletion on chromosome 8p.
Birth weight was 1835 grams (25th centile), length was 44.5 cm (50th centile), and frontooccipital circumference (OFC) was 29 cm (10th centile). At birth, examination revealed a holosystolic murmur, mild dysmorphic facial features, minor digital anomalies, and a sacral dimple. Spinal cord tethering was ruled out by spinal ultrasound.
Postnatal cardiac evaluation revealed a balanced type A complete AVSD with mild AV regurgitation, moderate atrial septal defect (ASD), hypoplastic aortic arch, a narrow left ventricular outflow tract, a large PDA, and good biventricular function. At surgery, he was found to have a right atrium with two appendages and absence of a bridging innominate vein.
Shortly after birth, the patient was transferred to the neonatal intensive care with tachypnea and desaturations. Further evaluation revealed evidence of a large anterior congenital diaphragmatic hernia with herniation of the liver, levoposition of the heart and a hypoplastic, posteriorly rotated left lung.
The patient remained hospitalized until approximately 12 weeks of age and is presently 5-months-old.
Patient 4
Patient 4 is the male monozygotic twin of patient 3. Prenatally, he was found to a balanced complete AVSD, a small aortic arch, and a large PDA. An amniocentesis was performed and aCGH and chromosome analyses revealed a deletion on chromosome 8p.
He was born at 2020 grams (25th centile), with a length of 44.8 cm (30th centile), and frontooccipital circumference (OFC) of 29.5 cm (10th centile). Additional findings on postnatal cardiac evaluation included a small aortic valve, bilateral peripheral pulmonary stenosis, and mild right ventricular dilatation. He was also found to have a small right choroid plexus cyst and a sacral dimple. Spinal cord tethering was ruled out by spinal ultrasound.
He was discharged home on day of life 38, with a discharge weight of 2960 g, a length of 48.5 cm and an OFC of 33 cm.
MATERIALS AND METHODS
Patients
All four patients were referred for clinical cytogenetic testing. Institutional review board approved informed consent was obtained for high-resolution studies.
Clinical Cytogenetic Tests
Standard laboratory procedures were used for metaphase preparations. G-banded chromosome analysis and FISH analysis were performed in either Medical Genetics Laboratories at Baylor College of Medicine or by the referring center. DNA was extracted from peripheral blood [Ou et al., 2008] and amniotic fluid samples [Bi et al., 2008] as previously described. Patients 1 and 2 were examined in the neonatal period and Patients 3 and 4 in the prenatal period. In the case of Patients 1, 3, and 4, aCGH testing and chromosome analysis were performed concurrently at the Medical Genetics Laboratories at Baylor College of Medicine. For Patient 2, aCGH analysis was performed following normal chromosome analysis results. All clinical aCGH analyses were performed using Baylor College of Medicine (BCM) Chromosomal Microarray version 6 (CMA V6) manufactured by Agilent Technologies (Santa Clara, CA) according to the manufacturer's instructions with modifications [Ou et al., 2008].
High-Resolution aCGH Analysis
DNA from Patients 1 and 4 was further evaluated by high resolution genome-wide aCGH using Human Genome CGH 244K Oligo Microarray Kits G4411B (Agilent Technologies, Santa Clara, CA) according to the manufacturer's protocol version 2.0. Arrays were scanned using an Agilent DNA Microarray Scanner (Agilent Technologies, Santa Clara, CA). Data extracted using Feature Extraction Software Version 9.1.3 (Agilent Technologies, Santa Clara, CA) was analyzed using CGH Analytics 3.4.40 Software (Agilent Technologies, Santa Clara, CA) with copy number changes identified with the assistance of the Aberration Detection Method 2 algorithm (threshold 6.0). Control DNA consisted of DNA from a healthy gender-matched reference individual with no personal or family history of heart or diaphragm defects.
Sequencing Analysis
PCR amplification of genomic fragments was performed using previously described primer pairs [Okubo et al., 2004]. PCR-fragments were cleaned and sequenced commercially (Agencourt Bioscience, Beverly, MA) and DNA changes were identified by comparing the published GATA4 sequence to sequence data obtained from patient samples using Sequencher 4.5 software (GeneCodes, Ann Arbor, MI).
RESULTS
Visible deletions of 8p were identified on the chromosome analysis for Patients 1, 3 and 4 (Fig 1). Chromosome analysis and FISH for 22q11.2 deletions performed on a peripheral blood sample from Patient 2 at an outside facility were reported as normal.
Figure 1.
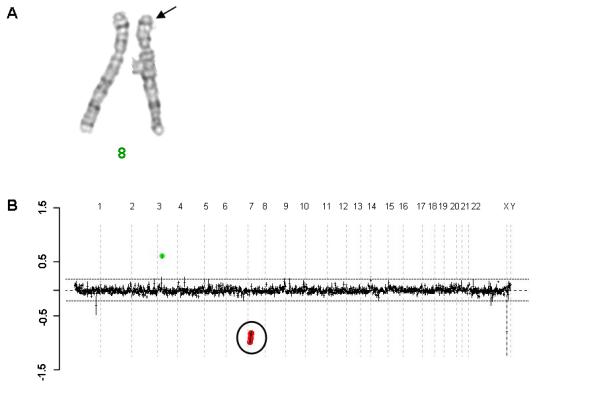
Partial karyotype and clinically based array comparative genomic hybridization results for Patient 4. A) Chromosome 8 partial karyotype showing an interstitial deletion of 8p23.1. B) Prenatal clinical array (BCM CMA V6) confirming deletion of chromosome 8p23.1 with the oligonucleotide probes detecting the copy number loss marked in red. This array also detected a gain in copy number of four genes (SLC2A9, WDR1, ZNF518B, MIST) on chromosome 4 (marked in green) all of which are lie partially or completely within known copy number variants.
Clinical aCGH detected an identical 8p23.1 deletion in all four patients with a minimal deletion size of 2.945 Mb (8,850,913 to 11,796,333) and a maximal deletion size of 6.352 Mb (6,436,314 to 12,788,647) (Fig 1). Parental studies confirmed that all of the deletions were de novo.
High resolution aCGH analyses confirmed the deletions previously seen in the clinical studies. In each case the deletion was flanked by the low copy repeats 8p-OR-REPD and 8p-OR-REPP with differences in apparent deletion size between Patients 1 and 4 being attributable to known copy number variant regions within and/or directly adjacent to the low copy repeats. The minimal deleted interval encompassed twenty-two known genes/open reading frames including GATA4 (Fig 2).
Figure 2.
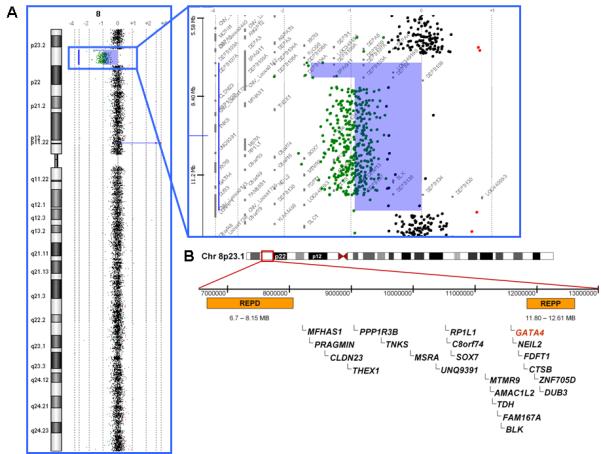
A) High resolution array comparative genomic hybridization analysis of Patient 4 showing an interstitial deletion of chromosome 8p23.1. B) Graphical representation of unique genes (RefSeq reviewed or validated) located between segmental duplications 8p-OR-REPD and 8p-OR-REPP which are deleted in all of the patients reported in this study. Defensin genes, which are present in multiple copies within segmental duplications, are not shown.
To determine whether disruption of both GATA4 alleles could explain the severe cardiac defects and CDH seen in our patients, we screened the entire GATA4 coding sequence and intron/exon boundaries of the remaining GATA4 allele in Patients 1 and 4. No variations from the published GATA4 reference sequence were identified in the coding sequence or the intron/exon boundaries of these DNA samples.
DISCUSSION
We have described the phenotype associated with the recurrent deletion of 8p23.1 in four patients with prenatally diagnosed complex congenital heart defects. The GATA4 gene resides within this recurrently deleted interval and has been implicated as the gene responsible for heart defects associated with 8p23.1 deletions. The role of GATA4 in heart development is supported by mouse models [Kuo et al., 1997; Molkentin et al., 1997; Jay et al., 2007] and studies of patients with GATA4 gene mutations.
