

Abstract
Introduction of lipoamino acid (LAA), Lys-palmitoyl, and cationization into a series of galanin analogs yielded systemically-active anticonvulsant compounds. To study the relationship between the LAA structure and anticonvulsant activity, orthogonally protected LAAs were synthesized in which the Lys side chain was coupled to fatty acids varying in length from C8 to C18, or to a monodispersed polyethylene glycol, PEG4. Galanin receptor affinity, serum stability, lipophilicity (logD) and activity in the 6 Hz mouse model of epilepsy of each of the newly synthesized analogs was determined following systemic administration. The presence of various LAAs or Lys(MPEG4) did not affect the receptor binding properties of the modified peptides, but their anticonvulsant activities varied substantially, and were generally correlated with their lipophilicity. Our results suggest that varying the length or polarity of the LAA residue adjacent to positively-charged amino acid residues may effectively modulate the antiepileptic activity of the galanin analogs.
Introduction
When delivered directly into the brain, many neuropeptides, including galanin, neuropeptide Y, somatostatin, neurotensin or opioid peptides can modulate excitatory or inhibitory circuits and suppress seizures and/or pain sensation.1-4 Despite a considerable interest in generating blood-brain barrier (BBB) permeable analogs of neuropeptides, only a few successful examples have been reported to date.4-12 To improve central nervous system (CNS) bioavailability of neuroactive peptides, various strategies have been explored; including lipidization, cationization or glycosylation.13-15 Banks and Kastin showed that the lipophilicity of peptides improved their permeability through the BBB.16 Despite the finding that only small amounts of the peptides entered the brain, the authors noted a direct correlation between the logD values and blood-to-brain ratio. Reversible lipidization of opioid peptides also improved centrally-mediated analgesic effects.17 It is also important to note that oral bioavailability of the somatostatin analog TT-232 was achieved by coupling various LAAs to either N- or C-terminal components of the analog.18 Although lipophilicity has been acknowledged to play an important role in structure-bioavailability relationships,16, 19 very few examples are available where lipidization of neuropeptides has been found to improved their activity in the brain.17, 20, 21 As pointed out in a review by Witt et al,13 “lipidization of peptides may increase plasma protein binding, systemic elimination and intracellular sequestration; thus hampering their efficient penetration into the CNS.” Indeed, when just the lipidization strategy was applied to galanin, the truncated galanin analog containing a Lys-palmitoyl residue was inactive as an anticonvulsant, despite displaying high affinity toward galanin receptors and a high logD.22
In contrast, lipidization appeared very effective in improving the CNS bioavailability of the truncated galanin analogs when the Lys(palmitoyl) residue was introduced in concert with cationization.22 The most active analog, Gal-B2, contained the C-terminal “-Lys-Lys-Lys(palmitoyl)-Lys-NH2” motif (Figure 1). The combined lipidization and cationization resulted in very potent antiepileptic compounds; e.g., the anticonvulsant ED50 of Gal-B2 was found to be 0.8 mg/kg when tested intraperitoneally (i.p.) in the 6 Hz pharmacoresistant model of epilepsy. Since galanin suppresses seizures by activating GalR1 and GalR2 located in the hippocampus and other limbic structures, our data suggest that the combination of cationization and lipidization is effective in improving the BBB penetration of Gal-B2 without negatively affecting receptor affinity. Furthermore, our results suggest that the sequence position of LAAs is important for the systemic activity of Gal-B2, suggesting that the lipidization/cationization motif was not simply producing the additive effect by combining hydrophobicity and positive charges.
Figure 1.

Structures of Gal-B2 and LAAs used in this SAR study. Note that MPEG4 has an identical number of atoms as the C16 (palmitoyl), but is significantly less lipophilic.
In order to define the structural requirements of the LAA residue for improving the anticonvulsant activity of Gal-B2, we synthesized a series of LAAs in which the Lys side chain was coupled to either fatty acids, varying in length from C8 to C18, or to a monodispersed PEG4 (MPEG4). These LAAs were used to replace the Lys-palmitoyl residue in position 16 of Gal-B2 (Figure 1). The binding affinity, octanol/water partitioning coefficient, and ability to suppress seizures in the in vivo 6 Hz model of epilepsy was determined following i.p. administration. Our findings suggest that the length and polarity of the LAAs are important for maintaining antiepileptic activity of the galanin analogs.
Results
Design and chemical synthesis
To study the role of Lys-palmitoyl, residue 16, in the activity of Gal-B2, we designed a series of analogs where we systematically changed the length of the fatty acid or replaced the palmitoyl motif with a MPEG4 (Figure 1). The length of the LAAs varied from C8 to C18, by two carbon atom increments (Table 1). The MPEG4-containing Gal-B2 analog was designed to dissect the lipophilic contribution of the Lys-palmitoyl residue, since MPEG4 is significantly more polar than C16 despite having an identical number of atoms. To synthesize these new structure activity relationship (SAR) analogs of Gal-B2, we applied a direct coupling of the orthrogonally protected Nε-lipolysine intermediates (Nε-palmitoyl-Nα-Fmoc-lysine is commercially available). For the synthesis of Nε-octanoyl 1 (C8), -decanoyl 2 (C10), -lauryl 3 (C12), -myristoyl 4 (C14) and -stearoyl 5 (C18) substituted Nα-Fmoc-lysine, a convenient method was applied using Na2CO3 as a coupling reagent with a corresponding acid halide in dioxane/water solvent system (Scheme 1). Synthesis of Fmoc-Lys-(MPEG4)-OH 6, was achieved by coupling Fmoc-Lys-OH with perfluorophenol activated MPEG4-acetic acid 7. Compound 7 was synthesized from the substitution reaction of MPEG4-OH with ethyl bromoacetate followed by ester hydrolysis (Scheme 2). The modified amino acids were purified by flash chromatography and used for solid-phase peptide synthesis (SPPS).
Table 1.
Structures of LAAs used for SPPS of the systemically-active galanin analogs.
| LAA | Name | Structure |
|---|---|---|
| 1 | Fmoc-Lys(octanoyl)-OH | |
| 2 | Fmoc-Lys(decanoyl)-OH | |
| 3 | Fmoc-Lys(lauroyl)-OH | |
| 4 | Fmoc-Lys(myristoyl)-OH | |
| 5 | Fmoc-Lys(stearoyl)-OH | |
| 6 | Fmoc-Lys(MPEG4)-OH |
Scheme 1. Synthesis of the Fmoc-protected LAAsa.

aKey: (a) Na2CO3, H2O/dioxane, rt, overnight, 64~87%.
