

Abstract
With an anisole containing polypyridylamine potential tetradentate ligand OL, a μ-1,2-peroxo-dicopper(II) complex [{OLCuII}2(O22-)]2+ forms from the reaction of the mononuclear compound [CuI(OL)(MeCN)]B(C6F5)4(OLCuI) with O2 in non-coordinating solvents at -80 °C. Thermal decay of this peroxo complex in the presence of toluene or ethylbenzene leads to rarely seen C-H activation chemistry; benzaldehyde and acetophenone/1-phenylethanol mixtures, respectively, are formed. Experiments with 18O2 confirm that the oxygen source in the products is molecular O2 and deuterium labeling experiments indicate kH/kD = 7.5 ± 1 for the toluene oxygenation. The O2-reaction of [CuI(BzL)(CH3CN)]+ (BzLCuI) leads to a dicopper(III)-bis-μ-oxo species [{BzLCuIII}2(μ-O2-)2]2+ at -80 °C and from such solutions, very similar toluene oxygenation chemistry occurs. Ligand BzL is a tridentate chelate, possessing the same moiety found in OL, but without the anisole O-atom donor. In these contexts, the nature of the oxidant species in or derived from [{OLCuII}2(O22-)]2+ is discussed and likely mechanisms of reaction initiated by toluene H-atom abstraction chemistry are detailed. To confirm the structural formulations of the dioxygen-adducts, UV-vis and resonance Raman spectroscopic studies have been carried out and these results are reported and compared to previously described systems including [{CuII(PYL)}2(O2)]2+ (PYL =TMPA = tris(2-methylpyridyl)amine). Using (L)CuI, CO-binding properties (i.e., νC-O values) along with electrochemical property comparisons, the relative donor abilities of OL, BzL and PYL are assessed.
Introduction
The topic of dioxygen activation by copper enzymes has garnered considerable recent attention due to the relevance of copper(I)-dioxygen interactions and derived species to effect or alter critical biological functions.1-6 For instance, a dicopper(II)-μ-η2:η2-(side-on)-peroxo is the structure formed in hemocyanin (Hc), the O2-carrier for arthropods and mollusks;7 a similar structure or possibly an isomeric bis-μ-oxo-dicopper(III) form is implicated in tyrosinase (Tyr), a ubiquitous binuclear copper enzyme that catalyzes the hydroxylation of phenols to catechols and/or the oxidation of catechols to quinones.1,8 Enzymatic copper-dioxygen derived species also effect aliphatic C-H bond hydroxylations, for example in dopamine β-monooxygenase (DβM) and peptidylglycine α-amidating monooxygenase (PHM), neurosecretory vesicular copper enzymes which play a crucial role in the biogenesis of neurotransmitters and peptide hormones.9,10 DβM and PHM possess two active site copper ions, separated by about 11 Å, too far to accommodate a bridging dioxygen derived species.11 Instead, a mononuclear copper(II)-superoxide (CuII-O2-) or copper(II)-hydroperoxide (CuII-OOH) entity is believed to be the active species which initiates C-H bond hydroxylation reactions via initial hydrogen atom abstraction chemistry.9,12,13 X-ray crystallographic analysis has also revealed that particulate methane monooxygenase (p-MMO), an enzyme which catalyzes the conversion of methane to methanol in bacterial methanotrophs, possesses both mono and binuclear copper centers;14 while a CuIn-dioxygen derived entity is likely responsible for aliphatic C-H bond oxidation chemistry, there is as yet little insight concerning the active site identity or chemistry.
Biomimetic studies have proven to be very helpful in the development of fundamental chemistry involving copper(I)/O2 interactions by elucidating the nature of possible Cun-O2 (n = 1, 2) species as well as for clarifying their corresponding substrate reactivity mechanisms.2-4 There has been very recent activity involving mono-copper O2-derived complexes and reactivity.6,15 Of primary interest here are copper-O2 derived species which are binuclear and which have been demonstrated to initiate aliphatic C-H bond oxidations; four notable examples follow (a-d): Itoh and co-workers (a) discovered and then elucidated a DβM model system wherein a dicopper(III)-bis-μ-oxo {CuIII2-(O2-)2} complex possessing pyridylalkylamine ligands effects an internal (i.e., ligand) benzylic hydroxylation reaction.16 Tolman and coworkers (b) detailed intramolecular ligand oxidative N-dealkylation chemistry with CuIII2-(O)2 species that consist of alkyl substituted triazacyclononane ligands.17 Karlin and coworkers (c) described the μ-η2:η2-peroxodicopper(II)/bis-μ-oxo-dicopper(III) oxidative N-dealkylation of exogenous substrate N,N-dimethylanilines; the same complexes effect tetrahydrofuran 2-position hydroxylation.18,19 Lastly, Suzuki and coworkers (d) reported a dicopper(II)-μ-hydroxo-μ-hydroperoxo entity that effects intramolecular methylene hydroxylation of a coordinated ligand (i.e. an Ar-CH2NR2 group) and subsequent N-dealkylation.20 A structurally similar dicopper(II)-μ-phenoxo-μ-hydroperoxo from Karlin and coworkers was found to not only effect ligand aliphatic -CH2- group oxidation, but also α-cyanide C-H hydroxylation of RCH2CN solvent molecules present in solution.21
In the present report, a ligand-copper-complex with striking copper-dioxygen mediated C-H activation is described by utilizing a new tetradentate anisole containing ligand (OL; Chart 1). Generally, tetradentate chelates lead to dicopper(II)-μ-1,2-end-on-peroxide species which are oxidatively inert and nucleophilic in their reactivity, see Chart 1.2,3,22 Via a designed variation of a well known tetradentate ligand, TMPA (PyL; Chart 1), we here continue our broad efforts to elucidate new copper-dioxygen chemistry, unearth new structures and reactive intermediates, deduce structure-spectroscopic correlations and determine scope and mechanisms of substrate reactions.2,23,24 The copper(I) complex of OL, [CuI(OL)(CH3CN)]+ (OLCuI) reacts with O2 at low temperature (-80 °C) in non-coordinating and non-ether containing solvents (toluene or CH2Cl2) leading to the formation of a dicopper(II)-μ-1,2-end-on-peroxide species [{OLCuII}2(O22-)]2+. The spectroscopic properties of [{OLCuII}2(O22-)]2+ are compared and contrasted to those of the related and well established dicopper(II)-μ-1,2-(end-on)-peroxo complex [{PyLCuII}2(O22-)]2+ (Chart 1), that has also been structurally characterized.25-27
Chart 1.