Some of the most compelling evidence that mutations in GATA4 are associated with heart defects in humans come from studies of seven families in which ASD was found to segregate with heterozygous GATA4 mutations with varying levels of penetrance (Table I) [Garg et al., 2003; Hirayama-Yamada et al., 2005; Sarkozy et al., 2005; Okubo et al., 2004]. The GATA4 gene has also been screened in individual patients with ASD, VSD, AVSD, and tetralogy of Fallot (TOF). Although a number of non-synonymous changes were identified in these studies, it was often difficult to conclude whether these changes were causal due to a lack of parental/population controls and/or functional studies. This is particularly true of changes that are inherited from a non-affected parent. The most convincing of these changes include a de novo E216D missense mutations in two individuals with TOF that was shown to have reduced transcriptional activity in a reporter assay, and a frameshift mutation (Pro226fs) and a premature stop codon mutation (Arg266Ter)—which severely truncate the protein—each found in a patient with atrioventricular septal defects (AVSD) (Table II) [Nemer et al., 2006; Reamon-Buettner et al., 2007].
Table I.
GATA4 Alterations in Familial Heart Defects
| Reference | Alteration (AA change) | Phenotype | Penetrance | Families | Control chromosomes | Functional studies |
|---|---|---|---|---|---|---|
| Garg et al., 2003 | Gly296Ser | Non-syndromic ASD (+/- VSD, PVS, cardiac valve insufficiency, AVSD) | 100% of clinically evaluated affected individuals | 1 Family | 0/6000 (and 0/10 evaluated unaffected family members) | Disrupted interaction with TBX5, decreased DNA binding affinity, hypomorphic transactivation ability |
| Glu359fs | Non-syndromic ASD | 100% of evaluated affected individuals | 1 family | 0/6000 (and 0/2 evaluated unaffected family members) | Transcriptionally inactive | |
| Okubo et al., 2004 | Ser358fs | ASD +/- PS | 100% of clinically evaluated affected individuals | 1 family | 0/200 (and 0/13 evaluated unaffected family members) | Frameshift & premature STOP codon at amino acid 403, likely results in haploinsufficiency, similar to 1075delG described by Garg et al., 2003 |
| Sarkozy et al., 2005b | Gly296Ser | ASD, PVS, no conduction abnormalities | 2/2 Affected individuals (family 1), 3/3 clinically evaluated affected individuals (family 2) | 2/16 Families total | NR | See Garg et al., 2003 |
| Hirayama-Yamada et al., 2005 | Glu359fs | ASD (5 individuals) dextrocardia (1 individual) | 6/7 Affected individuals (family 1) | 1 Family/16 families | NR | |
| Ser52Phe | ASD (3 individuals) | 3/3 Affected individuals (family 2) | 1 Family/16 families | 0/202 Control chromosomes from healthy Japanese individuals | Decreased transcriptional activity (Schluterman et al., 2007), normal subcellular localization & DNA binding activity, Ser52 located in transactivation domain 1 (TAD1) |
ASD, atrial septal defect; AVSD, atrioventricular septal defect; NR, not reported; PS, pulmonary stenosis; PVS, pulmonary valve stenosis; VSD, ventricular septal defect.
Table II.
GATA4 Alterations in Sporadic Heart Defects
| Reference | Nucleotide Change¥ | AA Change | Phenotype | Frequency | Inheritance pattern | Control Chromosomes | Functional studies |
|---|---|---|---|---|---|---|---|
| Poirier et al., 2003 | NR | Ala411Val§ | Cardiac hypertrophy | Unclear (95 individuals screened) | NR | NR | NR |
| Poirier et al., 2003 | NR | Ser377Gly | Cardiac hypertrophy | Unclear (95 individuals screened) | NR | NR | NR |
| Reamon-Buettner et al., 2005 | 874T>C | Cys292Arg | VSD, ASD, AVSD | NR | 0/40 | NR | |
| 687G>T | Arg229Ser | VSD, ASD, AVSD | NR | 0/130 | NR | ||
| 631T>C | Phe211Leu | VSD, AVSD | NR | 0/130 | NR | ||
| 731A>G | Tyr244Cys | VSD, AVSD | NR | 0/130 | NR | ||
| 743A>G | Asn248Ser | ASD, AVSD | NR | 0/2 | NR | ||
| 782T>C | Leu261Pro | VSD, ASD | NR | NR | NR | ||
| 677delC | Pro226fs | AVSD | NR | 0/2 | NR | ||
| 700G>A | Gly234Ser | AVSD | NR | 0/4 | NR | ||
| 755G>C | Arg252Pro | AVSD | NR | 0/4 | NR | ||
| 796C>T | Arg266Ter | AVSD | NR | 0/2 | NR | ||
| 818A>G | Asn273Ser | AVSD | NR | 0/2 | NR | ||
| 830C>T | Thr277Ile | AVSD | NR | NR | NR | ||
| 848G>A | Arg283His | AVSD, overriding aorta, ASD | NR | 0/2 | NR | ||
| 855T>G | Asn285Lys | AVSD | NR | NR | NR | ||
| 905A>G | His302Arg | AVSD | NR | NR | NR | ||
| 622T>C | Phe208Leu | VSD | NR | 0/2 | NR | ||
| 640G>A | Gly214Ser | VSD | NR | NR | NR | ||
| 668T>C | Met223Thr | VSD | NR | 0/2 | NR | ||
| 715A>G | Asn239Asp | VSD | NR | NR | NR | ||
| 716A>G | Asn239Ser | VSD | NR | NR | NR | ||
| 779G>A | Arg260Gln | VSD | NR | NR | NR | ||
| 779G>A | Ile255Thr | ASD | NR | NR | NR | ||
| 881C>T | Ala294Val | ASD | NR | NR | NR | ||
| Nemer et al., 2006 | 648C>G | Glu216Asp | TOF | 2/26 Patients with TOF | De novo in both patients | 0/446 | Reduced transcriptional activity in reporter assay |
| Reamon-Buettner et al., 2007 | 1288C>G | Leu430Val | AVSD | NR | NR | NR | |
| 1081A>G | Met361Val | VSD | NR | NR | NR | ||
| 1130G>A | Ser377Asn | VSD | NR | NR | NR | ||
| 1324G>A | Ala442Thr | VSD | NR | NR | NR | ||
| 1295T>C | Leu432Ser | ASD | NR | NR | NR | ||
| 1288C>G | Leu430Val | ASD | NR | NR | NR | ||
| Rajagopal et al., 2007 | 487C>T | Pro163Ser | ECD | 1/43 Patients with ECD | Paternal, unaffected father | 0/600 | NR |
| 1037C>T | Ala346Val | ECD | 1/43 Patients with ECD | Maternal, unaffected mother | 0/600 | NR | |
| 886G>T | Gly296Cys | Secundum ASD, PVS | 1/8 Patients with septal defects | Paternal, affected father (persistent left superior vena cava) | 0/500 | NR | |
| 1207C>A | Leu403Met | Hypoplastic RV, sinus venosus ASD | 1/9 Patients with RV hypoplasia | Unknown, no parental DNA available | 0/500 | NR | |
| Tomita-Mitchell et al., 2007 | 278G>C§ | Gly93Ala§ | Secundum ASD | 1/222 ASD patients | Mother carrier, father unknown | 0/318 | NR |
| 946C>G§ | Gln316Glu§ | ASD, small muscular VSDs, mild PVS | 1/137 VSD patients | Both parents unknown | 0/318 | NR | |
| 1232C>T§ | Ala411Val§ | VSD | 1/137 VSD patients | Both parents unknown | 0/318 | NR | |
| 1273G>A§ | Asp425Asn§ | ASD | 1/222 ASD patients | Mother carrier, father not carrier | 0/528 | NR | |
| 1273G>A§ | Asp425Asn§ | TOF | 1/201 TOF patients | Both parents unknown | 0/528 | NR | |
| Posch et al., 2008 | 1232C>T§ | Ala411Val§ | Cribriform ASD and PAPVR | 1/205 patients with congenital septal defects | unknown | 0/600 | NR |
ASD, atrial septal defect; AVSD, atrioventricular septal defect; ECD, endocardial cushion defect; NR, not reported; PAPVR, partial anomalous pulmonary venous return; PVS, pulmonary valve stenosis; RV, right ventricle; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
Documented in SNP database but no further details available.
Nucleotide +1 is the A of the ATG translation initiation codon.
The complex cardiac phenotypes seen in our patients led us to question whether the spectrum of heart defects associated with 8p23.1 deletions was more severe than that seen in patients with GATA4 mutations. With this in mind we reviewed previous reports of interstitial, terminal and complex deletions of 8p23.1 (Table III) to determine the spectrum of cardiac phenotypes associated with these deletions. Previously reported patients with interstitial deletions were found to have a spectrum of cardiac defects that included ASD, VSD, AVSD, pulmonary stenosis, pulmonary valve stenosis, tetralogy of Fallot, and/or combinations of these defects. Cardiac defects in patients with terminal deletions extending to at least 8p23.1 included AVSD, hypoplastic left heart, hypoplastic right ventricle, pulmonary atresia/stenosis, pulmonary valve stenosis, partial anomalous pulmonary venous return, subaortic stenosis, transposition of the great arteries, double inlet/double outlet right ventricle, double inlet left ventricle and tetralogy of Fallot.