Scheme 2. Synthesis of Fmoc-Lys(MPEG4)-OHa.

a Key: (a)NaH 0 °C, 1 h; Ethyl bromoacetate, 50 °C, 16 h; (b) 1M LiOH/CH3OH 6h, 59% (from two steps); (c) Perfluorophenol, DCC, overnight; (d) Fmoc-L-lysine, DIPEA, 0 °C, 60% (from two steps).
The SPPS was carried out using an automated peptide synthesizer at 25 μmol scale on preloaded Fmoc-Lys(Boc)-Rink amide AM resin. Five-fold Fmoc-amino acids/PyBop/DIPEA (1:1:2) were used in peptide synthesis. Two-fold of the Fmoc protected LAAs or MPEG4-Lysine were used in manual coupling synthesis. The synthesized peptides were cleaved from the resin using reagent K (trifluoroacetic acid (TFA)/water/thioanisole/phenol/ethanedithiol; 90/5/5/7.5/2.5 v/v), followed by precipitation and washing with ice cold methyl tert-butyl ether (MTBE). Crude peptides were purified by preparative reversed-phase HPLC using diphenyl column with a linear gradient of water/acetonitrile (both buffered with 0.1% TFA). The purities of peptides were greater than 95% by analytical HPLC analyses (Supplemental Figure S1, Supplemental Table S1). Purified analogs were quantified by UV absorbance (λ = 279.8 nm, ε = 7000 cm-1 M-1), and their monoisotopic mass was confirmed by MALDI-TOF mass spectrometry (Table 2).
Table 2.
Structure and properties of the galanin analogs. All analogs are amidated at the C-terminus.
| Analog | Structure | HPLC retention timea | α-helixb | Mass spec. (MALDI, MH+) Calcd/found |
|---|---|---|---|---|
| Gal-(K)4c | (Sar)WTLNSAGYLLGPKKKK | 15.13 ± 0.42 | 14% | 1873.09/1873.14 |
| Gal-B2-C8 | (Sar)WTLNSAGYLLGPKK(Lys-octanoyl)K | 20.13 ± 0.17 | 9% | 2001.21/2001.17 |
| Gal-B2-C10 | (Sar)WTLNSAGYLLGPKK(Lys-decanoyl)K | 21.49 ± 0.05 | 6% | 2029.24/2029.45 |
| Gal-B2-C12 | (Sar)WTLNSAGYLLGPKK(Lys-lauroyl)K | 22.51 ± 0.03 | 16% | 2057.28/2057.26 |
| Gal-B2-C14 | (Sar)WTLNSAGYLLGPKK(Lys-myristoyl)K | 23.76 ± 0.18 | 16% | 2085.31/2085.33 |
| Gal-B2c | (Sar)WTLNSAGYLLGPKK(Lys-palmitoyl)K | 25.84 ± 0.10 | 23% | 2112.31/2112.33 |
| Gal-B2-C18 | (Sar)WTLNSAGYLLGPKK(Lys-stearoyl)K | 26.73 ± 0.18 | 5% | 2141.33/2141.48 |
| Gal-B2-MPEG4 | (Sar)WTLNSAGYLLGPKK(Lys-MPEG4)K | 17.52 ± 0.03 | 18% | 2123.23/2123.29 |
A linear gradient of water/90% acetonitrile (buffered with 0.1% TFA) on a Vydac diphenyl column, starting from 20% acetonitrile to 100% acetonitrile in 40 min.
Determined in the presence of 50% v/v 2,2,2-trifluoroethanol.
Data from 22.
Physicochemical and structural properties
To determine the lipophilicity of the analogs, logD values were calculated using the combination of the HPLC retention times and the logD determined from the classical shake-flask method, as previously described.22 Calculations of logD values are provided in Supplemental Table S2. For the LAA containing analogs, the logD values ranged from 0.78 for Gal-B2-C8 to 1.35 for Gal-B2-C18. As shown in Figure 2, the logD values linearly correlated with the carbon chain length of the LAA (linear fit yielded R2 = 0.98). Since the logD values increased four-fold by replacing one Lys residue with Lys-stearoyl (compare logD of Gal-(K)4 (0.34)22 with Gal-B2-C18, 1.35), and almost doubled by changing the LAA from C8 to C18, these results emphasize the effectiveness of the LAAs to increase the lipophilicity of short peptides. The analog with the MPEG4-amino acid isostere had approximately two-fold lower logD (0.57), as compared to the Lys-palmitoyl residue (1.24).
Figure 2.
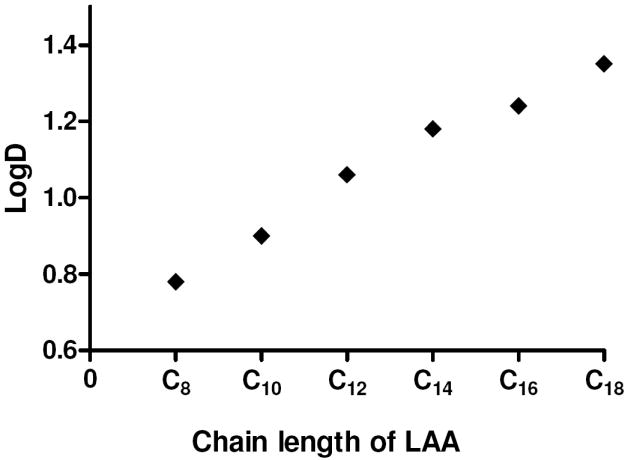
Relationships between the aliphatic chain length of the LAAs in Gal-B2 analogs and the octanol/water partition coefficient (logD). Linear fit yielded R2=0.9839, P<0.0001.
Gal-B2 is largely unstructured in water, but contains 23% α-helical content in the presence of 50% trifluoroethanol (TFE).22 In our previous work,22 we hypothesized that the presence of LAA could stabilize the helical conformation of the systemically-active galanin analogs by hydrophobic interactions with Tyr9 and/or Trp2. To test this hypothesis, the conformational properties of the Gal-B2 analogs were investigated by circular dichroism (CD). In the presence of 5 mM potassium phosphate containing 150 mM NaF (pH 7.5), the helical content varied among the analogs from 1% to 7% (only Gal-B2-C8 had 15%). The helical content in 50% TFE increased for most analogs as compared to that determined in “buffer only” conditions (Table 2). No apparent correlations were observed between the length of the LAA and percentage of helical content in the analogs.