As already suggested, the most striking element of [{OLCuII}2(O22-)]2+ chemistry is its efficient reactivity with exogenously added alkylbenzenes. Although aliphatic C-H bond oxidations of toluene or ethylbenzene have been observed in an Fe2-O2 or Fe(IV)-oxo system,28,29 to the best of our knowledge, this is the first time that such chemistry has been observed by starting with a dicopper(II)-μ-1,2-end-on-peroxide species. Other unobservable Cu2O2 adducts are considered as possible active species based on parallel experiments conducted on an analogous tridentate ligand system, BzPy1 (BzL; Chart 1). The O2-reaction of the copper(I) adduct [CuI(BzL)(CH3CN)]+ (BzLCuI) leads to a dicopper(III)-bis-μ-oxo species [{OLCuIII}2(μ-O2-)2]2+ that also effects C-H activation, i.e. the complex possesses electrophilic reactivity, Chart 1. Such reactivity for dicopper(III)-bis-μ-oxo species was expected based on analogous work utilizing sterically hindered tetradentate ligands that exhibit toluene oxidation chemistry.30 The possible involvement of a mononuclear copper(II)-superoxide species [(L)CuII-(O2·-)]+ is also considered.
Experimental Section
Materials and Methods
Unless otherwise stated all solvents and chemicals used were of commercially available analytical grade. Dioxygen was dried by passing it through a short column of supported P4010 (Aquasorb, Mallinkrodt). Propionitrile was distilled over CaH2 and acetone was distilled over Drierite. Diethyl ether, acetonitrile, toluene, THF, and methylene chloride were obtained by passing the solvents through a 60 cm long column of activated alumina under N2 (Innovative Technology, Inc.). Toluene was dried further by distillation over lumps of sodium under an argon atmosphere. Preparation and handling of air sensitive compounds were performed under an argon atmosphere using standard Schlenk techniques or in an MBraun Labmaster 130 inert atmosphere (< 1 ppm O2, < 1 ppm H2O) glovebox filled with nitrogen. Deoxygenation of solvents was effected by either repeated freeze/pump/thaw cycles or bubbling with argon for 45 - 60 minutes. Elemental analyses were performed by Quantitative Technologies, Inc. (QTI, Whitehouse, NJ) or Desert Analytics (Tucson, AZ). 1H NMR spectra were recorded at 400 MHz on a Bruker AMX-400 instrument. Chemical shifts were reported as δ values relative to an internal standard (Me4Si) and the residual solvent proton peak.
X-ray Crystallography
Crystals of each compound were placed in the N2 cold stream at 110 K of an O.D. Xcaliber3 system equipped with a graphite monochromator and an Enhance (Mo) X-ray Source (λ = 0.71073 Å) operated at 2 kW power (50 kV, 40 mA). The detector was placed at a distance of 50 mm from the crystal. The frames were integrated with the Oxford Diffraction CrysAlisRED software package. All structures were solved and refined using the Bruker SHELXTL (v6.1) software package. Analysis of the data showed negligible decay.
UV-visible Spectroscopy
Room-temperature UV-visible spectra were recorded with a Varian Cary-50 spectrophotometer. Low-temperature UV-visible spectra were recorded on a Hewlett-Packard Model 8453A diode array spectrophotometer equipped with a two-window quartz H.S. Martin Dewar filled with cold MeOH (25 °C to -85 °C) that was maintained and controlled by a Neslab VLT-95 low temperature circulator. Spectrophotometer cells (2 mm) were purchased from Quark Glass and equipped with a column and pressure/vacuum stopcock. The procedures are also described elsewhere.31 Reaction solutions were introduced to molecular oxygen via bubbling through an 18-gauge, 24- inch-long stainless steel syringe needle.
Resonance Raman (rR) Spectroscopy
rR spectra were obtained using a Princeton Instruments ST-135 back-illuminated CCD detector on a Spex 1877 CP triple monochromator with 1200, 1800, and 2400 grooves/mm holographic spectrograph gratings. Excitation was provided by Coherent 190C-K Kr+ and Innova Sabre 25/7 Ar+ CW ion lasers. Laser lines were chosen to coincide with the intense absorption transitions of [{OLCuII}2(O22-)]2+. The spectral resolution was < 2 cm-1. Samples were run at 77 K in a liquid N2 finger dewar (Wilmad). Isotopic substitution was achieved by oxygenating with 18O2 (Icon).
Electrochemistry
Cyclic voltammetry was carried out with a Bioanalytical Systems BAS-100B electrochemistry analyzer. The cell was a standard three-electrode system with platinum wire auxiliary as the counter electrode. A glassy carbon electrode (GCE, BAS MF 2012) was used as the working electrode. The reference electrode was Ag/Ag+. Measurements were performed at room temperature in DMF solvent containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAHP) and 1 - 0.1 mM copper complex. Ferrocene (1 mM) was added as the internal standard. Solutions were deoxygenated by directly bubbling with argon.
Electron Paramagnetic Resonance (EPR) spectroscopy
EPR spectra were recorded on a Bruker EMX spectrometer controlled with a Bruker ER 041 XG microwave bridge operating at X-band (~9.4 GHz). The low-temperature EPR measurements were conducted by using either a continuous-flow liquid-helium cryostat and ITC503 temperature controller made by Oxford Instruments, Inc., or a liquid-nitrogen finger dewar.
GC and GC-MS Analysis; Determination of Solvent Oxidation Product Yields
Analysis of the toluene and/or ethylbenzene oxidation products were confirmed by comparison to commercially available standards, specifically benzaldehyde and benzyl alcohol or acetophenone and 1-phenylethanol, respectively. The yields of the corresponding product signals were calculated based on comparisons to a calibration curve; externally added decane was used as an internal standard. 18O-incorporation was determined by RCuI oxygenation using 18O2.
All GC experiments were carried out and recorded using a Hewlett-Packard 5890 Series II Gas Chromatograph. The GC conditions for the analysis of the toluene oxidation products (benzaldehyde, benzyl alcohol) were: Injector Port Temperature: 250 °C; Detector Temperature 250 °C; Column Temperature: Initial Temperature 45 °C; Initial Time, 2min; Final Temperature 250 °C, Final Time, 2 min, Gradient Rate 30 °C/min; Flow Rate: 51 ml/min. The GC conditions for the analysis of the ethylbenzene oxidation products (acetophenone, 1-phenylethanol) were as follows: Injector Port Temperature: 250 °C; Detector Temperature 250 °C; Column Temperature: Initial Temperature 50 °C; Initial Time, 2min; Final Temperature 90 °C, Final Time, 1 min, Gradient Rate 1 °C/min; Flow Rate: 51 mL/min. All GC/MS experiments were carried out and recorded using a Shimadzu GC-17A/GCMS0QP5050 Gas Chromatograph/Mass Spectrometer. The GC-MS conditions for the product analysis following toluene and ethylbenzene oxidation were: Injector Port Temperature: 220 °C; Detector Temperature 280 °C; Column Temperature: Initial Temperature 80 °C; Initial Time, 3min; Final Temperature 250 °C, Final Time, 5 min, Gradient Rate 10 °C/min; Flow Rate: 16 mL/min; Ionization voltage: 1.5 kV.
Synthetic Procedures for Ligands and CuI/II complexes
The following compounds were synthesized following procedures previously described:25,27,32 [CuI(MeCN)4]B(C6F5)4, PyL, [CuI(PyL)(CH3CN)]B(C6F5)4 (PyLCuI), [CuII(PyL)(H2O)](ClO4)2 (PyLCuII), [{CuII(PyL)}2(O2)]2+ [{PyLCuII}2(O22-)]2+.
OL
In a 125 mL air-free addition funnel, 2-picolyl chloride (4.4 g, 34.6 mmol) in 50 mL of methanol containing triethylamine (4.2 g, 41.6 mmol) was deaerated with argon for 20 min. The solution was then added dropwise to a 250 mL 3-neck flask containing a portion of 5-tert-butyl-o-anisidine (2.7 g, 15.1 mmol) dissolved in 100 mL methanol. The reaction solution was stirred for 3 days under an argon atmosphere at ~65 °C. The crude product was purified via column chromatography utilizing silica gel. Elution with ethyl acetate removed the unreacted 2-picolyl chloride and elution with ethyl acetate/hexane (1:1) yielded the desired OL product (3.7 g, 68%) as a brown-yellow solid, (Rf = 0.25, silica gel, ethyl acetate/hexane = 1:1). 1H NMR (CDCl3, 300 MHz): δ 1.10 (s, 9H), 3.83 (S,3H), 4.53 (s, 4H), 6.77-6.90 (m, 3H), 7.06-7.10 (m, 2H), 7.48-7.59 (m, 4H), 8.48-8.51 (m, 2H). Elemental Analysis (C24H29N3O): Calculated; C, 76.76; H, 7.78; N, 11.19; Found; C, 76.20; H, 7.81; N, 11.39.
BzL
A portion of 2-picolyl chloride (1.79 g, 14.0 mmol) was dissolved in 50 mL dichloromethane. Upon stirring, 50 mL of saturated NaOH solution was added followed by dropwise addition of benzylamine (0.5 g, 4.67 mmol). After stirring for 4 days at room temperature under an argon atmosphere, the organic layer was separated and dried with MgSO4. The crude product was treated with 2 g phthalic anhydride in order to remove any primary or secondary amines. After four hours, the crude product was filtered and purified by column chromatography (silica gel). Elution with ethyl acetate removed the unreacted 2-picolyl chloride yielding the desired BzL product (1.1 g, 61 %) as a brown-yellow solid, (Rf = 0.25, silica gel, ethyl acetate). 1H NMR (CDCl3, 400 MHz): δ 3.69 (s, 2H), 3.81 (s, 4H), 7.11-7.68 (m, 11H), 8.51 (d, 2H). Elemental Analysis (C19H19N3): Calculated; C, 78.86; H, 14.52; N, 6.62. Found; C, 78.11; H, 14.46; N, 6.79.
[CuI(OL)(MeCN)]B(C6F5)4; OLCuI
Dissolution of OL (109 mg, 0.3 mmol) in 8 mL of O2-free diethyl ether followed by slow addition under an argon atmosphere to [CuI(MeCN)4]B(C6F5)4 (273 mg, 0.3 mmol) afforded a bright yellow solution. Precipitation by 100 ml O2-free pentane resulted in a yellow powder. The supernatant was decanted and the yellow powder was dissolved under vacuum. The dissolution and re-precipitation process in Et2O and pentane and vacuum drying process was repeated 3 to 5 times until a yellow microcrystalline solid was observed giving a final yield of 83 % (280 mg; 0.25 mmol). Single crystals suitable for X-ray structural analysis were obtained by recrystallization from Et2O/Pentane. 1H NMR (DMSO-d6, 300 MHz): δ 1.22 (s, 9H), 2.20 (s, 3H), 4.10 (s, br, 3H), 4.87 (s, br, 4H), 6.60-6.90 (br, 3H), 7.07 (br, 2H), 7.60-7.90 (m, 4H), 8.68 (br, 2H). Elemental Analysis (C49H30BCuF20N4O): C, 51.39; H, 2.64; N, 4.89. Found: C, 50.66; H, 2.60; N, 4.96.
[CuI(BzL)(MeCN)]B(C6F5)4; BzLCuI
Under an argon atmosphere, BzL (46 mg, 0.16 mmol) was dissolved in 8 mL of diethyl ether solution and slowly added to a 50 mL Schlenk flask containing [CuI(MeCN)4]B(C6F5)4 (144 mg, 0.16 mmol). Pentane (O2-free, 100 ml) was added to the bright yellow solution obtained affording a yellow solid. The supernatant was removed via cannula and the solid was dried under vacuum. Repeated (2-3 times) dissolution and re-precipitation followed by vacuum drying afforded a fluffy yellow solid, 76 % yield (131 mg; 0.25 mmol). Single crystals suitable for X-ray crystallographic analysis were obtained by recrystallization from Et2O/Pentane. 1H NMR (DMSO-d6, 300 MHz): δ 1.98 (s, 3H), 3.84 (s, 4H), 3.89 (s, 2H) 7.17 7.24 (m, 5H), 7.31 (br, 2H), 7.61 (br, 2H), 7.97 (br, 2H), 8.56 (br, 2H). Elemental Analysis (C45H22BCuF20N4): Calculated; C, 50.37; H, 2.07; N, 5.22. Found; C, 50.65; H, 1.89; N, 4.85.