TABLE III.
Summary of Reported Cases Involving Deletion of 8p23.1
| Reference | Cytogenetic findings | Heart | Diaphragm/Lungs | Other |
|---|---|---|---|---|
| INTERSTITIAL DELETIONS | ||||
|
| ||||
| Present study, Patient 1 | 46,XY,del(8)(p23.1p23.1) | Complete AVSD (unbalanced), complex single ventricle, double outlet right ventricle, pulmonary atresia, left atrioventricular valve atresia, hypoplastic LV, D-transposition of the great vessels | Possible thinning of corpus callosum, large anterior fontanelle, dysmorphic features, 2-vessel umbilical cord | |
| Present study, Patient 2 | 46,XY,del(8)(p23.1p23.1) | AVSD (unbalanced), left atrioventricular valve atresia, hypoplastic LV, severe tricuspid regurgitation, absent intrahepatic inferior vena cava, hemiazygos continuation to left SVC | Symmetrical intrauterine growth retardation, familial postaxial polydactyly of the left hand | |
| Present study, Patient 3 | 46,XY,del(8)(p23.1p23.1) | Complete AVC (balanced type A), mild AV regurgitation, hypoplastic aortic arch, narrow left ventricular outflow tract | Large anterior CDH with left lobe of liver in anterior mediastinum, left lung hypoplasia | Small sacral dimple but no evidence of spinal abnormalities |
| Present study, Patient 4 | 46,XY,del(8)(p23.1p23.1) | Complete AVSD (balanced), small aortic valve, bilateral peripheral pulmonary stenosis, mild right ventricular dilatation | Small sacral dimple but no evidence of spinal abnormalities | |
| Orye and Craen, 1976 | 46,XY,del(8)(pter→p23∷p21→qter)* | MR, slow growth, dysmorphic features, broad chest, puffy hands and feet | ||
| Hutchinson et al., 1992; Giglio et al., 2000 | 46,XY,del(8)(pter→p23.2∷p22→qter) | PS, PFO | Partial agenesis of corpus callosum | |
| Digilio et al., 1998 | del(8)(p21p23) | AVSD, subaortic stenosis, persistent left SVC | Slow growth, microcephaly, MR, dysmorphic features, broad chest, hypospadias | |
| Digilio et al., 1998 | del(8)(p21p23) | Complete AVSD, PS, persistent left SVC | ||
| Digilio et al., 1998, 1999 | del(8)(p21p23) | AVSD, PS, dextrocardia, persistent left SVC, hypoplastic RV | Slow growth, microcephaly, MR, dysmorphic features, broad chest, hypospadias, cryptorchidism | |
| Pehlivan et al., 1999 | 46,XX,del(8)(p23.1p23.2) | Secundum ASD, PVS, left SVC, cardiomyopathy | Basal ganglia calcification, arched palate, hypothyroidism, atrophic pancreas, lordosis, renal artery stenosis | |
| Pehlivan et al., 1999 | 46,XX,del(8)(p23.1p23.2) | AVSD, TOF | Language delay, MR, dysmorphic features, esotropia, scoliosis | |
| Reddy, 1999 | 46,XY,del(8)(p23.1p23.1) or add(8)(p23.1)[20] | ASD, VSD, PS | Hepatosplenomegaly, feet deformity | |
| Reddy, 1999 | 46,XY,del(8)(p23.1p23.1). ish del(8)(p23.1p23.1)(wcp8+, 8ptel+) | ASD | Developmental delay, behavioral disorder | |
| Shimokawa et al., 2005 | 46,XY,del(8)(p23.1p23.1) | ASD | Left CDH with herniation of abdominal organs into left thorax, left pulmonary hypoplasia | Bilateral cryptorchidism |
| Slavotinek et al., 2005; 2006 | 46,XY,del(8)(p23.1p23.1) | Large ASD, VSD | Left CDH, pulmonary hypoplasia, pleural effusions | Ascites, polyhydramnios |
| Lopez et al., 2006 | 46,XY,del(8)(p23.1p23.1) | AVSD | Left CDH, severe pulmonary hypoplasia, Incomplete lobation of right lung | Dysmorphic features, intestinal malrotation |
| Paez et al., 2008 | del(8)(p22p23.3) | Ebstein anomaly, hypoplastic RV, PVS, mild regurgitation of mitral valve due to stenosis, pulmonary artery HTN, ASD, VSD | Mild MR, microcephaly | |
| Paez et al., 2008 | del(8)(p23.1p23.1) | ASD, PVS, VSD, AVSD, persistent left SVC | Gonadotropin-dependent early expression of puberty, moderate MR, mild microcephaly, facial dysmorphic features, developmental delay | |
| Totals for this subgroup | Heart defects: 17/18 (94.4%) | CDH: 4/18 (22.2%) | ||
|
| ||||
| TERMINAL DELETIONS | ||||
|
| ||||
| Lubs and Lubs 1973; Digilio et al., 1998 | del(8)(p21) | ASD, VSD, persistent left SVC | ||
| Tallemite et al., 1975; Digilio et al., 1998 | del(8)(p21) | VSD, PS | ||
| Guanti et al., 1976; Digilio et al., 1998 | del(8)(p21) | Single ventricle, PS, transposition of great arteries | ||
| Leisti and Aula 1977; Digilio et al., 1998 | del(8)(p21) | VSD | ||
| Rodewald et al., 1977; Digilio et al., 1998 | 46,XY,del(8)(p21) | PS, ASD | MR, behavior anomalies, craniofacial dysmorphic features, barrel chest, phalangeal abnormalities, clinodactyly | |
| Reiss et al., 1979; Digilio et al., 1998 | 46,XY,del(8)(p22) | AVSD with L-R shunt and pulmonary HTN | Developmental delay, dysmorphic features, bilateral inguinal hernias | |
| Patil and Hanson, 1980 | del(8)(p21) | CNS and craniofacial abnormalities, spasticity with hyperreflexia | ||
| Marino et al 1982; Digilio et al., 1999 | de(8)(p21) | Complete AVSD, pulmonary stenosis | ||
| Dobyns et al., 1985; Digilio et al., 1998 | del(8)(p21) | AVSD, PS, persistent left SVC, double-orifice mitral valve | ||
| Brocker-Vriends et al., 1986; Digilio et al., 1998 | (half of 8p deleted)** | TOF, double-outlet RV, left persistent SVC | Dysmorphic features | |
| Brocker-Vriends et al., 1986; Digilio et al., 1998 | del(8)(p21) | Complete AVSD, double-outlet RV, PDA, persistent left SVC | Dysmorphic features | |
| Kiss and Osztovics, 1987 | 46,XY,del(8)(p21) | Craniofacial dysmorphic features | ||
| Fryns et al., 1989; Devriendt et al., 1989 | 46,XY,del(8)(p23.1) | Delayed psychomotor and speech development, behavioral problems, two truncal café-au-lait spots, cryptorchidism | ||
| Ostegaard and Tommerup, 1989 | 46,XY,del(8)(p21.3) | TOF | Microcephaly, facial dysmorphic features, puffy feet, cryptorchidism | |
| Blennow and Brondum-Nielsen, 1990 | 46,XX,del(8)(p23) | Developmental delay, moderate MR, strabismus | ||
| Pecile et al., 1990; Digilio et al., 1998 | 46,XY,del(8)(p22) | SM | Dysmorphic features, talipes equinovarus, puffy hands and feet | |
| Pecile et al., 1990; Digilio et al., 1998 | 46,XX,del(8)(p23.1) | AVSD, PS | Left CDH | Dysmorphic features, puffy hands and feet |
| Fraer et al., 1992 | 46,XY,del(8)(p23.1) | AVSD | Left CDH, pulmonary hypoplasia | Dysmorphic features |
| Hutchinson et al., 1992 | del(8)(p23) | Premature adrenarche, aggressive behavior, visual abnormalities, dysmorphic features | ||
| Hutchinson et al., 1992 | del(8)(p23) | Esophageal atresia/tracheoesophageal fistula, dysmorphic features, preaxial polydactyly, 13 pairs of ribs and extra thoracic vertebrae, left cryptorchidism, horseshoe kidneys | ||
| Hutchinson et al., 1992; Digilio et al., 1998 | del(8)(p23) | PFO, mild PS | CNS abnormalities, dysmorphic features | |
| Hutchinson et al., 1992; Digilio et al., 1998 | del(8)(p23) | VSD's, Ebstein's anomaly, PDA, pulmonary hypertension | Developmental delay, strabismus, dysmorphic features, right hydronephrosis | |
| Hutchinson et al., 1992; Digilio et al., 1998 | del(8)(p23) | Complete AVSD, common AV valve, double-outlet RV, small LV, severe PS | Dysmorphic features | |
| Marino et al 1992; Giglio et al., 2000 | 46,XY,del(8)(p21) | PS, Persistent left SVC, AVSD, dextrocardia, hypoplastic RV | Severe MR, craniofacial dysmorphic features, finger anomalies, hypospadias, cryptorchidism | |