Metabolic stability
To investigate how the length of the LAA residue might affect resistance of the galanin analogs to proteolytic degradation, we employed the in vitro metabolic stability assay. The half-life of the analogs was determined in buffered 25% rat blood serum incubated at 37 °C. The amount of each analog remaining after an appropriate time interval was quantified by HPLC and the time-course of disappearance of the analogs is shown in Figure 3A. Calculated half-live values for the analogs are summarized in Figure 3B. All analogs exhibited pronounced resistance to proteolytic degradation (for comparison, the unmodified Gal(1-16) analog had 7.8 min half-time under these conditions). 22 Interestingly, the galanin analogs containing the shortest or the longest aliphatic chain were the least stable, whereas the analog containing C14 had the longest half-life of 12 h (Figure 3B). This correlation suggested a rather complex mechanism by which the LAAs might increase the stability of galanin analogs in plasma. We hypothesized that the metabolic stability of the galanin analogs might be correlated with their serum albumin binding properties (e.g., binding properties of long-chain carboxylic acids to albumin are dependent on aliphatic chain length).23 However, our efforts in determining the protein binding properties of Gal-B2 by microdialysis failed due to possible micelle formation.22 The Gal-B2-MPEG4 analog exhibited a very long half-life (12.4 ± 0.65 h), suggesting that the extended amino acid residue might provide steric hindrance for access of proteinases.
Figure 3.
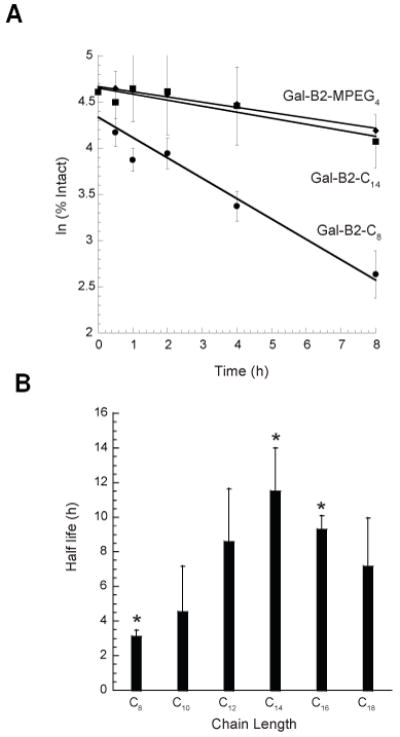
(A) In vitro serum stability assay for the galanin analogs. Representative plots showing a time-course of degradation for C8 (●), C14 (■), and MPEG4 (◆)-containing galanin analogs in the presence of 25% rat serum at 37 °C. Degradation of analogs was monitored by analytical HPLC. Data points were obtained at 0, 0.5, 1, 2, 4 and 8 h from the average of at least three independent experiments. (B) Relationships between the length of the LAAs and the in vitro metabolic stability of the galanin analogs. The half-lives were determined by incubating the peptides in 25% diluted rat serum at 37 °C. The concentrations of the remaining analogs were determined by HPLC. Data were obtained from at least three independent experiments. ANOVA single factor analysis yielded P-value of 0.004 for C8, C14, and C16-containing species (*).
In vitro and in vivo pharmacology
Since two galanin receptors, GalR1 and GalR2, are known to be involved in controlling seizures in the brain,24, 25 we investigated the interactions of the galanin analogs with both receptor subtypes using a competitive binding assay. As described in our previous work, Ki values were determined using a time-resolved fluorescence binding assay with a europium-labeled galanin.22 The GPCR membrane preparations used in the assay were commercially available (Perkin-Elmer or Millipore) and were derived from recombinant human GalR1 or GalR2 gene sequences. As shown in Table 3, when compared to each other, none of the analogs studied here exhibited significantly different affinities toward either receptor subtype. This suggested that the relatively large LAA moiety did not affect the affinity of the analogs towards the galanin receptors. All analogs maintained several-fold preference in binding to GalR1 subtype, as compared to GalR2, similar to that of unmodified Gal(1-16).22
Table 3.
In vitro and in vivo pharmacological properties of the galanin analogs
| In vitro assay (receptor binding) | In vivo assay (anticonvulsant activity) | ||||||
|---|---|---|---|---|---|---|---|
| Analog | GalR1 Ki (nM) | GalR2 Ki (nM) | (6 Hz, 32 mA, 4 mg/kg) | ||||
| 15’ | 30’ | 1h | 2h | 4h | |||
| Gal-(K)4 a | 0.4 ± 0.1 | 24.0 ± 9.9 | 0/4 | 3/4 | 1/3 | 0/3 | 0/4 |
| Gal-B2-C8 | 0.7 ± 0.1 | 14.9 ± 0.6 | 0/4 | 0/4 | 0/4 | 0/4 | 1/4 |
| Gal-B2-C10 | 1.3 ± 0.7 | 14.4 ± 0.6 | 2/4 | 2/4 | 0/4 | 0/4 | 0/4 |
| Gal-B2-C12 | 1.4 ± 0.4 | 16.1 ± 8.5 | 1/4 | 3/4 | 2/4 | 0/4 | 0/4 |
| Gal-B2-C14 | 2.6 ± 0.1 | 18.2 ± 1.6 | 2/4 | 3/4 | 2/4 | 0/4 | 0/4 |
| Gal-B2 a | 3.5 ± 1.0 | 51.5 ± 34.4 | 3/4 | 4/4 | 4/4 | 4/4 | 0/4 |
| Gal-B2-C18 | 4.0 ± 2.0 | 15.0 ± 1.0 | 4/4 | 3/4 | 4/4 | 2/4 | 0/4 |
| Gal-B2-MPEG4 | 0.5 ± 0.1 | 20.5 ± 6.5 | 1/4 | 1/4 | 1/4 | 2/4 | 0/4 |
Data from 22.
The anticonvulsant activity of the analogs was studied in the 6 Hz (32 mA) model of pharmacoresistant epilepsy following i.p. administration of a bolus dose of 4 mg/kg. The analogs were evaluated for their ability to suppress seizures at various times (0.25 to 4 h) after peptide administration. The time-response curves were then integrated to provide a qualitative measurement of efficacy; i.e., area under the curve (AUC) values (Table 3, Figure 4). The analogs differed significantly in their ability to protect mice from seizures. Based on the AUC values, the most active galanin analogs contained Gal-B2-C16 and Gal-B2-C18, whereas the least active analogs included C8 and C10. Interestingly, the Lys-MPEG4-containing analog was also active as an anticonvulsant, albeit significantly less when compared to Gal-B2.
Figure 4.