[CuII(OL)(H2O)](ClO4)2·H2O; OLCuII
Under an argon atmosphere, a 10 mL EtOH solution of OL (0.159 g, 0.44 mmol) was added dropwise to a 10 mL aqueous solution of Cu(ClO4)2·6H2O (170 mg, 0.44 mmol). A blue crystalline solid was easily obtained by slow evaporation under vacuum (0.27 g, 78%). UV-vis: (methanol) 681 nm (80 M-1 cm-1); EPR (DMF/Toluene 1:1) g⊥ = 2.05, g∥ = 2.27, A∥ = 169 × 10-4 cm-1. Elemental Analysis (C23H31Cl2CuN3O11): Calculated; C, 41.86; H, 4.73; N, 6.37; Found: C, 42.27; H, 4.50; N, 6.24. Warning: While we have experienced no problems in working with perchlorate compounds, they are potentially explosive, and care must be taken not to work with large quantities.
[CuII(BzL)(MeOH)](ClO4)2; BzLCuII
A 15 mL EtOH solution was deoxygenated and added to a 50 mL Schlenk flask containing BzL (45 mg, 0.44 mmol) and Cu(ClO4)2·6H2O (58 mg, 0.44 mmol). The bright blue solution was slowly stirred under vacuum resulting in a blue powder. EPR (DMF/Toluene 1:1) g⊥ = 2.05, g∥ = 2.27, A∥ = 169 × 10-4 cm-1. Elemental Analysis (C20H22Cl2CuN3O9): Calculated; C, 41.21; H, 3.80; N, 7.21; Found; C, 40.88; H, 3.60; N, 7.30. Crystalline material suitable for X-ray structural analysis was obtained by repeating the slow vacuum drying process following dissolution of BzLCuII in EtOH.
[{CuII(OL)}2(O22-)][B(C6F5)4]2; [{OLCuII}2(O22-)]2+
Under a nitrogen atmosphere within the drybox, OLCuI (11 mg, 0.01 mmol) was dissolved in 10 ml O2-free solvent (CH2Cl2, toluene, ethylbenzene) giving a bright yellow solution. The reaction flask (or cuvette) was quickly removed from the dry-box and cooled to -80 °C in a dry ice/acetone bath and O2 was gently bubbled through the reaction solution. A purple solution of complex [{OLCuII}2(O22-)]2+ was formed in less than one minute. Excess dioxygen was removed by argon bubbling and the solvent oxidation products were analyzed as described above. Also, the copper(II) decay products in methylene chloride, OLCuII-Cl, and in toluene, [{OLCuII}2(μ-OH)2]2+, were characterized as given below.
[CuII(OL)(Cl-)]B(C6F5)4·CH2Cl2; OLCuII-Cl
OLCuI (46 mg, 0.04 mmol) was dissolved in 10 ml O2-free CH2Cl2 (under Ar) giving a bright yellow solution and this was cooled to -80 °C in a dry ice/acetone bath. Dioxygen was gently bubbled through the reaction solution resulting in a purple solution of [{OLCuII}2(O22-)]2+ within a few seconds. Excess dioxygen was removed from the reaction system by purging with argon for 2 to 3 minutes. Since [{OLCuII}2(O22-)]2+ was not stable at -80 °C the purple color as well as the characteristic absorption gradually bleached during the purging process. The resulting solution was then warmed to ambient temperature and the thermal decay product was kept under argon overnight resulting in a blue crystalline solid (22 mg, 45%). X-ray crystallographic analysis confirmed the identity of the cupric-chloride species, OLCuII-Cl. UV-vis: (Methanol) 718 nm (85 M-1 cm-1); EPR (DMF/Toluene 1:1) g⊥ = 2.05, g∥ = 2.26, A∥ = 167 × 10-4 cm-1. Elemental Analysis (C48H29BCl3CuF20N3O): C, 47.08; H, 2.39; N, 3.43. Found: C, 47.46; H, 2.16; N, 3.28.
[{CuII(OL)}2(HO-)2][B(C6F5)4]2·0.8(C7H8); [{OLCuII}2(μ-OH)2]2+
Within the drybox, OLCuI (46 mg, 0.04 mmol) was dissolved in 8 ml of O2-free toluene giving a bright yellow solution. The reaction solution was removed from the drybox and cooled to -80 °C in a dry ice/acetone bath. Dioxygen was gently bubbled through the reaction solution until full formation of [{OLCuII}2(O22-)]2+ (~1 min) was achieved. The solubility of [{OLCuII}2(O22-)]2+ at high concentrations is poor in non-polar solvents. As a result, some of the complex existed as a purple solid that precipitated at the bottom of the Schlenk flask. Remaining excess O2 was removed by quickly purging the solution with argon for 2 - 3 minutes. The resulting solution was then allowed to decay by warming to ambient temperature and the purple color, solution and solid, ceased to exist in ~10 second following removal from the cold bath. The resulting reaction mixture was filtered and the green precipitate subsequently obtained was characterized as [{OLCuII}2(μ-OH)2]2+ (74 % yield). Crystalline material suitable for X-ray structural analysis were obtained by allowing the dilute toluene supernatant to remain a few days under argon until crystals fell out of solution. Elemental Analysis: Calculated; C, 51.66; H, 2.72; N, 3.64; Found: C, 51.65; H, 2.64; N, 3.19.
[{CuIII(BzL)}2(O2-)2][B(C6F5)4]2; [{BzLCuII}2(μ-O2-)2]2+
Under a nitrogen atmosphere within the drybox, BzLCuI (25 mg, 0.024 mmol) was dissolved in 8 ml O2-free solvent (toluene, Et2O, THF, acetone) giving a bright yellow solution. The reaction flask (or cuvette) was removed from the drybox and cooled to -80 °C in a dry ice/acetone bath and O2 was gently bubbled through the reaction solution. A yellow-brown solution of complex [{BzLCuII}2(μ-O2-)2]2+ was fully formed in 5 to 30 minutes depending on the solvent. Excess dioxygen was removed by argon bubbling and the solvent oxidation products in toluene were analyzed as described above; corresponding product yields are reported within the text.
Ligand Analysis after [{OLCuII}2(O22-)]2+ Decomposition in CH2Cl2
A 10 equivalent excess of KCN (520 mg, 8 mmol) in aqueous solution (10 mL) was added to the pale green reaction solution that was produced following thermal transformation of [{OLCuII}2(O22-)]2+ (35 mg, 0.08 mmol) in CH2Cl2.33 A biphasic mixture resulted that consisted of the desired yellow organic product layer and the colorless aqueous solution layer. The products were passed through a plug of silica gel. The original, unreacted OL ligand was recovered (27 mg, 93%). 1H-NMR spectroscopy also supported that the ligand remained intact.
Isotope Effect Determination for the Toluene Oxygenation Reaction
Under an argon atmosphere OLCuI (23 mg, 0.02 mmol) was dissolved in an O2-free C6H5CH3 (860 mg, 9.33 mmol)/C6D5CD3 (918 mg, 9.2 mmol) solvent mixture. The yellow solution was then cooled to -80 °C in a dry ice/acetone bath and O2 was gently bubbled through the reaction solution (~1 min) to allow full formation of the purple colored [{OLCuII}2(O22-)]2+. Any remaining O2 was removed by purging argon through the solution and the resulting suspension was allowed to decay by warming to ambient temperature. The reaction mixture was then filtered and the filtrate was analyzed by GCMS. The isotope effect for the formation of benzaldehyde was calculated by comparison of the signal intensity ratios of C6H5CHO and C6D5CDO obtained by mass spectrometric analysis. Under the experimental conditions employed, product analysis thus gives an `apparent' isotope effect kH/kD = 7.5 ± 1.
Results and Discussion
Syntheses of Ligands, and Copper Complexes
The potential tetradentate N3O ligand OL and the purely N3 tridentate ligand BzL were straightforwardly synthesized by addition of commercially available 2-picolyl chloride to the appropriate precursors, 5-tert-butyl-o-anisidine and benzylamine, respectively. Following column purification, metallation of the ligands proceeded by dissolution of [CuI(CH3CN)]B(C6F5)4 and the respective ligand in deoxygenated diethyl ether (Et2O) and subsequent precipitation in pentane. Repeated dissolution and precipitation afforded yellow crystalline material that was characterized by 1H-NMR spectroscopy and C-, H-, N- elemental analysis as [CuI(OL)(CH3CN)]B(C6F5)4 (OLCuI) and [CuI(BzL)(CH3CN)]B(C6F5)4 (BzLCuI). Slow diffusion of 20 equivalents by volume of pentane over an Et2O solution of the ligand-copper(I)-complexes leads to products amenable to X-ray crystallographic analysis as will be discussed in the following section. See the Experimental Section for more details.
OL as well as an unmethylated phenol analogue were originally designed and synthesized to study their relationship to a binucleating phenol analogue referred to as PD'OH.21,34 The dicopper(I) complex [CuI2(PD'OH)(MeCN)2]2+ reacts with O2 to give a hydroperoxide species which effects RCN oxidation (to an aldehyde and cyanide);21 the dicopper(II) complex [CuII2(PD'O-)(H2O)2]3+ can be made to mediate specific nucleobase guanine oxidation for certain DNA duplex constructs.10,35 As stated in the Introduction, OL is utilized in this study to examine the ability of the N3O chelate to elucidate C-H bond activation, i.e. toluene and ethylbenzene oxidation, following formation of the Cu2O2 species; more details follow. The reactivity characteristics are compared to copper-complex systems utilizing an N3 (BzL) and N4 (TMPA; PyL) ligand, see Chart 1. For the purpose of structural and electrochemical investigations, the copper(II) complexes, [CuII(OL)(H2O)](ClO4)2 (OLCuII) and [CuII(BzL)(ROH)](ClO4)2 (BzLCuII; ROH is EtOH or MeOH), were prepared. The ligand-copper(II)-complexes were synthesized by the slow addition of aqueous Cu(ClO4)2•6H2O to the appropriate ligand in protic solvents (EtOH, MeOH), see the Experimental Section.
X-ray Crystallographic Analysis of the Copper(I) and Copper(II) Complexes
A summary of numerical crystal and refinement data for OLCuI, BzLCuI, and BzLCuII is given in Table 1, with full details given in the Supporting Information. Representative ORTEP diagrams of the copper(I) complexes, OLCuI and BzLCuI, are shown in Figures 1 and 2, respectively, and the ORTEP diagram of BzLCuII is shown in Figure 3; selected bond distances and angles given in the Figure captions. The coordination spheres of all three complexes consists of the three nitrogen atoms of the bis(2-pycolyl)amine (PY1) moiety of the ligands and an exogenous solvent molecule occupies the fourth position. Tetra-coordination is achieved via ligation from an acetonitrile molecule for the copper(I) complex and a neutral EtOH molecule for the cupric species.
Table 1.
Numerical Crystal and Refinement Data for the X-ray Crystal Structures of BzLCuI, OLCuI, BzLCuII
| Complex | BzLCuI | OLCuI | BzLCuII |
|---|---|---|---|
| formula (sum) | C48.5 H30 B Cu F20 N4 | C49 H30 B Cu F20 N4 O | C20.24H23.48 Cl2 Cu N3 O9 |
| fw | 1123.11 | 1145.12 | 587.22 |
| crystal system | triclinic | triclinic | monoclinic |
| space group | P1- | P1- | P21/n |
| a (Å) | 13.023(3) | 8.0704(10) | 9.1046(5) |
| b (Å) | 13.409(4) | 17.337(3) | 30.057(7) |
| c (Å) | 15.048(3) | 17.499(2) | 9.6380(5) |
| α (deg) | 112.14(3) | 107.807(12) | 90 |
| β (deg) | 104.071(19) | 90.416(10) | 91.630(4) |
| γ (deg) | 97.69(2) | 91.680(11) | 90 |
| V(Å3) | 2285.5(12) | 2329.8(6) | 2636.4(7) |
| Z | 2 | 2 | 4 |
| μ/mm-1 (Mo Kα) | 0.600 | 0.593 | 1.082 |
| reflections collected (total) | 19 421 | 27 377 | 37 457 |
| Rint (no. of equiv. reflections) | 7713 | 11 504 | 6088 |
| observed reflections [I/s(I) > 2] | 4168 | 7952 | 4353 |
| Final R, Rw [I/σ(I) > 2] | 0.0612, 0.1076 | 0.0412, 0.1195 | 0.0565, 0.1433 |
Figure 1.