| Digilio et al, 1993 | 46,XY,del(8)(p21) | Complete AVSD, PS, hypoplastic RV | Microcephaly, dysmorphic features, scrotal hypoplasias | |
| Digilio et al., 1993, 1999 | del(8)(p23) | Complete AVSD, pulmonary stenosis | ||
| Digilio et al., 1993; Giglio et al., 2000 | 46,XY,del(8)(p21) | PS, AVC | Slight MR, scrotal hypospadias | |
| Devriendt et al., 1995; Digilio et al., 1998 | 46,XX,del(8)(p23.1) | ASD, PS | Aggressive behavior, mild MR, immobile soft palate, facial dysmorphic features | |
| Wu et al., 1996; Digilio et al., 1998 | del(8)(p23.1) | ASDs, PVS, AV regurgitation | Mild MR, speech delay, behavioral and attention disorders, post-traumatic seizures, abnormal EEG, craniofacial dysmorphic features | |
| Wu et al., 1996; Digilio et al., 1998 | del(8)(p23.1) | HLHS, mitral/aortic stenosis, hypoplastic aortic arch | Dysmorphic features, puffy feet, bilateral hydroureternephrosis | |
| Wu et al., 1996 | del(8)(p23.1) | Mild motor delays, speech delay, behavioral problems | ||
| Claeys et al., 1997 | 46,XX,del(8)(p22.1) | Hypotonia, mild MR, delayed development, corpus callosum hypoplasia, dysmorphic features, puffy lower limbs | ||
| Claeys et al., 1997; Devriendt et al., 1999 | 46,XY,del(8)(p23.1) | AVSD, isomerism with azygos continuation, hypoplastic RV, PS, AS | Absence epilepsy, mild MR, microcephaly, hyperactivity, dysmorphic features, partial small bowel malrotation, bilateral cryptorchidism | |
| Claeys et al., 1997; Devriendt et al., 1999 | del(8)(p23.1) | ASD, PS | Mild MR, microcephaly, aggression, hypernasal speech | |
| Claeys et al., 1997; Devriendt et al., 1999 | 46,XY,del(8)(p23.1) | AVSD, univentricular heart with hypoplastic LV, double inlet RV, double outlet RV, PS, dominance of right coronary artery | Developmental delay, dysmorphic features, hypertrichosis (forehead and arms), mildly swollen hands and feet | |
| Claeys et al., 1997; Devriendt et al., 1999 | del(8)(p23.1) | Mild MR, behavioral problems, epilepsy, cryptorchidism | ||
| Johnson et al., 1997 | 46,XX,del(8)(p23.1) | TOF, AVSD | Developmental delay, scoliosis | |
| Johnson et al., 1997 | 46,XY,del(8)(23.1) | ASD, VSD | Developmental delay | |
| Devriendt et al., 1998 | 46,XY,del(8)(p21.3) | AVSD, hypoplastic right ventricle, double-outlet RV, malposition of large vessel side to side, associated PVS | Growth retardation, delayed psychomotor development, hypospadias | |
| Devriendt et al., 1998; 1999 | del(8)(p21.3) | AVSD, double-outlet RV, hypoplastic RV, PS, left isomerism | IUGR, delayed development, hypospadias | |
| Digilio et al., 1998 | del(8)(p21) | AVSD, PS, hypoplastic RV | Slow growth, microcephaly, MR, dysmorphic features, hypospadias | |
| Digilio et al., 1998 | del(8)(p23) | ASD, VSD, PAPVR, persistent left SVC | Microcephaly, MR, dysmorphic features, broad chest, hypospadias | |
| Digilio et al., 1998 | del(8)(p23) | ASD, absent left mediastinal pleura and pericardium | MR, seizures, dysmorphic features, broad chest, hypospadias | |
| Digilio et al., 1998 | del(8)(p23) | AVSD, PS | Microcephaly, MR, dysmorphic features, broad chest | |
| Digilio et al., 1998 | mos del(8)(p23pter) | ASD, VSD, PS | Slow growth, MR, seizures, dysmorphic features, broad chest | |
| Digilio et al., 1998 | del(8)(p21) | AVSD, subaortic stenosis | Microcephaly, MR, dysmorphic features, broad chest, hypospadias, cryptorchidism | |
| Faivre et al., 1998 | del(8)(p23.1) | AVSD, ASD | Posterior left CDH, hypoplastic left lung | Dolichocephaly, edematous eyelids |
| Pehlivan et al., 1999 | 46,XY,del(8)(p23.1) | Secundum ASD, VSD | Language delay, microcephaly, abnormal facies | |
| Pehlivan et al., 1999 | 46,XX,del(8)(p23.1) | Secundum ASD, PVS, mitral valve abnormalities | MR, scoliosis | |
| Pehlivan et al., 1999 | 46,XX,del(8)(p23.1) | Microcephaly, dysmorphic features | ||
| Bhatia et al., 1999 | 46,XX, del(8)(p23.1) | AVSD, persistent left SVC, hypoplastic right ventricle, pulmonary atresia, tricuspid atresia | Mildly dysmorphic features, nuchal edema | |
| Devriendt et al., 1999 | del(8)(p23.1) | ASD, PS | Mild MR, behavioral problems, epilepsy, external ear anomaly | |
| Devriendt et al., 1999 | del(8)(p23.1) | ASD, PS | Mildly delayed development, slight microcephaly, behavioral problems | |
| Reddy, 1999 | 46,XY,del(8)(p23.1)[22]. ish del(8)(p23.1)(8ptel-) | Normal adult | ||
| Reddy, 1999 | 46,XX,del(8)(p23.1)[15 colonies]. ish del(8)(p23.1)(8p tel-) | Normal at birth | ||
| Reddy, 1999 | mos 46,XX,del(8)(p23.1)[11]/46,XX[39]. ish mos del(8)(p23.1)(8p tel-)[12/25] | Recurrent spontaneous abortions as an adult | ||
| Reddy, 1999 | 46,XX,del(8)(p23.1)[20] | Hypotonia, cerebral palsy, microcephaly, developmental delay, craniofacial abnormalities, eye abnormalities, pectus excavatum | ||
| Gilmore et al., 2001 | del(8)(p23) | Gross motor development delay, moderate delays in language and social skills until 6 years old | ||
| Borys and Taxy, 2004 | 46,XX,del(8)(p23.1) | ASD | Left CDH, pulmonary hypoplasia | Accessory spleen |
| Baynam et al., 2008 | 46,XY,del(8p23.1) | ASD | Left sided CDH and pulmonary hypoplasia, persistent pulmonary hypertension | Cornelia de Lange-like features, CNS abnormalities, dysmorphic features, chylothorax, reduced muscle bulk, cryptorchidism |
| Totals for this subgroup | Heart Defects: 45/60 (75%) | CDH: 5/60 (8.3%) | ||
|
| ||||
| INVERTED DUPLICATION DELETION | ||||
|
| ||||
| Weleber et al., 1976 | inv dup del(8)(p11.2→23.1∷p23.1→qter) | CNS abnormalities, lacrimal sac fistula, dysmorphic features | ||
| Mattei et al., 1980 | inv dup del (8)(p11→p23.2∷p23.2→qter) | Hypotonia, slow growth, corpus callosum agenesis, dysmorphic features | ||
| Jensen et al., 1982 | 46,XY,inv dup del(8)(p21.2→p23.1∷p23.1→qter) | Cerebral atrophy, dysmorphic features, asymmetric chest, restricted articular function in all major joints, thoracolumbar scoliosis | ||
| Jensen et al., 1982 | 46,XX,inv dup(8)(p21.1→23.3∷p23.3→qter) | Spastic paraparesis, epilepsy, abnormal EEG, dysmorphic features, coloboma, umbilical hernia, supernumerary finger | ||
| Dill et al., 1987 | 46,XX,inv dup del(8)(p12→p23.1∷p23.1→qter) | Profound MR, hypotonia, dysmorphic features, scoliosis, long & hyperextensible fingers and toes, partial 2-3 syndactyly of toes | ||
| Henderson et al., 1992 | 46,XX,inv dup del(8)(p12→p23.1∷p23.1→qter) | Severe MR | ||
| Minelli et al., 1993 | inv dup del(8)(p12→p22∷p22→qter) | Hypotonia, brain anomalies, MR, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del(8)(p12→p22∷p22→qter) | Heart malformation | Hypotonia | |
| Minelli et al., 1993 | inv dup del(8)(p21.1→p22.1∷p22.1→qter) | Hypotonia, brain anomalies, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del(8)(p12→p22∷p22→qter) | Hypotonia, brain anomalies, MR, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del(8)(p12→p22∷p22→qter) | Hypotonia, brain anomalies, MR, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del(8)(p12→p22∷p22→qter) | MR, short stature, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del(8)(p12→p22∷p22→qter) | Hypotonia, MR, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del (8)(p12→p22∷p22→qter) | Hypotonia, brain anomalies, MR, short stature, dysmorphic facies | ||