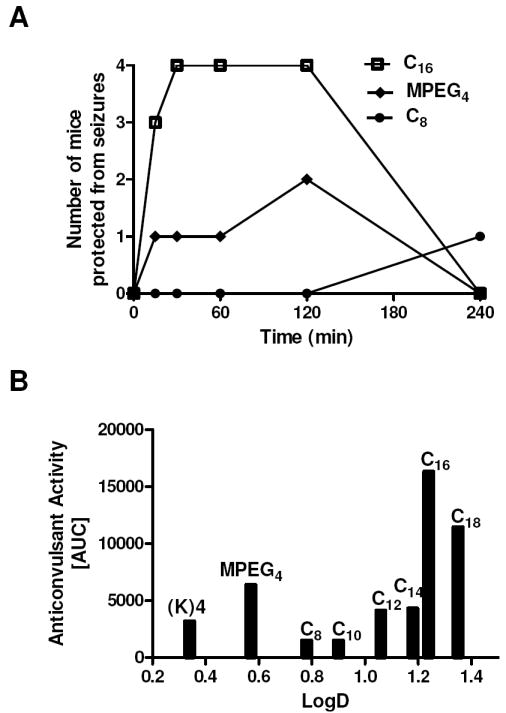
Anticonvulsant activity of the galanin analogs. The analogs were administered i.p. as a bolus dose of 4 mg/kg. (A) time-response studies ranging from 15 min to 4 h were used to calculate area under the curve values (AUC). (B) Relationships between logD and the anticonvulsant activity (AUC values) of the galanin analogs in the 6 Hz model of epilepsy.
Discussion
To study the structural requirements for the LAA residue 16 in mediating the antiepileptic activity of the systemically-active galanin analogs,22 we synthesized and characterized a new series of analogs that differed in their length/polarity of the fatty acid moiety coupled to the Lys16 residue. The major finding of this work was that, the six galanin analogs (despite having comparable affinities towards the galanin receptors and differing in the length of the fatty acid from C8 to C18), exhibited pronounced differences in their anticonvulsant activities following i.p. administration. The analogs containing shorter fatty acids were less active as compared to those with longer chains. These results suggest that the increased lipophilicity provided by the longer fatty acids is an important factor in improving the anticonvulsant activity of galanin analogs following systemic administration. Further work will be required to determine whether the resulting compounds are agonists, partial agonists, or antagonists. Interestingly, both Gal-B2 and Gal-B2-C8 showed comparable analgesic activities in a mouse pain assay following i.p. administration (E. Adkins-Scholl, H.S. White, G. Bulaj, unpublished results), suggesting that systemically-active galanin analogs varying in the length of a LAA may control seizures or pain via galanin receptors in the CNS or peripheral nerves, respectively.26-28 Our previous data indicated that the position of the LAA residue appeared to be important for the anticonvulsant activity of Gal-B2,22 thus implying that lipophilicity alone (which would be expected to increase passive diffusion) is not the sole important factor for improving seizure suppression by this analog. Although the “-Lys-Lys-Lys(palmitoyl)-Lys-NH2” motif appeared to be the most effective in increasing the anticonvulsant activity of the galanin analogs following i.p. administration. Clearly, more SAR studies, or perhaps even a combinatorial approach, will be needed to further improve the potency of the Gal-B2 related analogs.22
Introduction of the LAA to the galanin analogs significantly improved their in vitro metabolic stability; this effect may be, at least in part, accounted for by steric effects of the long side chain in position 16 of the analogs. This finding is not surprising in light of previous reports with lipopeptides, in which prolonged in vitro and/or in vivo half-lives were observed,29 including: gonadotropin-releasing hormone analogs,30 glucagon-like peptide 1 analogs,31 insulin,32 and octreotide.20 The metabolic stability of lipopeptides may also be affected by their interactions with fatty acid binding sites in serum albumin.32 As shown in Figure 3B, the longest serum half-lives were observed for Gal-B2-C14 and Gal-B2-C16, whereas the shortest were observed for Gal-B2-C8. Interestingly, the C8 (octanoic acid) moiety binds with 11-fold higher affinity to human serum albumin compared to the C14 (myristic acid).23
Another aspect of this work is our less conventional approach of using orthogonally protected LAAs and MPEG4-amino acids as building blocks for SPPS. Introduction of LAA into peptides have been reported using three distinct strategies: 1) on-resin fatty acid acylation of Nα-amino acids,33, 34 or Nε-lysine,35 2) esterification of fatty acids,36 and 3) conjugation at the C-terminus of peptide.18, 37 Our strategy of using the Fmoc protected LAAs directly coupled during the solid-phase peptide synthesis offers apparent advantages in applications such as routine synthesis of lipopeptide analogs for SAR studies or synthesis of peptide-based combinatorial libraries directed toward optimization of the chain length of the LAAs in the cationic/lipidic motif.
To what extent the systemically-active galanin analogs may have improved the BBB permeability remains unanswered. However, since some of the modified galanin analogs possess pronounced antiepileptic activity, believed to be mediated by the galanin receptors located in the limbic system and thus, behind the blood-brain-barrier, these data suggest that sufficient quantities of the analogs must penetrate into the brain. As we have suggested previously,22 lipidization may improve CNS bioavailability of Gal-B2 via a combination of adsorptive-mediated endocytosis and passive diffusion.38, 39 More mechanistic studies assessing the mechanism of transport of galanin analogs across the cell-based models of BBB are underway. Regardless of the mechanism by which these analogs penetrate into the brain, our data suggest that changing the length and/or polarity of the LAA residue in the context of cationization may offer a useful strategy to modulate CNS bioavailability of galanin analogs. In summary, we believe that the variations in lipidization and cationization appear to provide a useful strategy for modulating CNS bioavailability of not only the galanin analogs described here, but perhaps even other neuropeptides.
Experimental section
General synthetic procedures
Reagent chemicals were obtained from Aldrich Chemical Corporation and were used without prior purification. Reactions were performed under N2 atmosphere, unless otherwise indicated. Chromatography refers to flash chromatography on silica gel (Whatman 230-400 mesh ASTM silica gel). Preparative HPLC was performed on a Waters 600 pump system equipped with a Waters 2487 dual wavelength detector (λ1 = 220 nm, λ2 = 280 nm) and a preparative Vydac diphenyl column (219TP101522); analytical HPLC used an analytical Vydac diphenyl column (219TP54). The HPLC mobile phases are Buffer A, 100% water (0.1% TFA) and Buffer B, 90% acetonitrile (0.1% TFA). Metabolic stability used a Waters Alliance 2695 system equipped with an autosampler, dual wavelength detector, and a Waters YMC ODS-A diphenyl column (AA125052503WT). NMR spectra were recorded at 400 MHz (1H), 101 MHz (13C) at 25 °C. Proton and carbon chemical shifts are given in ppm relative to TMS internal standard. MALDI-TOF MS were determined at the University of Utah Core Facility. CD spectra were obtained on an Aviv 62DS CD spectropolarimeter at room temperature. Optical rotations were measured on a Perkin–Elmer polarimeter (model 343) using a 1 mL capacity quartz cell with a 10 cm path length.