Representative diagram showing the cationic portion of [CuI(OL)(CH3CN)]B(C6F5)4, OLCuI; hydrogen atoms and the B(C6F5)4 counter-anion has been omitted for clarity. Selected bond distances are: Cu1-N1, 2.010(2) Å; Cu1-N2, 2.286(3) Å; Cu1-N3, 2.063(1) Å; Cu1-N4, 1.911(2) Å; Cu1...O1, 5.072(5) Å. Selected bond angles are: N1-Cu1-N4, 126.79(8)°; N1-Cu1-N2, 79.32(7)°; N1-Cu1-N3, 115.38(8)°; N2-Cu1-N3, 79.68(7)°; N2-Cu1-N4, 129.57(7)°; N3-Cu1-N4, 113.52(8)°.
Figure 2.

Representative diagram showing the cationic portion of [CuI(BzL)(CH3CN)]B(C6F5)4, BzLCuI; the B(C6F5)4 counteranion has been omitted for clarity. Selected bond distances are: Cu1-N1, 1.990(4) Å; Cu1-N2, 2.309(4) Å; Cu1-N3, 2.042(4) Å; Cu1-N4, 1.900(4) Å. Selected bond angles are: N1-Cu1-N4, 123.83(2)°; N1-Cu1-N2, 79.24(1)°; N1-Cu1-N3, 123.77(1)°; N2-Cu1-N3, 79.63(1)°; N2-Cu1-N4, 133.31(2)°; N3-Cu1-N4, 108.39(2)°.
Figure 3.

Representative diagram showing the cationic portion of [CuII(BzL)(EtOH)](ClO4)2, BzLCuII the two ClO4- counteranions has been omitted for clarity. Selected bond distances are: Cu-N1, 1.979(3) Å; Cu-N2, 2.035(3) Å; Cu-N3, 1.988(3) Å; Cu-O, 1.965(3) Å. Selected bond angles are: N1-Cu-N3, 165.74(1)°; N1-Cu-N2, 82.85(1)°; N2-Cu-N3, 83.09(1)°; N1-Cu-O, 96.33(1)°; N2-Cu-O, 172.94(1)°; N3-Cu-O, 97.34(1)°.
As shown in Figure 1, the overall geometry of OLCuI is highly distorted from tetrahedral. The copper ion is nearly co-planar with the Nacetonitrile and two NPy atoms, sitting slightly above the triangular plane defined by these three N-atoms in the direction of the bridgehead nitrogen. The interaction between the copper(I) ion and the central amine nitrogen atom is the weakest, as indicated by the longer bond length of 2.286(3) Å versus an average Cu-NPy bond distance of 2.036(6) Å. The most noteworthy feature of the OLCuI structure is that exogenous MeCN coordination is preferred over the OL ether oxygen atom, thus the tethered anisole moiety is rotated away from the copper(I) ion, placing the O-atom out of bonding distance at 5.072(5) Å. Of all the ligand donors, the exogenous MeCN molecule is coordinated the strongest to the copper(I) ion of OLCuI as indicated by the bond length of 1.911(2) Å.
The complex PyLCuI and close analogue [CuI(PyL')(MeCN)]+ possess pseudo penta-coordination, with a distorted trigonal bipyramidal geometry; one of the three pyridyl groups of the yL' ligand has a 3-C(O)OCH3 substituent.36,37 The interaction between the copper ion and the bridgehead nitrogen is the weakest as evidenced by the ~ 2.4 Å Cu-Nbridgehead bond length. The four-coordinate structure of OLCuI is quite different, of course due to the dangling anisole group. In fact previous work showed that for the adducts [CuI(PyL)(D)]+ (D = CO or PPh3), one of the pyridine arms dissociates as supported by solution 1H-NMR data and an X-ray crystal structure of [CuI(PyL)(PPh3)]+.36 The copper ion of OLCuI may adopt a similar coordination in solution, as demonstrated by copper(I)-carbon monoxide binding studies which are discussed in a later section.
The overall coordination environment of BzLCuI is analogous to that in OLCuI, yet further distorted from a tetrahedral geometry, see Figure 2. The Cu1-N2 bond distance (2.309(4) Å) in BzLCuI, N2 being the central amine nitrogen atom, is longer than the corresponding value in OLCuI. The average Cu-NPy bond distance is shorter, 2.016(4) Å, along with the Cu-Nacetonitrile bond distance of 1.900(4) Å. The benzyl moiety is positioned somewhat (by comparison) towards or over the copper(I) ion.
By contrast, as shown in the ORTEP diagram for BzLCuII (Figure 3), the benzyl arm is swung away from the copper(II) ion. The Cu-Namine bond distance for the cupric species is 2.035(3) Å, approximately 0.27 Å shorter than in the copper(I) structure (vide supra). The average Cu-NPy bond distance is about the same, 1.983(8) Å, only about 0.03 Å shorter. The copper(II) ion is very close to the plane which includes the ethanol O-atom and other nitrogen moieties, making for an overall square planar complex.
Itoh and coworkers have investigated how cuprous ions interact with aromatic π-systems of pyridylalkylamine tridentate ligands with a phenylethylene appendage.38,39 The copper(I)-arene π-interactions of η2-type were found to alter or diminish the reactivity of the ligand-copper(I) species toward dioxygen. No copper-arene interaction is exhibited in the solid-state X-ray structure of BzLCuI or BzCuII because the benzylamine linker is shorter than the aforementioned Itoh species.
Structural Characterization of the Reaction Products following CH2Cl2 and Toluene Oxidation by [{CuII(OL)}2(O22-)]2+
ORTEP diagrams of [CuII(OL)(Cl)]+ and [{OLCuII}2(μ-OH)2]2+ are given in Figures 4 and 5 respectively, with selected bond distances and angles given in the Figure captions. The complexes were obtained as reaction products following the thermal transformation of the dicopper(II)-μ-1,2-(end-on)-peroxo species [{CuII(OL)}2(O22-]2+ that forms upon oxygenation of OLCuI in CH2Cl2 or toluene appropriately; further details are given below. A summary of crystal and refinement data for the solvent oxidation reaction products are given in Table 2 with full details given in the Supporting Information.
Figure 4.

ORTEP diagram showing views of the cationic portion of [CuII(OL)(Cl)]+. Selected bond distances are: Cu-O1, 2.317(5) Å; Cu-Cl1, 2.242(6) Å; Cu1-N1, 1.984(0) Å; Cu1-N2, 2.073(6) Å; Cu1-N3, 1.996(3) Å. Selected bond angles are: N1-Cu-N3, 165.70(8)°; N1-Cu-N2, 82.65(7)°; N3-Cu-N2, 83.61(8)°; N1-Cu-Cl, 96.92(6)°; N3-Cu-Cl, 96.87(6)°; N2-Cu-Cl, 179.01(6)°; N1-Cu-O, 96.76(7)°; N3-Cu-O, 84.26(7)°; N2-Cu-O, 78.22(6)°; Cl-Cu-O, 100.95(4)°.
Figure 5.