| Minelli et al., 1993 | inv dup del (8)(p12→p22∷p22→qter) | Brain anomalies, MR, dysmorphic features | ||
| Minelli et al., 1993 | inv dup del (8)(p12→p22∷p22→qter) | Hypotonia, brain anomalies, MR, dysmorphic facies | ||
| Engelen et al., 1994; de Die-Smulders et al., 1995 | inv dup del(8)(p12→p23.1∷p23.1→qter) | Hypotonia, psychomotor retardation, facial dysmorphic features | ||
| Engelen et al., 1994; de Die-Smulders et al., 1995 | inv dup del(8)(p21.1→p22∷p22→qter) | Dextrocardia | Severe psychomotor retardation, dysmorphic facies, left kidney hypoplasia | |
| Engelen et al., 1994; de Die-Smulders et al., 1995 | inv dup del(8)(p12→p23.1∷p23.1→qter) | Severe MR, dysmorphic facies, contractures of extremities, severe scoliosis, left cryptorchidism | ||
| Engelen et al., 1994; de Die-Smulders et al., 1995 | inv dup del(8)(p12→p23.1∷p23.1→qter) | Profound MR, dysmorphic features, severe scoliosis, contractures of lower extremities | ||
| Mitchell et al., 1994 | 46,XY,inv dup del(8)(p12→p23.1∷p23.1→qter) | CNS abnormalities, MR, dysmorphic features, bilateral clinodactyly of fifth fingers | ||
| de Die-Smulders et al., 1995 | inv dup del(8)(p21.1→p22∷p22→qter) | CNS abnormalities, dysmorphic features, hearing loss | ||
| de Die-Smulders et al., 1995 | inv dup del(8)(p11.2→p23.1→p23.1 →qter) | ASD | Agenesis of corpus callosum, psychomotor delay, dysmorphic features | |
| de Die-Smulders et al., 1995 | inv dup del(8)(p12→p23.1∷p23.1→qter) | Hypotonia, severe MR, developmental delay, facial dysmorphic features | ||
| Guo et al., 1995 | inv dup del(8)(p11.2→p23.1∷p23.1→qter) | ASD, VSD | MR, hypotonia, agenesis of corpus callosum, facial dysmorphic features, umbilical hernia, hydronephrosis | |
| Guo et al., 1995 | inv dup del(8)(p11.2→p23.1∷p23.1→qter) | MR, hypotonia, developmental delay, partial agenesis of corpus callosum, dysmorphic facies | ||
| Guo et al., 1995 | inv dup del(8)(p11.2→p23.1∷p23.1→qter) | MR, developmental delay, hypotonia, agenesis of corpus callosum, cranial asymmetry, dysmorphic facies | ||
| Floridia et al., 1996; Giglio et al., 2000 | 46,XX,dic inv dup del(8)(cen→p23.1∷p23.1→qter) | Agenesis of corpus callosum, severe MR | ||
| Floridia et al., 1996; Giglio et al., 2000 | 46,XY,inv dup del(8)(p21→p23.1∷p23.1→qter) | Hypoplasia of corpus callosum, severe MR | ||
| Yenamandra et al., 1999 | 46,XX,inv dup del(8)(p12→p23.1∷p23.1→qter) | Mild motor delays, speech and language delay, corpus callosum hypoplasia, dysmorphic features, hypoplastic distal phalanges of first toes | ||
| Giglio et al., 2000 | 46,XX,inv dup del(8)(p11.2→p23.1∷p23.1→qter) | Severe MR | ||
| Macmillin et al., 2000 | inv dup del(8)(p11.2→23.1∷p23.1→qter) | Limited heart exam: frequent bradycardia | Lumbar and sacral spinal abnormalities, Dandy-Walker variant, bladder distension with bilateral hydronephrosis | |
| Macmillin et al., 2000 | inv dup del(8)(p11.2→23.1∷p23.1→qter) | Small ASD, multiple small VSDs, mildly depressed LV function, aortic coarctation | CNS abnormalities, Dandy-Walker variant, hypotonic with poor suck, possible gastrointestinal malrotation | |
| Shimokawa et al., 2004 | inv dup del(8)(p11.22→23.1∷p23.1→qter) | cardiac anomalies | MR, craniofacial, ocular, skeletal anomalies | |
| Shimokawa et al., 2004 | inv dup del(8)(p11.1→23.1∷p23.1→qter) | cardiac anomalies | MR, craniofacial, ocular, skeletal anomalies | |
| Shimokawa et al., 2004 | inv dup del(8)(p11.23→23.1∷p23.1→qter) | cardiac anomalies | MR, craniofacial, ocular, skeletal anomalies | |
| Shimokawa et al., 2004 | inv dup del(8)(p?12→23.1∷p23.1→qter) | cardiac anomalies | MR, craniofacial, ocular, skeletal anomalies | |
| Shimokawa et al., 2004 | inv dup del(8)(p12→23.1∷p23.1→qter) | cardiac anomalies | MR, craniofacial, ocular, skeletal anomalies | |
| Sherr et al., 2005 | 46,XX, der(8)del(8)(p23.1)dup(8)(p23.1p12) | Hypotonia, global developmental delay, CNS abnormalities, cerebral palsy | ||
| Totals for this subgroup | Heart Defects: 11/39 (28.2%) | CDH: 0/39 (0%) | ||
|
| ||||
| COMPLEX CHROMOSOMAL REARRANGEMENTS | ||||
|
| ||||
| Mattei et al., 1980 | t(8;14)(8pter→8p11→8p23.1∷8p23.1→8p11∷14q13→14qter) | Hypotonia, CNS abnormalities, dysmorphic features, phalangeal abnormalities | ||
| Brocker-Vriends et al., 1985 | 46,XY,der(8),t(8;13)(p21;q22),t(2;19)(q21;q13) | Mild dysmorphic features, laryngomalacia (infant) | ||
| Brocker-Vriends et al., 1985 | 46,XX,der(8),t(8;13)(p21;q22) | Pyloric stenosis, elbow hyperextensibility | ||
| van Balkom et al., 1992 | 46,XX,del(8)(p21)/46,XX,dup(8)(p21pter) | Agenesis of corpus callosum, short first metacarpals, short first metatarsal | ||
| Scarbrough et al., 1987 | 46,XX,der(8),t(3;8)(p21;p23) | 2/6 systolic murmur | Hypotonia, dysmorphic features, long trunk, camptodactyly | |
| Sujansky et al., 1993; Giglio et al., 2000 | 46,XX,rec(8)(qter→q22∷p23.1→qter) | PS, PDA, ASD, VSD | MR | |
| Digilio et al., 1994; Ciccone et al., 2006 | mos 46,XX,psu dic(8)(p23.2)/46,XX,del(8)(p23.1) | Severe MR, dysmorphic features, kyphoscoliosis, globous abdomen, premature grey hair (at 14 years old) | ||
| Zahed et al., 1998 | 46,XX,rec(8)t(8;9)(p23.1;q34)mat | Hypotonia, developmental delay, hyperactivity, dysmorphic features | ||
| Giglio et al., 2000 | 46,XY,der(8)t(1;8)(q42;p23.1)pat | PS, PFO | MR, seizures, pyloric stenosis | |
| Giglio et al., 2000 | 46,XY,der(8)t(8;16)(p21.3;q24) | PS, VSD, right aortic arch | Slight MR | |
| Giglio et al., 2000 | 46,XY,der(8)t(8;16)(p21.3;q24) | PS | Slight MR | |
| de Vries et al., 2001 | 46,XY.ish der(8)t(8;20)(p23;p13) (D8S2333-) | Murmur but no structural abnormalities | Delayed speech & language, dysmorphic features, blocked tear ducts, cryptorchidism | |
| de Vries et al., 2001 | 46,XY.ish der(8)t(8;20)(p23;p13) (D8S2333-) | Mild MR, behavioral disturbances, slightly delayed motor development, dysmorphic features | ||
| Bosse et al., 2004 | 45,X,dic(Y;8)(q11.23;p23.1) | Mild MR, psychomotor delay, speech delay, aggression, dysmorphic features, talipes planus | ||
| Ozkinay et al., 2006 | 46,XY,der(8)t(8p;21q)dn | IUGR, CNS abnormalities, pelvicaliectasis | ||
| Totals for this subgroup | Heart Defects: 6/15 (40%) | CDH: 0/15 (0%) | ||
ASD, atrial septal defect; AVC, atrioventricular canal; AVSD, atrioventricular septal defect; CDH, congenital diaphragmatic hernia; CNS, central nervous system; EEG, electroencephalogram,; HLHS, hypoplastic left heart syndrome; HTN, hypertension; IUGR, intrauterine growth retardation; LV, left ventricle; L-R, left-right; MR, mental retardation; PFO, patent foramen ovale; PDA, patent ductus arteriosus; PS, pulmonary stenosis; PVS, pulmonary valve stenosis; RV, right ventricle; SVC, superior vena cava; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
interstitial deletion, but insufficient resolution to determine if it deletes region of interest.
karyotype nomenclature not provided.