Fmoc-Lys(octanoyl)-OH (1)
Fmoc-L-lysine (0.737 g, 2.0 mmol) was added to dioxane (10 mL), and then a solution of Na2CO3 (0.636 g, 6.0 mmol) in H2O (12.6 mL) was added drop wise at 0 °C. After 5 min, a solution of octanoyl chloride (0.325 g, 2.0 mmol) in dioxane (10 mL) was added to the mixture. The solution was allowed to warm to room temperature and stirred overnight. The solvents were extracted with CH2Cl2 (250 ml), washed with saturated NaHCO3 (20 mL), 1M HCl (2 ml) and brine (20 mL), dried over Na2SO4 and concentrated. Flash chromatography (CH2Cl2/CH3OH, 6:1) gave a white amorphous solid 1 (0.86 g, 87%). TLC Rf = 0.29 (CH2Cl2/CH3OH, 10:1); [α]20D +10.2° (c 1, CHCl3); 1H NMR (CDCl3): δ (ppm) 7.68 (d, J = 7.6 Hz, 2H), 7.53 (m, 2H), 7.32 (m, 2H), 7.23 (m, 2H), 5.67 (m, 1H), 4.29 (m, 2H), 4.13 (m, 1H), 3.19 (m, 2H), 2.10 (tm, 2H), 1.84-1.17 (m, 16H), 0.78 (m, 3H); 13C NMR (CDCl3): δ (ppm) 174.40, 143.95, 141.40, 127.90, 127.29, 125.39, 120.12, 67.15, 47.30, 39.42, 36.84, 31.94, 29.52, 29.27, 26.07, 22.84, 14.31; HRMS (MALDI) (m/z) (MNa+): found 517.2673; Calcd for C29H38N2O5Na 517.2678.
Fmoc-Lys(decanoyl)-OH (2)
Compound 2 was submitted to the same procedure described above for the preparation of 1 and gave a white amorphous solid (yield, 78%); TLC Rf = 0.40 (CH2Cl2/CH3OH, 8:1); [α]20D +12.0° (c 1, CHCl3); 1H NMR (CDCl3): δ (ppm) 7.67 (d, J = 7.6 Hz, 2H), 7.52 (m, 2H), 7.30 (m, 2H), 7.21 (m, 2H), 5.78 (d, J = 7.6 Hz,1H), 4.27 (m, 2H), 4.12 (m, 1H), 3.17 (m, 1H), 2.10 (tm, 2H), 1.83-1.14 (m, 20H), 0.78 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3): δ (ppm) 174.69, 156.64, 144.14, 143.91, 141.45, 127.94, 127.31, 125.38, 120.17, 98.99, 67.27, 66.36, 61.17, 54.04, 47.31, 39.41, 36.87, 32.26, 32.09, 29.72, 29.60, 29.53, 29.04, 26.08, 22.90, 22.54, 14.36; HRMS (MALDI) (m/z) (MNa+): found 545.2978; Calcd for C31H42N2O5Na 545.2991.
Fmoc-Lys(lauroyl)-OH (3)
Compound 3 was submitted to the same procedure described above for the preparation of 1 and gave a white solid (yield, 64%); mp 126.0-128.0 °C; TLC Rf = 0.76 (CH2Cl2/CH3OH, 4:1); [α]20D +14.3° (c 1, CHCl3); 1H NMR (CDCl3): δ (ppm) 7.67 (d, J = 7.6 Hz, 2H), 7.52 (m, 2H), 7.30 (m, 2H), 7.21 (m, 2H), 5.82-5.77 (m, 2H), 4.26-4.39 (m, 2H), 4.12 (t, J = 7.2 Hz, 1H), 3.17 (m, 2H), 2.09 (m, 2H), 1.83-1.35 (m, 6H), 1.48 (m, 17H), 0.79 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3): δ (ppm) 175.11, 174.81, 156.56, 143.93, 141.49, 127.93, 127.31, 125.38, 120.18, 67.32, 53.76, 47.34, 39.36, 36.94, 32.13, 29.84, 29.72, 29.56, 29.14, 26.03, 22.91, 22.34, 14.35; HRMS (MALDI) (m/z) (MNa+): found 573.3299, Calcd for C33H46N2O5Na 573.3304.
Fmoc-Lys(myristoyl)-OH (4)
Compound 4 was submitted to the same procedure described above for the preparation of 1 and gave a white solid (yield, 64%); mp 132.0-134.0 °C; TLC Rf = 0.81 (CH2Cl2/CH3OH, 3:1); [α]20D +13.4° (c 1, CHCl3); 1H NMR (CDCl3): δ (ppm) 7.73 (d, J = 7.6 Hz, 2H), 7.59 (m, 2H), 7.37 (m, 2H), 7.27 (m, 2H), 5.97 (br, 1H), 5.79 (d, J = 7.6 Hz, 1H), 4.41-4.33 (m, 2H), 4.18 (t, J = 7.6 Hz, 1H), 3.23 (m, 2H), 2.15 (m, 2H), 1.89-1.41 (m, 6H), 1.21 (m, 22H), 0.87 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3): δ (ppm) 175.27, 174.86, 156.58, 144.11, 143.92, 141.48, 127.94, 127.32, 125.39, 120.18, 67.37, 53.78, 47.33, 39.39, 36.91, 32.16, 29.93, 29.89, 29.75, 29.59, 29.53, 29.08, 26.06, 22.93, 22.40, 14.36; HRMS (MALDI) (m/z) (MNa+): found 601.3623, Calcd for C35H50N2O5Na 601.3617.
Fmoc-Lys(stearoyl)-OH (5)
Compound 5 was submitted to the same procedure described above for the preparation of 1 and gave a white solid (yield, 68%); mp 133.0-135.0 °C; TLC Rf = 0.62 (CH2Cl2/CH3OH, 5:1); [α]20D +11.8° (c 1, CHCl3); 1H NMR (CDCl3): δ (ppm) 7.68 (d, J = 7.6 Hz, 2H), 7.52 (m, 2H), 7.31 (m, 2H), 7.22 (m, 2H), 5.73 (d, J = 7.2, Hz, 1H), 4.28 (m, 2H), 4.12 (m, 1H), 3.19 (m, 2H), 2.09 (m, 2H), 1.84-1.18 (m, 37H), 0.80 (m, 3H); 13C NMR (CDCl3): δ (ppm) 175.27, 174.69, 156.54, 144.13, 143.94, 141.41, 127.94, 127.31, 125.40, 120.19, 67.32, 53.75, 47.35, 39.29, 36.97, 32.15, 29.93, 29.58, 29.10, 29.19, 26.03, 22.92, 22.33, 14.35; HRMS (MALDI) (m/z) (MNa+): found 657.4240; Calcd for C39H58N2O5 Na 657.4243.