ORTEP diagrams showing views of the cationic portion of [{OLCuII}2(m-OH)2]2+; hydrogen atoms (except for HO-) and the B(C6F5)4 counteranion have been omitted for clarity. Selected bond distances are: Cu1-N1, 2.245(8) Å; Cu1-N2, 2.123(3) Å; Cu1-N3, 2.002(9) Å; Cu1-O1, 2.369(2) Å; Cu1-O2, 1.954(5) Å; Cu1-O2A, 1.944(1) Å; Cu1...Cu1A, 2.889(6) Å. Selected bond angles are: O1-Cu-N1, 152.66(3)°; O1-Cu-N2, 75.23(7)°; O1-Cu-N3, 80.42(2)°; O1-Cu-O2, 86.02(3)°; N1-Cu-N3, 96.23(8)°; O2-Cu-N1, 95.93(3)°; O2-Cu-N2, 93.47(5)°; O2-Cu-N3, 166.44(2)°; N1-Cu-N2, 77.42(7)°; N3-Cu-N2, 83.37(9)°.
Table 2.
Numerical Crystal and Refinement Data for the X-ray Crystal Structures of Solvent Oxidation Products.
| Complex | OLCuII-Cl | [{OLCuII}2(μ-OH)2]2+ | [{OLCuII}4(CO3)2]4+ |
|---|---|---|---|
| formula (sum) | C47.47 H27.94 Cl1.94 Cu F20 N3 O | C94 H56 B2 Cu2 F40 N6 O4 | C195.6 H122 B4 CU4 F80 N12 O11.4 |
| fw | 1179.43 | 1179.43 | 4640.05 |
| crystal system | Triclinic | Triclinic | Triclinic |
| space group | P1- | P1- | P1- |
| a (Å) | 12.1990(5) | 12.330(2) | 17.232(3) |
| b (Å) | 12.4156(5) | 12.7247(17) | 18.005(6) |
| c (Å) | 16.1018(7) | 14.7999(18) | 18.767(6) |
| α (deg) | 73.056(4) | 74.404(11) | 115.41(3) |
| β (deg) | 82.456(4) | 84.914(12) | 109.99(2) |
| γ (deg) | 85.222(4) | 80.369(12) | 94.716(19) |
| V (Å3) | 2310.08(17) | 2202.7(6) | 4759(2) |
| Z | 2 | 1 | 1 |
| μ/mm-1 (Mo Kα) | 0.77167 | 0.626 | 0.585 |
| reflections collected (total) | 43 084 | 22 767 | 40 552 |
| Rint (no. of equiv. reflections) | 11 737 | 10 667 | 16 536 |
| observed reflections [I/s(I) > 2] | 9297 | 7840 | 5665 |
| Final R, Rw [I/σ(I) > 21 | 0.0500, 0.1173 | 0.0375, 0.1050 | 0.0883, 0.2072 |
In contrast to the four-coordinate structure of OLCuI but common in copper(I) ion chemistry, the copper atom in [CuII(OL)(Cl)]+ is pentacoordinate, see Figure 4. The structure of [CuII(OL)(Cl)]+ is distorted square pyramidal where τ = 0.22, the latter value is based on a method defined by Reedijk and Addison for structural comparison of five-coordinate copper(II) species; τ = 0.0 for a square pyramidal geometry and τ = 1.0 for a trigonal bipyramidal geometry.40,41 The three nitrogen atoms of the PY1 unit and the ligated Cl- constitute the basal plane with the anisole oxygen atom Oanisole occupying the axial position. The average bond distance between the copper(II) center and the nitrogen atoms are 2.017(9) Å. The distances for Cu-Cl and Cu-Oanisole are longer, 2.242(6) Å and 2.317(5) Å, respectively, indicating a weak interaction between the copper(II) ion and the ether-oxygen ligand. The two pyridyl moieties from the PY1 unit of [CuII(OL)(Cl)]+ are nearly coplanar (Figure 4); the bridgehead nitrogen sits by ~ 0.5 Å above the best least-squares plane defined by N1, N2, N3, Cl and Cu, towards the anisole moiety. The chloride atom sits ~ 0.62 Å below the plane.
The Oanisole is also coordinated to the copper(II) ion of the toluene oxidation dicopperdihydroxy reaction product, [{OLCuII}2(μ-OH)2]2+, see Figure 5. The Cu-Oanisole bond distance is 2.369(2) Å, approximately 0.05 Å longer than that in the cupric-chloride species. The average Cu-Npy bond distance and the Cu-Namine bond distance of approximately 2.123 Å are all about the same, yet somewhat longer then in the cupric chloride species. The increased bond distance is expected because [{OLCuII}2(μ-OH)2]2+ possesses hexacoordinate copper(II), unlike that in [CuII(OL)(Cl)]+ (vide supra).
If the dicopper-dihydroxy species is stored for over one-week then the binuclear structure falls apart, likely forming the mononuclear copper(II)-hydroxy species [{OLCuII}(OH)]2+, as indicated by a change in the EPR signal from silent to an intensity of ~100 times greater (but still ~15 times smaller than OLCuII, see Experimental Section for values); further characterization was not carried out. If a toluene solution of [{OLCuII}2(μ-OH)2]2+ was opened to air then after a few days the green color changed to blue, and a tetranuclear carbonate species [{OLCuII}4(CO3)2]4+, formed. See Table 2 and the Supporting Information for details of the X-ray structure.
CO stretching Frequences for the Ligand-Copper(I)-Carbonyl Complexes
Copper(I)-CO complexes were obtained by dissolution of the corresponding copper(I) species in CO saturated diethyl ether and the products could be isolated as solids by addition of CO saturated pentane. Evidence for CO binding was confirmed by observation of a single IR-band (Table 3) within the expected range for synthetic copper(I)-carbonyl complexes (νCO: 2045 - 2110 cm-1).
Table 3.
Carbonyl stretching frequencies for the ligand-copper(I)-carbonyl complexes.
| Compound | Nujol (cm-1) | THF (cm-1) | CH3CN (cm-1) |
|---|---|---|---|
| [CuI(PyL)(CO)]+ | 2077 | 2090 | 2092 |
| [CuI(OL)(CO)]+ | 2093 | 2093 | 2095 |
| [CuI(BzL)(CO)]+ | 2093 | 2095 | 2094 |
The vCO values for ligand-copper(I)-carbonyl species are often used to deduce the complex coordination number as also related to the electron-donating ability of the corresponding ligand.46-49 It has been previously established for [CuI(PyL)(CO)]+ that the solid-state geometry is five-coordinate, however the predominate species in solution is four-coordinate, i.e. one pyridyl arm is “dangling” or de-ligated as shown in Chart 2.50 The CO vibrational frequency (νCO) of the solid-state five-coordinate species (thus with all N donors bound) is 2077 cm-1 and a shift to higher energy is observed in solution: νCO = 2090 cm-1 in THF and νCO = 2092 cm-1 in CH3CN.51 Dissociation of a pyridyl nitrogen-donor so only three N-donors are metal bound is consistent with weaker π-backbonding resulting in a stronger C-O bond and thus a higher νCO value. A solution-state structure similar to [CuI(PyL)(PPh3)+] is expected for [CuI(PyL)(CO)]+ as has also been exhibited by [CuI(PyLNMe2)(CO)]+; the PyLNMe2 ligand has NMe2-substituents at the para-position of all three pyridyl donors.25,51
Chart 2.

The CO stretching frequency of [CuI(OL)(CO)]+ and [CuI(BzL)(CO)]+ were investigated in CH3CN and THF, as having an average νCO value of 2094 cm-1, essentially the same as that found for the solid-state complex with νCO = 2093 cm-1. The similar CO stretching frequencies for [CuI(OL)(CO)]+, [CuI(BzL)(CO)]+ and the solution-state form of [CuI(PyL)(CO)]+ demonstrates that the Oanisole of ligand OL does not coordinate to copper(I) in [CuI(OL)(CO)]+. Similarly, the Oanisole likely does not coordinate in the solution-state form of OLCuI, consistent with the solid-state X-ray crystal structure, see Chart 2.
Electrochemical Investigation of the Ligand-Copper(II)-Complexes, LCuII
The variable dynamics of the tripodal ligand-copper systems, i.e., the fact that anisole `arm' of OL does not bind copper(I) in solution (Chart 2), precludes the use of the cuprous-carbonyl adducts in electrochemical studies for the purpose of delineating the difference in overall electron-donating ability of PyL vs. OL vs BzL. To obtain such insights, the RLCuII complexes were examined through electrochemical analyses employing cyclic voltammetry. The redox potentials of [CuII(PyL)(H2O)](ClO4)2 (PyLCuII), [CuII(OL)(H2O)](ClO4)2, (OLCuII), and [CuII(ZL)(EtOH)](ClO4)2 (BzLCuII) were measured in DMF solvent versus ferrocene/ferrocenium (Fc/Fc+), which was added as an internal standard. Both PyLCuII and OLCuII exhibited reversible CuII/I redox processes (ipc/ipa ≈ 1) with E1/2 values of -0.62 V and -0.51 V respectively, see Figure 6. However, BzLCuII exhibited irreversible behavior (data not shown) indicating that the anisole oxygen atom of OL does indeed affect the redox processes of OLCuII.
Figure 6.

Cyclic voltammograms of OLCuII (A) and PyLCuII in DMF with 0.1 M tetrabutylammonium hexafluorophosphate as supporting electrolyte. Ferrocene was added as an internal standard; the Fc/Fc+ redox waves are denoted by *.
Copper complex redox potentials are influenced by many factors, including the type and number of donor atoms as well as the coordination geometry.52-54 The copper(II) coordination geometry of OLCuII and PyLCuII likely contributes to the ΔE1/2 = 0.11 redox potential difference, assuming that the coordination environments of LCuII are analogous to their corresponding cupricchloride species. [CuII(OL)(Cl)]+ (τ = 0.23) adopts a slightly distorted square pyramidal geometry, see above, while [CuII(tmpa)(Cl)]+ exhibits a nearly perfect trigonal bipyramidal coordination (τ = 1.00).55 However, the primary effect is related to the difference in electron-donating ability of the Oether versus Npyridyl ligands within the two complexes.54 The higher (more positive) redox potential for OLCuII suggests that the Oether is a poor electron donor relative to the Npyridyl donor in PyLCuII.
UV-visible Spectroscopy in CH2Cl2; Generation of [{OLCuII}2(O22-)]2+
-Addition of O2 to OLCuI at -80 °C in non-coordinating and non-ether containing solvents such as CH2Cl2 and toluene generates an intense purple species formulated as a dicopper(II)-μ-1,2-(end-on)-peroxo species [{OLCuII}2(O22-)]2+. In methylene chloride, the LMCT absorption maxima (Figure 7) and `apparent' absorptivities (ε) were as follows: 415 nm (sh, 1100 M-1 cm-1), 508 nm (2000 M-1 cm-) and 630 nm (1250 M-1 cm-1). The ε values were calculated after about one to two minutes following introduction of O2 and they are based on the maximum absorbance values observed at 508 nm under the conditions of UV-visible bench-top oxygenation. [{OLCuII}2(O22-)]2+ begins decaying immediately upon reaching its maximum absorbance and has fully decayed after five minutes.
Figure 7.

UV-vis absorptions of [{OLCuII}2(O22-)]2+ (blue) at -80 °C in CH2Cl2. Addition of CO results in the quick formation of a new absorbance at 410 nm (red) before complete formation of the copper(I)-carbonyl adduct (green).
The absorptions of [{OLCuII}2(O22-)]2+ mimic in pattern and relative intensity the very distinctive UV-visible spectrum of the dicopper(II)-μ-1,2-(end-on)-peroxo species [{PyLCuII}2(O22-)]2+, which exhibits the following characteristics: λmax = 440 nm (sh, ε = 4000 M-1 cm-1), 525 nm (111500 M-1 cm-1) and 590 nm (sh, 7600 M-1 cm-1). Thus, by analogy, we assign the 415 and 630 nm absorptions as the triplet (O22-)π*ν → CuII(dz2) and singlet (O22-)π*σ → CuII(dz2) transitions, respectively, and the relatively intense 508 nm band as the (O22-)π*σ → CuII(dz2) transition.26 The absorptions of [{OLCuII}2(O22-)]2+ are of relatively low intensity in comparison to [{PyLCuII}2(O22-)]2+, likely because the dicopper(II)-dioxygen adduct does not fully form and/or that the Cu2O2 species quickly decays due to solvent oxidation.
The generation of [{OLCuII}2(O22-)]2+ following oxygenation of OLCuI indicates that the anisole-ether-oxygen atom is bound to the copper(II) ion. By analogy to [{PyLCuII}2(O22-)]2+, which has been structurally characterized by X-ray crystallography (Chart 1), formation of a dicopper(II)-end-on-peroxide coordination mode requires the ligand to bind to the copper(II) ion in an η4-tetradentate fashion (Scheme 1). As observed for other cases, tridentate ligation always leads to the formation of a dicopper(II)-η2-η2-side-on-peroxo or dicopper(III)-bis-μ-oxo complex, which have distinctly different UV-visible and resonance Raman (rR) spectroscopic characteristics.2,4,56 Further supporting that the Oanisole coordinates to the copper(II) ion within [{OLCuII}2(O22-)]2+ is that the X-ray crystal structure of [CuII(OL)(Cl)]+ indicates that the anisole-ether-oxygen can well bind to a copper(II) center, see above.
Scheme 1.