When combined with data from the four patients described in this report, it would appear that the spectrum of heart defects is more severe in interstitial and terminal deletions involving 8p23.1 when compared to defects seen in patients with heterozygous GATA4 mutations. Although this increase in severity could be due to deleterious mutations in the remaining GATA4 allele, we did not detect such mutations in Patients 1 and 4. A similar evaluation carried out by Paez et al. also failed to identify mutations in the remaining GATA4 allele in two patients with 8p23.1 deletions involving GATA4 who also presented with heart defects [Paez et al., 2008].
An alternative explanation for the increase in severity would be the existence of another gene(s) in the recurrent 8p23.1 deletion region that impact heart development. Of the genes deleted along with GATA4, SOX7 is one of the most likely candidate genes. SOX7 is expressed in mouse and human adult heart, and in the early cardiogenic region of Xenopus embryos [Takash et al., 2001; Taniguchi et al., 1999; Zhang et al., 2005]. SOX7 mRNA injection induced cardiogenic marker expression in Xenopus animal cap explants whereas knockdown of SOX7 using morpholinos decreased the expression of cardiogenic markers MHCα and Nkx2.5; and marker expression was rescued by injection of RNA encoding a SOX7 transcript [Zhang et al., 2005]. Interestingly, silencing of Sox7 in mouse F9 embryonal carcinoma cells blunts the increase in Gata4 mRNA levels seen after treatment with all trans-retinoic acid/dibuterol cAMP [Futaki et al., 2004]. In contrast, silencing of Gata4 did not result in decreased Sox7 expression in the same system. This suggests that Sox7 lies upstream of Gata4, and that haploinsufficiency of SOX7 may exacerbate the cardiac phenotype of individuals with GATA4 deletions.
Genes outside the GATA4 region may also be involved in abnormal cardiac development based on reports of patients with inverted duplication deletion events involving 8p. The mechanism involved in creation of the most common inverted duplication deletion events of 8p has been well described [Ciccone et al., 2006]. These events typically result in loss of copy number (deletion) of 8p distal to 8p-OR-REPD, normal copy number of the GATA4 region between 8p-OR-REPD and 8p-OR-REPP, and a variable region of increased copy number (duplication) proximal to 8p-OR-REPP. The number of cardiac malformations associated with patients carrying the inverted duplication deletion 8p was 11/39 (28.2%), a much lower proportion than 17/18 (94.4%) and 45/60 (75%) associated with interstitial and terminal deletions, respectively (Table III). In general, the spectrum of cardiac malformations in these cases is also milder but still includes ASD, VSD, right aortic arch, and pulmonary stenosis (Table III). If we assume normal expression of GATA4 and other genes between 8p-OR-REPD and 8p-OR-REPP, we are left to conclude that decreased expression of one or more genes distal to 8p-OR-REPD and/or over expression of one or more genes proximal to 8p-OR-REPP can also contribute to the development of some heart defects.
Congenital diaphragmatic hernia is also common in patients with a deletion encompassing 8p23.1 with, at least, nine previously reported cases [Holder et al 2007]. CDH is associated with 4/18 (22.2%) of reported interstitial deletions and 5/60 (8.3%) of terminal deletions but has not been described in patients with inverted duplication deletions. This is, presumably, due to the fact that terminal deletions may not always include the CDH minimal deleted interval and that this interval has a normal copy number in individuals with inverted duplication deletions.
The majority of CDH patients with 8p23.1 deletions have been described as having left sided (assumedly-posterior) CDH. Indeed, Patient 3 is the only individual described to date with an anterior CDH associated with an 8p23.1 deletion. This is surprising since the heterozygous Gata4 mice described by Jay et al. have anterior CDH similar to that seen in our patient [Jay et al., 2007]. Using array data presented here and previously reported molecularly-defined deletions associated with CDH, the minimal deleted region for CDH on 8p can be defined as the region bounded by 8p-OR-REPD distally and 8p-OR-REPP proximally (Fig 3) [Faivre et al., 1998; Slavotinek et al., 2004; Shimokawa et al., 2005].
Figure 3.

Molecularly defined deletions of 8p23.1 associated with CDH. Solid and dashed red lines indicate the minimally and maximally deleted regions, respectively, for each patient. The approximate location of the segmental duplications 8p-OR-REPD and 8p-OR-REPP are represented by green bars. GATA4, represented by the blue bar, is located within the minimal deleted region for CDH.
Although the use of array comparative genome hybridization can aid in the identification of 8p23.1 deletions in patients with heart and/or diaphragm defects, caution must be used in the interpretation of these findings. Chromosome 8p contains many copy number variant regions whose potential contribution to the development of birth defects has not been adequately studied. Recently Chen et al. [2007] described a patient with a Fryns-like phenotype including congenital diaphragmatic hernia, macrocephaly, brachytelephalangy, nail hypoplasia, short webbed neck with redundant posterior nuchal skin, coarse face, flat and broad nasal bridge, hypertelorism, macrostomia, microretrognathia, and low-set ears. The patient's phenotype was attributed to a de novo 0.7 MB deletion within 8p23.1 [Chen et al., 2007]. However, this deletion lies entirely within a known copy number variant region making it difficult to determine if this deletion is causal. Without careful evaluation, physicians may erroneously quote sibling and offspring recurrence risks that are either too low or too high based on an incorrect assumption of causality.
When used properly, aCGH detects detrimental submicroscopic changes that can be easily missed on routine chromosomal analysis especially in prenatal samples where the band resolution may be compromised [Pecile et al., 1990; Wu et al., 1996; Faivre et al., 1998]. This is illustrated well in Patient 2 where chromosome analysis failed to identify an 8p deletion which was easily detected by aCGH. This, and the high frequency of cardiac and diaphragmatic defects associated with 8p23.1 interstitial deletions, leads us to recommend that aCGH be performed on all prenatal and postnatal cases with congenital cardiac and/or diaphragm defects. At the present time we do not recommend that the GATA4 gene be sequenced in patients with cardiac defects since the percentage of cases with protein-altering changes in GATA4 is likely to be small— approximately 1% based on the articles reviewed—and the clinical significance of the majority of GATA4 changes remains unclear.
ACKNOWLEDGMENTS
The authors thank the patients and family members who participated in this study and Zhiyin Yu for her technical assistance. This study was funded in part by NIH grant HD-050583 (DAS).