MPEG4-acetic acid (7)
To a solution of MPEG4-OH (2.20 g, 10.6 mmol) in THF (20 mL) was added NaH (60%, 0.635 g, 15.9 mmol) at 0 °C. The solution was stirred for 1 h at room temperature, and then ethyl bromoacetate (1.4 mL, 12.6 mmol) was added dropwise. After 16 h at 50 °C, the solution was concentrated in vacuo. The residue was hydrolyzed with 1M LiOH (8 mL) and CH3OH (8 mL) for 6 h at room temperature. The solution was acidified with 1M HCl to pH 1, and then the solvents were removed under reduced pressure. The residue was purified by chromatography (CH2Cl2/CH3OH 10:1) to afford 7 (1.66 g, 59%) as a colorless oil. TLC Rf = 0.15 (CH2Cl2/CH3OH, 4:1); 1H NMR (CDCl3): δ (ppm) 9.10 (br, 1H), 4.03 (s, 2H), 73.57-3.44 (m, 16H), 3.25 (s, 3H); 13C NMR (CDCl3): δ (ppm) 172.94, 71.93, 71.09, 70.64, 70.58, 70.55, 70.51, 70.45, 68.61, 61.61, 59.02; HRMS (MALDI) (m/z) (MNa+): found 289.1248, Calcd for C11H22O7Na 289.1263.
Fmoc-Lys(MPEG4)-OH (6)
To a solution of MPEG4-acetic acid 7 (0.266 g, 1.0 mmol) and perfluorophenol (0.202 g, 1.1 mmol) in CH2Cl2 (5 mL) was added DCC (0.247 g, 1.2 mmol) at room temperature. After stirring overnight, the mixture was diluted with acetone (50 mL) and filtered; the residue was dried in vacuo to give the activated ester, which was used directly in the next step. The crude oil in DMF (5 mL) was added to a solution of Fmoc-L-lysine (0.442 g, 1.2 mmol) and DIPEA (0.7 mL, 4 mmol) in DMF (10 mL) at 0 °C. The solution was stirred overnight at room temperature, and then poured into brine (50 mL), extraction with CH2Cl2 (150 mL), washed with 1M HCl, brine, dried and purified by flash chromatography (CH2Cl2/CH3OH, 5:1) to afford an amorphous solid 6 (0.37 g, 60%). TLC Rf = 0.21 (CH2Cl2/CH3OH, 5:1); [α]20D +19.5 ° (c 3, CHCl3); 1H NMR (CDCl3): δ (ppm) 7.67 (d, J = 7.2 Hz, 2H), 7.52 (m, 2H), 7.29 (m, 2H), 7.21 (m, 2H) ,5.78 (d, J = 6.8 Hz, 1H), 4.29 (d, J = 6.8 Hz, 2H), 4.11 (m, 1H), 3.91 (s, 2H), 3.54-3.52 (m, 14H), 3.45-3.43 (m, 2H), 3.27 (s, 3H), 1.81-1.18 (m, 6H); 13C NMR (CDCl3): δ (ppm) 171.04, 156.36, 144.18, 144.03, 141.48, 127.90, 127.30, 125.39, 120.15, 72.07, 71.09, 70.69, 70.61, 70.27, 67.05, 59.15, 53.92, 47.38, 38.54, 31.78, 29.91, 29.00, 22.27; HRMS (MALDI) (m/z) (MNa+): found 639.2917, Calcd for C32H44N2O10Na 639.2893.
Peptide synthesis
All galanin analogs were synthesized on a Symphony peptide synthesizer (Protein Technologies Inc.) using Fmoc-based coupling protocols as previously described.22 Cleaved peptides were purified by reversed-phase HPLC using preparative HPLC column (Vydac diphenyl, 219TP1011522) and eluted with a linear gradient of acetonitrile (0.1% TFA). The flow rate was 10 mL/min, and the elution was monitored by UV detection at 220 nm and 280 nm. Buffer A (0.1% TFA in water) and buffer B (0.1% TFA, v/v, in 90% aqueous acetonitrile) were used to produce a linear gradient from 20 to 100% of buffer B over 40 min. Purified analogs were quantified by measuring UV absorbance at 279.8 nm (molar absorbance coefficient ε = 7,000 cm-1 M-1).
Partitioning coefficient, logD
LogD values were determined based on calculated capacity factors derived from HPLC retention times as previously described.22 The logD for five galanin analogs were determined using conventional shake flask methods, using 400 μg of peptide reconstituted in 1 mL of phosphate buffered saline. The aqueous peptide solution was shaken with an equal volume of water saturated octanol for 24 h. Concentration of the aqueous layer was determined by HPLC. Retention times for the analogs was determine on a linear gradient of 20% to 100% buffer B in 40 min, with immediate return to initial conditions and a 20 min re-equilibration. A linear correlation between logD and HPLC retention times gave calculated logD values.
Circular dichroism
α-Helical conformation of each analog was studied using CD as previously described.22 Peptides were reconstituted in NaF buffer to a concentration of 0.1 mg/mL. The peptides were scanned from 250 nm to 200 nm, with a 1 nm step size and 1.0 s dwell time. Data were averaged from 5 scans.
Metabolic stability assay
Peptide stability was assessed in a buffered 25% rat blood serum assay as previously described.22 To 200 μL of buffered serum, 2.5 μg of peptide was added and the solution was incubated at 37 °C. Reactions were quenched at select time points upon the addition of “quenching solution” (15% trichloroacetic acid in 40% isopropanol), and the remaining amount of intact peptide was determined by HPLC.
Pharmacological characterization
Galanin receptor binding assay and the anticonvulsant screening were carried out as previously described in detail.22 Briefly, the competitive binding assay was performed using human GalR1 and GalR2 receptor membrane preparations and europium-labeled galanin (Perkin-Elmer). Samples were incubated at room temperature for 90 min, followed by 4 washings with wash buffer (50 mM Tris-HCl pH 7.5 and 5 mM MgCl2) using a vacuum manifold. Enhancement solution (200 μL) was added, and the plates were incubated at room temperature for 30 min. The plates were read on a VICTOR3 spectrofluorometer. Competition binding curves were analyzed using GraphPad Prism software, using a nonlinear regression, sigmoidal dose-response (variable slope) curve with no constraints or weights, seen below. Top and Bottom are the plateaus on the y-axis. With this model, we hold the Kd and concentration of europium-labeled galanin constant (4.3 nM and 2 nM, respectively) and assume one binding site with reversible binding at equilibrium.