UV-visible Spectroscopy in Toluene; Generation of [{OLCuII}2(O22-)]2+ and [{BzLCuIII}2(μ-O2-)2]2+
Generation of [{OLCuII}2(O22-)]2+ in toluene (-80 °C) reveals LMCT absorption bands and ε values at 513 nm (8300 M-1 cm-1), 628 nm (5200 M-1 cm-1). The apparent molar extinction coefficients observed are much higher than those calculated in CH2Cl2, yet smaller than the molar absorptivities exhibited by [{PyLCuII}2(O22-)]2+26,36. As will be discussed in a later section, toluene oxidation also occurs during the thermal decay process of [{OLCuII}2(O22-)]2+, possibly accounting for the low ε values. For example, approximately 50 % of the decay process has occurred after two hours, whereas [{PyLCuII}2(O22-)]2+ is stable for weeks at -80 °C. In addition, the absorption bands of [{OLCuII}2(O22-)]2+ should be more intense than for [{PyLCuII}2(O22-)]2+ due to the poorer ligand donor ability of OL versus PyL. Generally, a poor ligand donor results in better orbital overlap between the peroxide and copper(II), which translates into greater charge-transfer band intensity.57
The low ε values might otherwise arise as a result of the likely small differences in coordination geometry exhibited by the copper(II) centers of [{OLCuII}2(O22-)]2+ and [{PyLCuII}2(O22-)]2+. X-ray structural characterization of the latter indicates that the dicopper(II)-end-on-peroxo species adopts a trigonal bipyramidal geometry (τ = 0.86), consistent with the structure of [CuII(PyL)(Cl)]+ (τ = 1.00). Assuming the same relationship exists between [{OLCuII}2(O22-)]2+ and the corresponding cupric-chloride analogue [CuII(OL)(Cl)]+ (τ = 0.23), the t value would suggest that [{OLCuII}2(O22-)]2+ adopts a square pyramidal geometry. In the trigonal bipyramidal coordination geometry (and dz2 ground-state), the (O22-)π* → CuII(dz2) orbital overlap is optimized. Although we lack X-ray structural insight for [{OLCuII}2(O22-)]2+ to fully support this assessment, if the copper(II) centers maintain a distorted square pyramidal geometry and dx2-y2 ground state, this could decrease the overlap of the O22- and CuII orbitals. Since epsilon intensity correlates to orbital overlap, the coordination geometry exhibited by [{OLCuII}2(O22-)]2+ may, in turn, lower the inherent intensity of its LMCT bands.
A reaction of OLCuI with O2 does not occur in nitrile solvents (EtCN, MeCN), consistent with the known affinity of nitriles to suppress or compete with O2 binding to copper(I) ions.46,58 Also, based on UV-visible spectroscopic criterion, a stable Cu2O2 species does not form in ether-containing solvents such as Et2O and THF, nor in solvents containing an oxygen-heteroatom such as acetone. Since these solvents all possess a potential oxygen donor ligand, they most likely compete with the OMe group in OL for binding to the copper(II) ion. As a result, the `end-on' CuII-O-O-CuII peroxide structure may no longer be stable due to the lack of full tetradentate chelate ligand coordination.
Dioxygen Reactivity of BzLCuI
Reaction of BzLCuI with O2 at -80 °C leads to the formation of a dicopper(III)-bis-μ-oxo species [{BzLCuIII}2(μ-O2-)2]2+ (see diagram below) in a number of solvents examined (toluene, acetone, and Et2O) (diagram) but not in the nitriles MeCN or EtCN.  The λmax absorption feature attributed to a σ* → dxy transition ranged in value between 385 nm to 392 nm within the various solvents and the molar absorptivities ranged between 6500 and 9500 M-1 cm-1, see Table 4. Dicopper(III)-bis-μ-oxo species typically exhibit more intense ε values due to the significant CuIII-O covalency that follows reduction of the peroxide ion and cleavage of the O-O bond. Since BzLCuI possesses the same PY1 moiety as OLCuI, similar competitive reactions with solvent (i.e., oxidation) may result upon addition of O2 to the reaction solution. In dry toluene, [{BzLCuIII}2(μ-O2-)2]2+ slowly forms, taking almost 30 minutes to reach λmax (385 nm; 7800 M-1 cm-1) but is then fully stable for up to three hours, see Figure 8.59
The λmax absorption feature attributed to a σ* → dxy transition ranged in value between 385 nm to 392 nm within the various solvents and the molar absorptivities ranged between 6500 and 9500 M-1 cm-1, see Table 4. Dicopper(III)-bis-μ-oxo species typically exhibit more intense ε values due to the significant CuIII-O covalency that follows reduction of the peroxide ion and cleavage of the O-O bond. Since BzLCuI possesses the same PY1 moiety as OLCuI, similar competitive reactions with solvent (i.e., oxidation) may result upon addition of O2 to the reaction solution. In dry toluene, [{BzLCuIII}2(μ-O2-)2]2+ slowly forms, taking almost 30 minutes to reach λmax (385 nm; 7800 M-1 cm-1) but is then fully stable for up to three hours, see Figure 8.59
Table 4.
UV-visible and Resonance Raman data for the Dicopper-Dioxygen (Cu2O2) Complexes.a
| Copper-Dioxygen Adducts | Solvent; Temp. | UV-visible Data | rR data; cm-1 (Δ18O2) | |
|---|---|---|---|---|
| LMCT; nm (M-1 cm-1) | νCu-O | νO-O | ||
| Dicopper(II) μ-1,2 Peroxo Species | ||||
| [{PyLCuII}2O22-)]2+ | THF; -80°C | 525 (11500) 590 (7600) | 561 (-26) | 831 (-44) |
| [{OLCuII}2O22-)]2+ | CH2Cl2; -80°C | 508 (2000) 630 (1250) | 571 (-26) | 837 (-45) |
| [{OLCuII}2O22-)]2+ | Toluene; -80°C | 513 (8300) 628 (5200) | ||
| Dicopper(III) Bis-μ-oxo Speciesa | ||||
| [{BzLCuIII}2O2-)2]2+ | Toluene; -80°C | 385 (7800) | 595 (-30) | |
| [{BzLCuIII}2O2-)2]2+ | THF; -80°C | 390 (8000) | ||
| [{BzLCuIII}2O2-)2]2+ | Acetone; -80°C | 392 (9500) | ||
| [{BzLCuIII}2O2-)2]2+ | Et2O; -80°C | 390 (6500) | ||
Dicopper(III)-bis-μ-oxo species do not possess O-O bonds, therefore such rR values are not listed.
Figure 8.

UV-visible absorption of [{BzLCuIII}2(μ-O2-)2]2+ (purple) at -80 °C in toluene following oxygenation of BzLCuI (blue).
Resonance Raman Spectroscopy of [{OLCuII}2(O22-)]2+ and [{BzLCuIII}2(μ-O2-)2]2+
In CH2Cl2 solvent, an O-O stretch at νO-O = 837 cm-1 was successfully observed in the rR spectrum for [{OLCuII}2(O22-)]2+; this downshifts by 45 cm-1 to 792 cm-1 upon 18O2 substitution (Figure 9). This O-O stretch usually falls in between 820 and 840 cm-1 region known for dicopper(II)-end-on-peroxo complexes; dicopper(II)-μ-η2:η2-side-on-peroxo species exhibit much lower stretching frequencies, typically in the 730 to 760 cm-1 range or lower, due to an additional back-bonding interaction.4,26,56 In addition, a Cu-O stretch was observed for [{OLCuII}2(O22-)]2+ at νCu-O = 571 cm-1, which shifted to 545 cm-1 upon 18O2 isotopic substitution. This finding also falls in line with the assignment of [{OLCuII}2(O22-)]2+ as a dicopper(II)-end-on-peroxo species, as such Cu-O stretches have not been observed in dicopper(II)-side-on-peroxo species.4 Therefore, the rR spectrum of [{OLCuII}2(O22-)]2+ is consistent with the observed UV-visible absorption features; together they clearly demonstrate that the copper-dioxygen adduct [{OLCuII}2(O22-)]2+ is a dicopper(II)-μ-1,2-end-on-peroxide species, with a dicopper-dioxygen coordination mode similar to that in [{PyLCuII}2(O22-)]2+ (Chart 1).
Figure 9.

rRaman spectrum of [{OLCuII}2(O22-)]2+ {B(C6F5)4- counterion} with 16O2 (red) and 18O2 (blue) isotopic substitution. The samples were run at 77 K in CH2Cl2 with 620 nm excitation. Solvent peaks denoted by *.
The νO-O and νCu-O stretches exhibited by [{OLCuII}2(O22-)]2+ are both centered at higher frequencies (6 and 10 cm-1, respectively) than those of [{PyLCuII}2(O22-)]2+, which has values at 831 and 561 cm-1, respectively.26 The rather small changes may be simply due to environmental effects, but could also be explained by the ligand donor atom change from NPy to Oanisole.60
In toluene solvent and with λex = 407 nm excitation, [{BzLCuIII}2(μ-O2-)2]2+ exibits a Cu=O stretch at νCu-O = 595 cm-1 in the rR spectrum that downshifted by 30 cm-1 to 565 cm-1 upon 18O2 substitution (Figure 10). The Cu-O stretch (σ* → dxy) for dicopper(III)-bis-μ-oxo complexes are usually within the range of 580 to 650 cm-1, a much higher frequency than dicopper(II)-peroxo species.4 Therefore, the rR spectroscopic properites of [{BzLCuIII}2(μ-O2-)2]2+ are consistent with the observed UV-visible spectroscopic features, conclusively demonstrating a dicopper-dioxygen coordination mode of a dicopper(III)-bis-μ-oxo species. Itoh and coworkers have previously demonstrated that copper(I) complexes derived from tridentate pyridyl alkylamine ligands similar to BzL (but with pyridyl 6-methyl substitutions) also react with dioxygen giving [(CuIII)2(μ-O2-)2]2+ complexes.45,62
Figure 10.