REFERENCES
- Baynam G, Goldblatt J, Walpole I. Deletion of 8p23.1 with features of Cornelia de Lange syndrome and congenital diaphragmatic hernia and a review of deletions of 8p23.1 to 8pter? A further locus for Cornelia de Lange syndrome. Am J Med Genet A. 2008;146A:1565–1570. doi: 10.1002/ajmg.a.32095. [DOI] [PubMed] [Google Scholar]
- Bhatia SN, Suri V, Bundy A, Krauss CM. Prenatal detection and mapping of a distal 8p deletion associated with congenital heart disease. Prenat Diagn. 1999;19:863–867. [PubMed] [Google Scholar]
- Blennow E, Brondum-Nielsen K. Partial monosomy 8p with minimal dysmorphic signs. J Med Genet. 1990;27:327–329. doi: 10.1136/jmg.27.5.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borys D, Taxy JB. Congenital diaphragmatic hernia and chromosomal anomalies: autopsy study. Pediatr Dev Pathol. 2004;7:35–38. doi: 10.1007/s10024-003-2133-7. [DOI] [PubMed] [Google Scholar]
- Bosse K, Eggermann T, Van der Ven K, Raff R, Engels H, Schwanitz G. Unbalanced translocation 8;Y (45,X,dic(Y;8)(q11.23;p23.1)): case report and review of terminal 8p deletions. Ann Genet. 2004;47:191–197. doi: 10.1016/j.anngen.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Brocker-Vriends AH, Mooij PD, van Bel F, Beverstock GC, van de Kamp JJ. Monosomy 8p: an easily overlooked syndrome. J Med Genet. 1986;23:153–154. doi: 10.1136/jmg.23.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker-Vriends AH, van de Kamp JJ, Geraedts JP, Bos SE, Nijenhuis TA. Unbalanced karyotype with normal phenotype in a family with translocation (8;13)(p21;q22) Clin Genet. 1985;27:487–495. doi: 10.1111/j.1399-0004.1985.tb00237.x. [DOI] [PubMed] [Google Scholar]
- Chen CP, Wang TH, Chen YJ, Chang TY, Liu YP, Tzen CY, Chern SR, Wang W. Prenatal diagnosis of Fryns syndrome associated with a microdeletion at 8p23.1. Prenat Diagn. 2007;27:967–969. doi: 10.1002/pd.1797. [DOI] [PubMed] [Google Scholar]
- Ciccone R, Mattina T, Giorda R, Bonaglia MC, Rocchi M, Pramparo T, Zuffardi O. Inversion polymorphisms and non-contiguous terminal deletions: the cause and the (unpredicted) effect of our genome architecture. J Med Genet. 2006;43:e19. doi: 10.1136/jmg.2005.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys I, Holvoet M, Eyskens B, Adriaensens P, Gewillig M, Fryns JP, Devriendt K. A recognisable behavioural phenotype associated with terminal deletions of the short arm of chromosome 8. Am J Med Genet. 1997;74:515–520. doi: 10.1002/(sici)1096-8628(19970919)74:5<515::aid-ajmg12>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Devriendt K, De Mars K, De Cock P, Gewillig M, Fryns JP. Terminal deletion in chromosome region 8p23.1-8pter in a child with features of velo-cardio-facial syndrome. Ann Genet. 1995;38:228–230. [PubMed] [Google Scholar]
- Devriendt K, Matthijs G, Van Dael R, Gewillig M, Eyskens B, Hjalgrim H, Dolmer B, McGaughran J, Brondum-Nielsen K, Marynen P, Fryns JP, Vermeesch JR. Delineation of the critical deletion region for congenital heart defects, on chromosome 8p23.1. Am J Hum Genet. 1999;64:1119–1126. doi: 10.1086/302330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devriendt K, Van Schoubroeck D, Eyskens B, Gewillig M, Vandenberghe K, Fryns JP. Prenatal diagnosis of a terminal short arm deletion of chromosome 8 in a fetus with an atrioventricular septal defect. Prenat Diagn. 1998;18:65–67. doi: 10.1002/(sici)1097-0223(199801)18:1<65::aid-pd207>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Marino B, Guccione P, Giannotti A, Mingarelli R, Dallapiccola B. Deletion 8p syndrome. Am J Med Genet. 1998;75:534–536. [PubMed] [Google Scholar]
- Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. Atrioventricular canal defect without Down syndrome: a heterogeneous malformation. Am J Med Genet. 1999;85:140–146. [PubMed] [Google Scholar]
- Dill FJ, Schertzer M, Sandercock J, Tischler B, Wood S. Inverted tandem duplication generates a duplication deficiency of chromosome 8p. Clin Genet. 1987;32:109–113. doi: 10.1111/j.1399-0004.1987.tb03335.x. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Dewald GW, Carlson RO, Mair DD, Michels VV. Deficiency of chromosome 8p21.1----8pter: case report and review of the literature. Am J Med Genet. 1985;22:125–134. doi: 10.1002/ajmg.1320220114. [DOI] [PubMed] [Google Scholar]
- Engelen JJ, de Die-Smulders CE, Fryns JP, Hoovers JM, Albrechts JC, Loots WJ, Jacobs ME, Hamers AJ. Partial trisomy and monosomy 8p due to inversion duplication. Clin Genet. 1994;45:203–207. doi: 10.1111/j.1399-0004.1994.tb04024.x. [DOI] [PubMed] [Google Scholar]
- Faivre L, Morichon-Delvallez N, Viot G, Narcy F, Loison S, Mandelbrot L, Aubry MC, Raclin V, Edery P, Munnich A, Vekemans M. Prenatal diagnosis of an 8p23.1 deletion in a fetus with a diaphragmatic hernia and review of the literature. Prenat Diagn. 1998;18:1055–1060. doi: 10.1002/(sici)1097-0223(1998100)18:10<1055::aid-pd405>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Fryns JP, Kleczkowska A, Vogels A, Van den Berghe H. Normal phenotype and slight mental retardation in de novo distal 8p deletion (8pter----8p23.1) Ann Genet. 1989;32:171–173. [PubMed] [Google Scholar]
- Futaki S, Hayashi Y, Emoto T, Weber CN, Sekiguchi K. Sox7 plays crucial roles in parietal endoderm differentiation in F9 embryonal carcinoma cells through regulating Gata-4 and Gata-6 expression. Mol Cell Biol. 2004;24:10492–10503. doi: 10.1128/MCB.24.23.10492-10503.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- Giglio S, Graw SL, Gimelli G, Pirola B, Varone P, Voullaire L, Lerzo F, Rossi E, Dellavecchia C, Bonaglia MC, Digilio MC, Giannotti A, Marino B, Carrozzo R, Korenberg JR, Danesino C, Sujansky E, Dallapiccola B, Zuffardi O. Deletion of a 5-cM region at chromosome 8p23 is associated with a spectrum of congenital heart defects. Circulation. 2000;102:432–437. doi: 10.1161/01.cir.102.4.432. [DOI] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet. 2001;68:874–883. doi: 10.1086/319506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore L, Cuskelly M, Jobling A, Smith S. Deletion of 8p: a report of a child with normal intelligence. Dev Med Child Neurol. 2001;43:843–846. doi: 10.1017/s0012162201001530. [DOI] [PubMed] [Google Scholar]
- Guo WJ, Callif-Daley F, Zapata MC, Miller ME. Clinical and cytogenetic findings in seven cases of inverted duplication of 8p with evidence of a telomeric deletion using fluorescence in situ hybridization. Am J Med Genet. 1995;58:230–236. doi: 10.1002/ajmg.1320580307. [DOI] [PubMed] [Google Scholar]
- Henderson KG, Dill FJ, Wood S. Characterization of an inversion duplication of the short arm of chromosome 8 by fluorescent in situ hybridization. Am J Med Genet. 1992;44:615–618. doi: 10.1002/ajmg.1320440517. [DOI] [PubMed] [Google Scholar]
- Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, Furutani M, Imamura S, Takao A, Nakazawa M, Matsuoka R. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A. 2005;135:47–52. doi: 10.1002/ajmg.a.30684. [DOI] [PubMed] [Google Scholar]
- Holder AM, Klaassens M, Tibboel D, de Klein A, Lee B, Scott DA. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. 2007;80:825–845. doi: 10.1086/513442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson R, Wilson M, Voullaire L. Distal 8p deletion (8p23.1----8pter): a common deletion? J Med Genet. 1992;29:407–411. doi: 10.1136/jmg.29.6.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, Wilson DB. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2007;301:602–614. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MC, Hing A, Wood MK, Watson MS. Chromosome abnormalities in congenital heart disease. Am J Med Genet. 1997;70:292–298. doi: 10.1002/(sici)1096-8628(19970613)70:3<292::aid-ajmg15>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Lopez I, Bafalliu JA, Bernabe MC, Garcia F, Costa M, Guillen-Navarro E. Prenatal diagnosis of de novo deletions of 8p23.1 or 15q26.1 in two fetuses with diaphragmatic hernia and congenital heart defects. Prenat Diagn. 2006;26:577–580. doi: 10.1002/pd.1468. [DOI] [PubMed] [Google Scholar]
- Macmillin MD, Suri V, Lytle C, Krauss CM. Prenatal diagnosis of inverted duplicated 8p. Am J Med Genet. 2000;93:94–98. doi: 10.1002/1096-8628(20000717)93:2<94::aid-ajmg3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Marino B, Reale A, Giannotti A, Digilio MC, Dallapiccola B. Nonrandom association of atrioventricular canal and del (8p) syndrome. Am J Med Genet. 1992;42:424–427. doi: 10.1002/ajmg.1320420404. [DOI] [PubMed] [Google Scholar]
- Mattei JF, Mattei MG, Ardissone JP, Coignet J, Giraud F. Clinical, enzyme, and cytogenetic investigations in three new cases of trisomy 8p. Hum Genet. 1980;53:315–321. doi: 10.1007/BF00287049. [DOI] [PubMed] [Google Scholar]