Anticonvulsant efficacy was assessed following i.p. administration to five groups of CF-1 mice (n = 4 mice) at a dose of 4 mg/kg. At various times (i.e., 0.25, 0.5, 1, 2, and 4 h) after administration, mice were challenged with a 6 Hz corneal stimulation (32 mA, 3 sec, 6 Hz). Mice not displaying a characteristic limbic seizure were considered protected. The percent of animals protected at each time-point was plotted against time, and the AUC values were calculated with GraphPad Prism.
Supplementary Material
Acknowledgments
This work was supported in part by the Epilepsy Therapy Grants Program from the Epilepsy Research Foundation, the University of Utah Startup Funds and the NIH grant R21 NS059669. We would like to thank the Anticonvulsant Screening Program (ASP) Project Officer James Stables and his group at the NIH/NINDS for their support with screening galanin analogs. Technical assistance of Dan McDougle is greatly appreciated.
Abbreviations
- AUC
area under the curve
- BBB
blood-brain barrier
- CD
circular dichroism
- CNS
central nervous system
- DCC
N,N’-dicyclohexylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- Fmoc
N-(9-fluorenyl)methoxycarbonyl
- GalR1
galanin receptors subtype 1
- GalR2
galanin receptors subtype 2
- GPCRs
G-protein coupled receptors
- i.p
intraperitoneally
- LAA
Lipoamino acid
- MPEG4
monodispersed polyethylene glycol PEG4
- PyBop
(Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- SAR
structure-activity relationship
- SPPS
solid phase peptide synthesis
- TFA
trifluoroacetic acid
- TFE
2,2,2-trifluoroethanol
Footnotes
Conflict of Interest Disclosure: GB and HSW are scientific co-founders of NeuroAdjuvants, Inc.
Supporting Information Available: Two supplemental tables and one supplemental figure are available as supporting information. Supplemental Tables S1 and S2 summarize the purity and logD calculations; whereas Supplemental Figure S1 shows the HPLC chromatograms obtained with the galanin analogs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tallent MK, Qiu C. Somatostatin: An endogenous antiepileptic. Mol Cell Endocrinol. 2008;286:96–103. doi: 10.1016/j.mce.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lerner JT, Sankar R, Mazarati AM. Galanin and epilepsy. Cell Mol Life Sci. 2008;65:1864–1871. doi: 10.1007/s00018-008-8161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci. 1999;22:25–30. doi: 10.1016/s0166-2236(98)01284-3. [DOI] [PubMed] [Google Scholar]
- 4.Boules M, Fredrickson P, Richelson E. Bioactive analogs of neurotensin: Focus on CNS effects. Peptides. 2006;27:2523–2533. doi: 10.1016/j.peptides.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 5.Polt R, Dhanasekaran M, Keyari CM. Glycosylated neuropeptides: a new vista for neuropsychopharmacology? Med Res Rev. 2005;25:557–585. doi: 10.1002/med.20039. [DOI] [PubMed] [Google Scholar]
- 6.Rousselle C, Clair P, Smirnova M, Kolesnikov Y, Pasternak GW, Gac-Breton S, Rees AR, Scherrmann JM, Temsamani J. Improved brain uptake and pharmacological activity of dalargin using a peptide-vector-mediated strategy. J Pharmacol Exp Ther. 2003;306:371–376. doi: 10.1124/jpet.102.048520. [DOI] [PubMed] [Google Scholar]
- 7.Prokai-Tatrai K, Prokai L, Bodor N. Brain-targeted delivery of a leucine-enkephalin analogue by retrometabolic design. J Med Chem. 1996;39:4775–4782. doi: 10.1021/jm960356e. [DOI] [PubMed] [Google Scholar]
- 8.Elmagbari NO, Egleton RD, Palian MM, Lowery JJ, Schmid WR, Davis P, Navratilova E, Dhanasekaran M, Keyari CM, Yamamura HI, Porreca F, Hruby VJ, Polt R, Bilsky EJ. Antinociceptive structure-activity studies with enkephalin-based opioid glycopeptides. J Pharmacol Exp Ther. 2004;311:290–297. doi: 10.1124/jpet.104.069393. [DOI] [PubMed] [Google Scholar]
- 9.Egleton RD, Mitchell SA, Huber JD, Janders J, Stropova D, Polt R, Yamamura HI, Hruby VJ, Davis TP. Improved bioavailability to the brain of glycosylated Met-enkephalin analogs. Brain Res. 2000;881:37–46. doi: 10.1016/s0006-8993(00)02794-3. [DOI] [PubMed] [Google Scholar]
- 10.Banks WA, Schally AV, Barrera CM, Fasold MB, Durham DA, Csernus VJ, Groot K, Kastin AJ. Permeability of the murine blood-brain barrier to some octapeptide analogs of somatostatin. Proc Natl Acad Sci U S A. 1990;87:6762–6766. doi: 10.1073/pnas.87.17.6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kokko KP, Hadden MK, Price KL, Orwig KS, See RE, Dix TA. In vivo behavioral effects of stable, receptor-selective neurotensin[8-13] analogues that cross the blood-brain barrier. Neuropharmacology. 2005;48:417–425. doi: 10.1016/j.neuropharm.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Banks WA, Wustrow DJ, Cody WL, Davis MD, Kastin AJ. Permeability of the blood-brain barrier to the neurotensin8-13 analog NT1. Brain Res. 1995;695:59–63. doi: 10.1016/0006-8993(95)00836-f. [DOI] [PubMed] [Google Scholar]
- 13.Witt KA, Gillespie TJ, Huber JD, Egleton RD, Davis TP. Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability. Peptides. 2001;22:2329–2343. doi: 10.1016/s0196-9781(01)00537-x. [DOI] [PubMed] [Google Scholar]
- 14.Egleton RD, Davis TP. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx. 2005;2:44–53. doi: 10.1602/neurorx.2.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banks WA. Delivery of peptides to the brain: Emphasis on therapeutic development. Biopolymers. 2008;90:589–594. doi: 10.1002/bip.20980. [DOI] [PubMed] [Google Scholar]