Resonance Raman (rR) spectrum of [{BzLCuIII}2(μ-O2-)2]2+ {B(C6F5)4- as counterion} with 16O2 (red) and 18O2 (blue) isotopic substitution. The samples were run at 77 K in toluene with 407 nm excitation.
Dichloromethane Degradation by [{OLCuII}2(O22-)]2+
The most striking characteristic of the dicopper(II)-μ-1,2-(end-on)-peroxide species of OL is its substrate/solvent oxidative reactivity. Warming [{OLCuII}2(O22-)]2+ to room-temperature leads to the immediate disappearance of its distinctive purple color accompanied by the formation of a blue complex OLCuII-Cl; see earlier section for its X-ray structural characterization. Isolation of this chloride complex in ~ 45 % yield suggests that CH2Cl2 solvent was oxidatively dechlorinated during the thermal transformation or decay of [{OLCuII}2(O22-)]2+; identification of CH2Cl2 derived oxidation products containing carbon have not been determined, but this was partially carried out in another study.63 After decay of [{OLCuII}2(O22-)]2+, demetallation and analysis of the product solution indicates that only the starting ligand OL is present and no degradation/oxidation (e.g., O-demethylation) had occurred.
PyLCuI and other copper(I) complexes are known to effect the reductive dechlorination of substrates such as dichloromethane, chloroform and benzyl and alkyl chlorides.36,64-66 Addition of CH2Cl2 to PyLCuI induces an immediate color change from yellow to dark brown; PyLCuII-Cl has previously been isolated in such reactions. However, OLCuI is stable in CH2Cl2 at room temperature for at least 10 minutes without obvious color change occurring, indicating that the isolation of OLCuII-Cl is unlikely to be a consequence of any copper(I) complex mediated reduction reaction.
Toluene Oxidation by [{OLCuII}2(O22-)]2+
As previously mentioned, generation of [{OLCuII}2(O22-)]2+ in toluene (C6H5CH3) also leads to solvent oxidation. By utilizing B(C6F5)4- as the complex counteranion, OLCuI is even soluble in such non-polar solvents as toluene. Lowering the temperature of solutions with high complex concentrations (> 2 mM) to -80 °C results in partial precipitation of the purple peroxide complex formed upon addition of O2 to OLCuI. Nonetheless, standing at -80 °C for a few hours results in full conversion of both the solution and solid state forms of [{OLCuII}2(O22-)]2+ to green decay products. The dicopper(II)-dihydroxy complex [{OLCuII}2(μ-OH)2)]2+, that described above, was produced in ~ 74% yield based on the dicopperperoxo complex.
Warming low-temperature toluene solutions of [{OLCuII}2(O22-)]2+ and carrying out subsequent GC product analyses discloses that ~ 40 % benzaldehyde and ~ 5 % benzyl alcohol are produced (Scheme 2). Product yields are based on the Cu2O2 species and excess O2 was removed via vacuum/argon-purge cycles following formation of [{OLCuII}2(O22-)]2+. Considering that the generation of benzaldehyde from toluene is a four-electron oxidation process and the peroxide [{OLCuII}2(O22-)]2+ is a two-electron oxidant (assuming no other oxidant, such as Cu(II) itself, is involved in this reaction), the products obtained account for an overall ~ 90 % reaction yield.
Scheme 2.

Benzyl alcohol to benzaldehyde oxidation chemistry has been studied by Stack and coworkers in a reactivity study of a bis-μ-oxo dicopper(III) complex.67 Therefore, benzyl alcohol, a two-electron toluene oxidation product, was probed as the possible initial product formed following solvent oxidation by [{OLCuII}2(O22-)]2+, which would perhaps be further oxidized to give benzaldehyde, the major product. To determine if [{OLCuII}2(O22-)]2+ could effect such a reaction, two equivalents of external benzyl alcohol was added to the reaction mixture. Instead of observing a 1:1 benzyl alcohol/benzaldehyde product mixture, GC analysis revealed the ratio as ~ 5:1, indicating that benzaldehyde has been formed in ~ 40 % yield, the same yield as when no external benzyl alcohol was added. Thus, [{OLCuII}2(O22-)]2+ does not convert benzyl alcohol to benzaldehyde. Instead, the formation of benzaldehyde and benzyl alcohol must occur at the same time during the decay process of [{OLCuII}2(O22-)]2+ and oxidation of toluene. Such an observation is not unprecedented, Que and coworkers have also found that benzaldehyde was the sole toluene oxidation product by an iron(IV) species.29 Thus, this four electron oxidation reaction may be due to the nature of the end-on peroxide species and its following chemistry, as discussed below.
To identify the source of the oxygen atom within the benzaldehyde product, 18O2 was used to generate [{OLCuII}2(O22-)]2+. After removing excess O2 and allowing [{OLCuII}2(O22-)]2+ to thermally decay, GC-MS analysis of the resulting toluene supernatant revealed that ~ 54 % of the generated benzaldehyde was labeled with 18O.68 This modest 18O-atom incorporation may be explained by the known facility of PhC(O)H to undergo oxygen atom exchange with residual water present in the gas or solvent medium.69,70 Despite this relatively low yield, this 18O labeling experiment strongly argues that the benzaldehyde oxygen atom originated from [{OLCuII}2(O22-)]2+ following addition of 18O2 to OLCuI.
When the formation and decay of [{OLCuII}2(O22-)]2+ was carried out in the presence of excess O2 the yield of benzaldehyde increased to ~ 75 %. The improved benzaldehyde yield not only suggests that molecular oxygen participates in the toluene oxidation reaction but also that a radical mechanism may be involved, such as toluene hydrogen atom abstraction. To garner more information about the possibility of hydrogen atom abstraction chemistry, a C6H5CH3/C6D5CD3 (1:1) mixture was adopted as the reaction solvent. Product analysis provided an apparent kH/kD = 7.5 ± 1 based on the yield of benzaldehyde formed, see the Experimental Section. This significant isotope effect clearly demonstrates that PhCH2-H bond cleavage is a major component of the rate-determining step for this toluene oxidation reaction.
Ethylbenzene Oxidation by [{OLCuII}2(O22-)]2+
If C-H bond cleavage plays a major role in the rate determining step, similar oxidation chemistry of ethylbenzene (PhCH2CH3) by [{OLCuII}2(O22-)]2+ should result under the same conditions. The toluene aliphatic C-H bond energy is 90 kcal/mol while that for the α-hydrogen aliphatic C-H bond energy of ethylbenzene is 87 kcal/mol.29,71,72 In fact, after generation of [{OLCuII}2(O22-)]2+ in ethylbenzene as solvent and thermal decay, GC-MS analysis indeed disclosed the formation of acetophenone and 1-phenylethanol in ~ 30 % and 25 % yield, respectively, based on the Cu2O2 species, see Scheme 3. Consistent with the above 18O-labeling experiments, the “corresponding” acetophenone and 1-phenylethanol were produced with greater than 95% 18O-atom incorporation. This confirms that [{OLCuII}2(O22-)]2+ is the oxygen source for the products in this aliphatic ethylbenzene oxidation reaction.
Scheme 3.

Toluene Oxidation by [{PyLCuII}2(O22-)]2+
Since [{OLCuII}2(O22-)]2+ exhibits an exceptional aliphatic alkylbenzene oxidation reactivity, it was of interest to see if similar reactions are induced by the analogous [{OLCuII}2(O22-)]2+ species. Therefore, [{PyLCuII}2(O22-)]2+ was generated in toluene by oxygenation of PyLCuI and warmed to ambient temperature to facilitate the thermal decay reaction. Surprisingly, [{PyLCuII}2(O22-)]2+ was very stable in toluene and took about one week for full completion of the thermal transformation or decay, as indicated by the disappearance of the distinctive purple color of the reaction solution and solid precipitate. This excellent thermal stability in toluene contrasts sharply with that observed for [{OLCuII}2(O22-)]2+, which decomposes in less than one minute upon warming. GC analysis of the reaction supernatant also led to the detection of benzaldehyde but in only ~ 8 % yield based on Cu2O2 species. This contrasts with the ~ 40 % yield observed following thermal decay and solvent oxidation induced by [{OLCuII}2(O22-)]2+ (Scheme 4).
Scheme 4.

Toluene Oxidation by [{BzLCuIII}2(O2-)2]2+
Both dicopper(III)-bis-μ-oxo and dicopper(II)-μ-η2:η2-(side-on)-peroxo species are known to mediate substrate hydrogen atom abstraction reactions.2-4 Therefore, the toluene oxidation chemistry of [{BzLCuIII}2(O2-)2]2+ was tested for comparison to [{OLCuII}2(O22-)]2+, as well as to indirectly probe the possible involvement of such a species derived from [{OLCuII}2(O22-)]2+ (Scheme 5). Warming the resulting reaction mixture to ambient temperature and analysis of the reaction supernatant as described in the Experimental Section resulted in yields almost identical to the products of the analogous reaction induced by [{OLCuII}2(O22-)]2+. GC analysis of the products disclose that if [{BzLCuIII}2(O2-)2]2+ is allowed to decay without excess O2 then ~ 40 % benzaldehyde and less than 5 % benzyl alcohol is produced. Thus, a putative dicopper(III)-bis-μ-oxo species derived from [{OLCuII}2(O22-)]2+ by de-ligation of the anisole ether oxygen ligand of OL is a likely possibility for the Cu2O2 species responsible for toluene and/or ethylbenzene oxygenation chemistry (Scheme 5).
Scheme 5.

Possible Reaction Mechanisms
Proposed mechanisms for the alkylbenzene oxidation following oxygenation of OLCuI or BzLCuI in toluene are shown in Scheme 6. Pathway A follows binuclear Cu2O2 species initiated chemistry; its detailed structure is left intentionally vague in order to account for all possible reactive binuclear species, such as a distorted dicopper(II)-end-on-peroxo species. However, based on the similar benzaldehyde product yields observed following thermal decay of either [{OLCuII}2(O22-)]2+ or [{BzLCuIII}2(μ-O2-)2]2+, a bis-μ-oxo-dicopper(III) complex [{OLCuIII}2(μ-O2-)2]2+ can be hypothesized to form during the thermal transformation process, proceeding via the equilibrium shown in Scheme 5. Another possibility for the responsible reactive species is a mononuclear copper(II)-superoxide complex [RCuII-O2-]+, a precursor to any Cu2O2 species formed following oxygenation of OLCuI or BzLCuI. Pathway B, Scheme 6 starts with chemistry initiated by this, [OCuII-O2-]+. Although neither [{OLCuIII}2(μ-O2-)2]2+ nor [RCuII-O2-]+ were detected in this work, their possible involvement can not be ruled out.73
Scheme 6.