- Minelli A, Floridia G, Rossi E, Clementi M, Tenconi R, Camurri L, Bernardi F, Hoeller H, Previde Re C, Maraschio P, et al. D8S7 is consistently deleted in inverted duplications of the short arm of chromosome 8 (inv dup 8p) Hum Genet. 1993;92:391–396. doi: 10.1007/BF01247342. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat. 2006;27:293–294. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, Yoshiura K, Kishino T, Ohta T, Niikawa N, Matsumoto N. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet. 2004;41:e97. doi: 10.1136/jmg.2004.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orye E, Craen M. A new chromosome deletion syndrome. Report of a patient with a 46,XY,8p- chromosome constitution. Clin Genet. 1976;9:289–301. doi: 10.1111/j.1399-0004.1976.tb01577.x. [DOI] [PubMed] [Google Scholar]
- Ostergaard GZ, Tommerup N. The 8p-syndrome. Ann Genet. 1989;32:87–91. [PubMed] [Google Scholar]
- Ozkinay F, Kanit H, Onay H, Cogulu O, Gunduz C, Ercal D, Ozkinay C. Prenatal diagnosis of de novo unbalanced translocation 8p;21q using subtelomeric probes. Genet Couns. 2006;17:315–320. [PubMed] [Google Scholar]
- Paez MT, Yamamoto T, Hayashi K, Yasuda T, Harada N, Matsumoto N, Kurosawa K, Furutani Y, Asakawa S, Shimizu N, Matsuoka R. Two patients with atypical interstitial deletions of 8p23.1: mapping of phenotypical traits. Am J Med Genet A. 2008;146A:1158–1165. doi: 10.1002/ajmg.a.32205. [DOI] [PubMed] [Google Scholar]
- Patil SR, Hanson JW. Partial 8p- syndrome. J Genet Hum. 1980;28:123–129. [PubMed] [Google Scholar]
- Pecile V, Petroni MG, Fertz MC, Filippi G. Deficiency of distal 8p--report of two cases and review of the literature. Clin Genet. 1990;37:271–278. doi: 10.1111/j.1399-0004.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS, Hing AV. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet. 1999;83:201–206. [PubMed] [Google Scholar]
- Pettenati MJ, Rao N, Johnson C, Hayworth R, Crandall K, Huff O, Thomas IT. Molecular cytogenetic analysis of a familial 8p23.1 deletion associated with minimal dysmorphic features, seizures, and mild mental retardation. Hum Genet. 1992;89:602–606. doi: 10.1007/BF00221946. [DOI] [PubMed] [Google Scholar]
- Poirier O, Nicaud V, McDonagh T, Dargie HJ, Desnos M, Dorent R, Roizès G, Schwartz K, Tiret L, Komajda M, Cambien F. Polymorphisms of genes of the cardiac calcineurin pathway and cardiac hypertrophy. Eur J Hum Genet. 2003;11:659–664. doi: 10.1038/sj.ejhg.5201023. [DOI] [PubMed] [Google Scholar]
- Posch MG, Perrot A, Schmitt K, Mittelhaus S, Esenwein EM, Stiller B, Geier C, Dietz R, Gessner R, Ozcelik C, Berger F. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am J Med Genet A. 2008;146A:251–253. doi: 10.1002/ajmg.a.32042. [DOI] [PubMed] [Google Scholar]
- Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V, Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB, Pu WT. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43:677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reamon-Buettner SM, Borlak J. GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J Med Genet. 2005;42:e32. doi: 10.1136/jmg.2004.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reamon-Buettner SM, Cho SH, Borlak J. Mutations in the 3'-untranslated region of GATA4 as molecular hotspots for congenital heart disease (CHD) BMC Med Genet. 2007;8:38. doi: 10.1186/1471-2350-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KS. A paternally inherited terminal deletion, del(8)(p23.1)pat, detected prenatally in an amniotic fluid sample: a review of deletion 8p23.1 cases. Prenat Diagn. 1999;19:868–872. doi: 10.1002/(sici)1097-0223(199909)19:9<868::aid-pd641>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Rodewald A, Stengel-Rutkowski S, Schulz P, Cleve H. New chromosomal malformation syndromes. I. Partial monosomy 8p. An attempt to establish a new chromosome deletion syndrome. Eur J Pediatr. 1977;125:45–57. doi: 10.1007/BF00470605. [DOI] [PubMed] [Google Scholar]
- Sarkozy A, Conti E, Neri C, D'Agostino R, Digilio MC, Esposito G, Toscano A, Marino B, Pizzuti A, Dallapiccola B. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 2005;42:e16. doi: 10.1136/jmg.2004.026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozy A, Esposito G, Conti E, Digilio MC, Marino B, Calabro R, Pizzuti A, Dallapiccola B. CRELD1 and GATA4 gene analysis in patients with nonsyndromic atrioventricular canal defects. Am J Med Genet A. 2005;139:236–238. doi: 10.1002/ajmg.a.31018. [DOI] [PubMed] [Google Scholar]
- Scarbrough PR, Carroll AJ, Finley WH, Bridges DR. A de novo 3p;8p unbalanced translocation resulting in partial dup(3p) and partial del(8p) J Med Genet. 1987;24:174–177. doi: 10.1136/jmg.24.3.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluterman MK, Krysiak AE, Kathiriya IS, Abate N, Chandalia M, Srivastava D, Garg V. Screening and biochemical analysis of GATA4 sequence variations identified in patients with congenital heart disease. Am J Med Genet A. 2007;143A:817–823. doi: 10.1002/ajmg.a.31652. [DOI] [PubMed] [Google Scholar]
- Sherr EH, Owen R, Albertson DG, Pinkel D, Cotter PD, Slavotinek AM, Hetts SW, Jeremy RJ, Schilmoeller G, Schilmoeller K, Wakahiro M, Barkovich AJ. Genomic microarray analysis identifies candidate loci in patients with corpus callosum anomalies. Neurology. 2005;65:1496–1498. doi: 10.1212/01.wnl.0000183066.09239.b6. [DOI] [PubMed] [Google Scholar]
- Shimokawa O, Kurosawa K, Ida T, Harada N, Kondoh T, Miyake N, Yoshiura K, Kishino T, Ohta T, Niikawa N, Matsumoto N. Molecular characterization of inv dup del(8p): analysis of five cases. Am J Med Genet A. 2004;128A:133–137. doi: 10.1002/ajmg.a.30063. [DOI] [PubMed] [Google Scholar]
- Shimokawa O, Miyake N, Yoshimura T, Sosonkina N, Harada N, Mizuguchi T, Kondoh S, Kishino T, Ohta T, Remco V, Takashima T, Kinoshita A, Yoshiura K, Niikawa N, Matsumoto N. Molecular characterization of del(8)(p23.1p23.1) in a case of congenital diaphragmatic hernia. Am J Med Genet A. 2005;136:49–51. doi: 10.1002/ajmg.a.30778. [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Lee SS, Davis R, Shrit A, Leppig KA, Rhim J, Jasnosz K, Albertson D, Pinkel D. Fryns syndrome phenotype caused by chromosome microdeletions at 15q26.2 and 8p23.1. J Med Genet. 2005;42:730–736. doi: 10.1136/jmg.2004.028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek AM, Moshrefi A, Davis R, Leeth E, Schaeffer GB, Burchard GE, Shaw GM, James B, Ptacek L, Pennacchio LA. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1-15q26.2. Eur J Hum Genet. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- Takash W, Canizares J, Bonneaud N, Poulat F, Mattei MG, Jay P, Berta P. SOX7 transcription factor: sequence, chromosomal localisation, expression, transactivation and interference with Wnt signalling. Nucleic Acids Res. 2001;29:4274–4283. doi: 10.1093/nar/29.21.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Hiraoka Y, Ogawa M, Sakai Y, Kido S, Aiso S. Isolation and characterization of a mouse SRY-related cDNA, mSox7. Biochim Biophys Acta. 1999;1445:225–231. doi: 10.1016/s0167-4781(99)00047-0. [DOI] [PubMed] [Google Scholar]
- Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J Med Genet. 2007;44:779–783. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weleber RG, Verma RS, Kimberling WJ, Fieger HG, Jr., lubs HA. Duplication-deficiency of the short arm of chromosome 8 following artificial insemination. Ann Genet. 1976;19:241–247. [PubMed] [Google Scholar]
- Wu BL, Schneider GH, Sabatino DE, Bozovic LZ, Cao B, Korf BR. Distal 8p deletion (8)(p23.1): an easily missed chromosomal abnormality that may be associated with congenital heart defect and mental retardation. Am J Med Genet. 1996;62:77–83. doi: 10.1002/(SICI)1096-8628(19960301)62:1<77::AID-AJMG16>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Yenamandra A, Perrone R, McLaughlin J, Mehta L. Inverted duplication/deletion of chromosome 8p: mild clinical phenotype. Am J Med Genet. 1999;82:91–93. doi: 10.1002/(sici)1096-8628(19990101)82:1<91::aid-ajmg19>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Zahed L, Der Kaloustian V, Batanian JR. Familial complex chromosome rearrangement giving rise to balanced and unbalanced recombination products. Am J Med Genet. 1998;79:30–34. [PubMed] [Google Scholar]
- Zhang C, Basta T, Klymkowsky MW. SOX7 and SOX18 are essential for cardiogenesis in Xenopus. Dev Dyn. 2005;234:878–891. doi: 10.1002/dvdy.20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuffardi O, Ciccone R, Giglio S, Pramparo T. Inversion Chromosomes. In: Lupski JR, Stankiewicz P, editors. Genomic Disorders: The Genomic Basis of Disease. Springer; New York: 2006. pp. 289–299. [Google Scholar]