- 16.Banks WA, Kastin AJ. Peptides and the blood-brain barrier: lipophilicity as a predictor of permeability. Brain Res Bull. 1985;15:287–292. doi: 10.1016/0361-9230(85)90153-4. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Hogenkamp DJ, Tran M, Li WY, Yoshimura RF, Johnstone TB, Shen WC, Gee KW. Reversible lipidization for the oral delivery of leu-enkephalin. J Drug Target. 2006;14:127–136. doi: 10.1080/10611860600648221. [DOI] [PubMed] [Google Scholar]
- 18.Toth I, Malkinson JP, Flinn NS, Drouillat B, Horvath A, Erchegyi J, Idei M, Venetianer A, Artursson P, Lazorova L, Szende B, Keri G. Novel lipoamino acid- and liposaccharide-based system for peptide delivery: application for oral administration of tumor-selective somatostatin analogues. J Med Chem. 1999;42:4010–4013. doi: 10.1021/jm9910167. [DOI] [PubMed] [Google Scholar]
- 19.Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J Med Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 20.Yuan L, Wang J, Shen WC. Reversible lipidization prolongs the pharmacological effect, plasma duration, and liver retention of octreotide. Pharm Res. 2005;22:220–227. doi: 10.1007/s11095-004-1189-z. [DOI] [PubMed] [Google Scholar]
- 21.Weber SJ, Abbruscato TJ, Brownson EA, Lipkowski AW, Polt R, Misicka A, Haaseth RC, Bartosz H, Hruby VJ, Davis TP. Assessment of an in vitro blood-brain barrier model using several [Met5]enkephalin opioid analogs. J Pharmacol Exp Ther. 1993;266:1649–1655. [PubMed] [Google Scholar]
- 22.Bulaj G, Green BR, Lee HK, Robertson CR, White K, Zhang L, Sochanska M, Flynn SP, Scholl EA, Pruess TH, Smith MD, White HS. Design, synthesis, and characterization of high-affinity, systemically-active galanin analogues with potent anticonvulsant activities. J Med Chem. 2008;51:8038–8047. doi: 10.1021/jm801088x. [DOI] [PubMed] [Google Scholar]
- 23.Kragh-Hansen U, Watanabe H, Nakajou K, Iwao Y, Otagiri M. Chain length-dependent binding of fatty acid anions to human serum albumin studied by site-directed mutagenesis. J Mol Biol. 2006;363:702–712. doi: 10.1016/j.jmb.2006.08.056. [DOI] [PubMed] [Google Scholar]
- 24.Mazarati A, Lundstrom L, Sollenberg U, Shin D, Langel U, Sankar R. Regulation of kindling epileptogenesis by hippocampal galanin type 1 and type 2 receptors: The effects of subtype-selective agonists and the role of G-protein-mediated signaling. J Pharmacol Exp Ther. 2006;318:700–708. doi: 10.1124/jpet.106.104703. [DOI] [PubMed] [Google Scholar]
- 25.Mazarati AM. Galanin and galanin receptors in epilepsy. Neuropeptides. 2004;38:331–343. doi: 10.1016/j.npep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Liu HX, Hokfelt T. The participation of galanin in pain processing at the spinal level. Trends Pharmacol Sci. 2002;23:468–474. doi: 10.1016/s0165-6147(02)02074-6. [DOI] [PubMed] [Google Scholar]
- 27.Flatters SJ, Fox AJ, Dickenson AH. In vivo and in vitro effects of peripheral galanin on nociceptive transmission in naive and neuropathic states. Neuroscience. 2003;116:1005–1012. doi: 10.1016/s0306-4522(02)00947-8. [DOI] [PubMed] [Google Scholar]
- 28.Wynick D, Thompson SW, McMahon SB. The role of galanin as a multi-functional neuropeptide in the nervous system. Curr Opin Pharmacol. 2001;1:73–77. doi: 10.1016/s1471-4892(01)00006-6. [DOI] [PubMed] [Google Scholar]
- 29.Toth I. A novel chemical approach to drug delivery: lipidic amino acid conjugates. J Drug Target. 1994;2:217–239. doi: 10.3109/10611869408996805. [DOI] [PubMed] [Google Scholar]
- 30.Blanchfield JT, Lew RA, Smith AI, Toth I. The stability of lipidic analogues of GnRH in plasma and kidney preparations: the stereoselective release of the parent peptide. Bioorg Med Chem Lett. 2005;15:1609–1612. doi: 10.1016/j.bmcl.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 31.Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thogersen H, Wilken M, Agerso H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 32.Havelund S, Plum A, Ribel U, Jonassen I, Volund A, Markussen J, Kurtzhals P. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm Res. 2004;21:1498–1504. doi: 10.1023/b:pham.0000036926.54824.37. [DOI] [PubMed] [Google Scholar]
- 33.Oh HS, Kim S, Cho H, Lee KH. Development of novel lipid-peptide hybrid compounds with antibacterial activity from natural cationic antibacterial peptides. Bioorg Med Chem Lett. 2004;14:1109–1113. doi: 10.1016/j.bmcl.2003.12.085. [DOI] [PubMed] [Google Scholar]
- 34.Rivett DE, Hewish D, Kirkpatrick A, Werkmeister J. Inhibition of membrane-active peptides by fatty acid-peptide hybrids. J Protein Chem. 1999;18:291–295. doi: 10.1023/a:1021035328105. [DOI] [PubMed] [Google Scholar]
- 35.Chicharro C, Granata C, Lozano R, Andreu D, Rivas L. N-terminal fatty acid substitution increases the leishmanicidal activity of CA(1-7)M(2-9), a cecropin-melittin hybrid peptide. Antimicrob Agents Chemother. 2001;45:2441–2449. doi: 10.1128/AAC.45.9.2441-2449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pignatello R, Puglisi G. Lipophilicity evaluation by RP-HPLC of two homologous series of methotrexate derivatives. Pharm Acta Helv. 2000;74:405–410. doi: 10.1016/s0031-6865(00)00021-2. [DOI] [PubMed] [Google Scholar]
- 37.Horvath A, Olive C, Wong A, Clair T, Yarwood P, Good M, Toth I. Lipoamino acid-based adjuvant carrier system: enhanced immunogenicity of group a streptococcal peptide epitopes. J Med Chem. 2002;45:1387–1390. doi: 10.1021/jm0110441. [DOI] [PubMed] [Google Scholar]
- 38.Tamai I, Sai Y, Kobayashi H, Kamata M, Wakamiya T, Tsuji A. Structure-internalization relationship for adsorptive-mediated endocytosis of basic peptides at the blood-brain barrier. J Pharmacol Exp Ther. 1997;280:410–415. [PubMed] [Google Scholar]
- 39.Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem. 2003;278:31192–31201. doi: 10.1074/jbc.M303938200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.