As suggested by the kinetic isotope effect results (vide supra), the first and most-likely rate determining step would be abstraction of the α-hydrogen atom of a PhCH2-H molecule to give a PhCH2• moiety. Following Pathway A, this benzyl radical would rapidly react with molecular O2 to give a benzyl peroxo radical (PhCH2OO•); via well known alkylperoxo radical chemistry (e.g., the Russell mechanism),74,75 this could disproportionate to yield a 1:1 ratio of non-radical products benzaldehyde and benzyl alcohol (Pathway A, Scheme 6). Excess or residual O2 present in the solution could originate from the reverse oxygenation reaction of the copper(I) complex (RLCuI). Such a disproportionation process well explains the nearly equal yield of acetophenone and 1-phenylethanol produced from the ethylbenzene oxidation. This reaction mechanism was also reported by Que and coworkers to interpret non-heme diiron catalyzed alkylbenzene oxidation reactions.28
However, as described above, benzaldehyde (the four-electron oxidation product) was found to be the dominant product following toluene oxidation. Therefore, a different process must occur in order to account for the majority benzaldehyde product formation. For example, reduction of the benzylperoxyl radical (PhCH2OO•) by RLCuI present in the system may occur prior to or parallel with the disproportionation process described above. Generation of [LCuII-OOCH2Ph]+ followed by elimination of [LCuII-OH]+ and PhC(O)H generation (Scheme 5) would in a relative manner greatly increase the benzaldehyde product yield, as is the observed situation. Support for this mechanistic pathway comes from our isolation of [{OLCuII}2(μ-OH)2]2+ (74 % yield), formally a dimer of [LCuII-OH]+.
Another pathway that would lead to only bsenzaldehyde product is if a copper(II)-superoxide species [RCuII-O2-]+ is the actual hydrogen atom abstractor. The resulting copper-hydroperoxide [RCuII-OOH]+ moiety could further react with the benzyl radical by a formal HOO• rebound reaction, releasing RCuI and benzylhydroperoxide product; the latter could eliminate water to give benzaldehyde (Scheme 6; Pathway B).76,77 Recently, experimental and theoretical mechanistic investigations of dopamine β-monooxygenase (DβM) and peptidylglycine α-amidating monooxygenase (PHM) C-H oxidation chemistry suggest a mononuclear copper(II)-superoxide species is the active hydrogen atom abstraction reagent.10,12,13,78 Further, recent investigations on synthetically derived copper(II)-superoxide species, [(ligand)CuII-(O2-)]+ {NMe2-tmpa, tris(4-dimethyl-aminopyrid-2-ylmethyl)amine); TMG3-tren, tris(2-(N-tetramethylguanidyl)-ethylamine}, demonstrate that they possess some H-atom abstraction capabilities.79
Possible Reactive Species Following Oxygenation of OLCuI
In prior work, the reactivity of different Cu2O2 species with exogenously added substrates was compared (Chart 3).22 The dicopper(II)-μ-1,2-end-on-peroxide complexes [{PyLCuII}2(O22-)]2+ and [CuII2(XYL-O-)(O22-)]2+ were found to exhibit nucleophilic character. As a result, interesting oxidative chemistry was not demonstrated, for instance, addition of PPh3 led to the release of O2 and formation of corresponding copper(I)-PPh3 complexes. By contrast, dicopper(II)-μ-η2:η2-(side-on)-peroxide species such as [CuII2(N4)(O22-)]2+ and [CuII2(XYL-H)(O22-)]2+ (Chart 3) exhibit electrophilic reactivity,2 for example the former oxidizes externally added PPh3 to give O=PPh3,22 while the latter undergoes internal hydroxylation of the xylyl ligand spacer.2,80 Thus, from this precedent, one may argue that a dicopper(II)-end-on-peroxide complex such as [{OLCuII}2(O22-)]2+ may not be a good enough oxidant or electrophilic enough to effect the toluene or ethylbenzene substrate oxidation discussed herein. We thus may consider three possibilities for the reactive copper-dioxygen species involved in the present chemistry: (1) a distorted square pyramidal dicopper(II)-μ-1,2-end-on-peroxide species [{OLCuII}2(O22-)]2+, (2) an unobservable dicopper(III)-bis-μ-oxo species [{OLCuII}2(O22-)]2+, or (3) a mononuclear copper(II)-superoxide species [OCuII-O2-]+.
Chart 3.
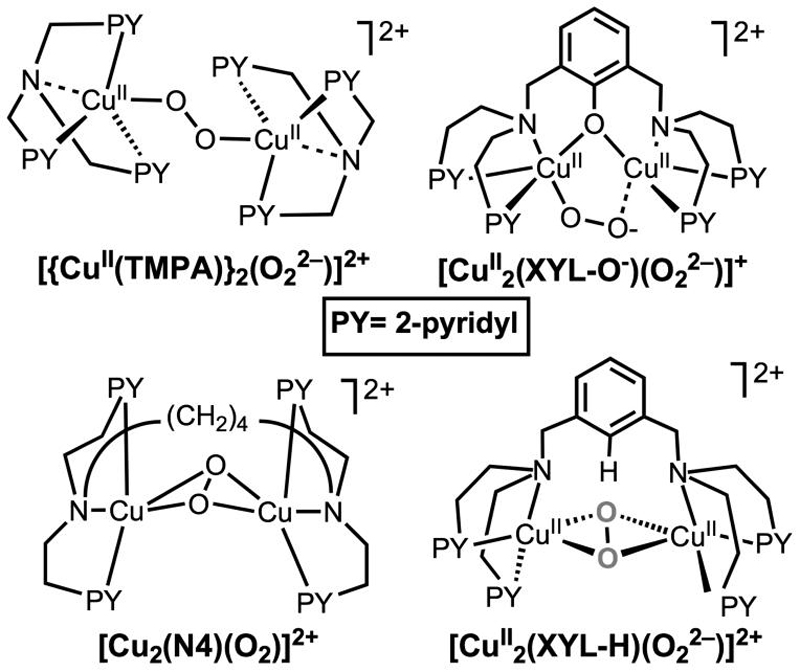
Addressing the first proposal, perhaps it is the modified copper coordination geometry present in [{OLCuII}2(O22-)]2+ that facilitates the remarkable exogenous substrate reactivity. It is informative to compare copper(II) ion geometries in peroxo CuII2-O2 compounds with simpler nonperoxidic analogues possessing the same ligand (Table 5). The O2-adduct coordination geometries (dicopper(II)-peroxo complexes) likely exhibit structures similar to corresponding stable mononuclear copper(II) analogs. The crystal structure of OLCuII-Cl indicates that within the ligand OL frame, a square pyramidal geometry is favored, and thus we suggest that such a distorted geometry (i.e., away from trigonal bipyramidal) would be retained in [{OLCuII}2(O22-)]2+.
Table 5.
Criterion τ value) for pentacoordinationa for select dicopper(II)-μ-1,2-end-on-peroxo species along with related (analog) ligand-copper(II)-complexes.
| Ligand | Copper(II) Complex | τ value | Ref |
|---|---|---|---|
| PyL (TMPA) | [{CuII(PyL)}2(O22-)]2+ | 0.86 | 27 |
| [CuII(PyL)(Cl-)]+ | 1.00 | 55 | |
| [CuII(PyL)(CN-)]+ | 0.90 | 81 | |
| [CuII(PyL)(ONO-)]+ | 0.74 | 82 | |
| LH,Bn | [{CuII(LH,Bn)}2(O22-)]2+ | 0.81 | 56 |
| [CuII(LH,Bn)(Cl-)]+ | 0.93 | 56 | |
| [CuII(LH,Bn)(H2O)]2+ | 0.78 | 83 | |
| LPy (Scheme 8) | [{CuII(LPy)}2(O22-)]2+ | not available | 84 |
| [CuII(LPy)(ONO-)]+ | 0.003 | 85 | |
| [CuII(LPy)(Cl-)]+ | 0.46 | 86 | |
| OL | [{CuII(OL)}2(O22-)]2+ | not available | -- |
| [CuII(OL)(Cl-)]+ | 0.23 | -- |
trigonal bypyramidal, τ = 1.00; square pyramidal, τ = 0.00.41
To date, two end-on Cu2O2 species have been structurally characterized, [{PyLCuII}2(O22-)]2+ (Chart 1)27 and Sukuzi's complex, [{CyII(LH,Bn)}2(O22-)] (LH,Bn = tris(N-benzylaminoethyl)amine.56 The copper(II) ions in both complexes adopt a nearly perfect trigonal bipyramidyl (TBP) geometry, with the apical amine nitrogen of the tetradentate ligand and the oxygen from the O22- occupying the axial positions. In contrast, the copper(II) ions of side-on bound peroxide complexes all adopt a (distorted) square pyramidal geometry with τ values varying from 0 to 0.44 (Table 6) based on their X-ray crystal structures. Copper(III)-bis-μ-oxo species possess structures that possess nearly perfect square pyramidal geometries, i.e., with τ approaching zero (Table 6).
Table 6.
| Copper(II) Complex | τ value | Cu2(O22-) species | Ref |
|---|---|---|---|
| [CuII(PyL)}2(O22-)]2+ | 0.86 | end-on peroxo | 27 |
| [{CuII(LH,Bn)}2(O22-)]2+ | 0.81 | end-on peroxo | 56 |
| [CuII(HB(3,5-i-Pr2pz)3)2](O22-) | 0.027 | side-on peroxo | 42,88 |
| [CuII2(O22-)(L)]2+ | 0.16,0.44 | side-on peroxo | 89 |
| [(CuIITpCF3,CH3)2(O22-)] | 0.002 | side-on peroxo | 90 |
| [CuII2(O22-)(iPr3TACD)]2+ | 0.30 (av) | side-on peroxo | 91 |
| [{CuIII(LBn3)}2(μ-O)2]2+ | 0.018 | bis-μ-oxo | 92,93 |
| [{CuIII(Me2-tpa)}2(μ-O)2]2+ | 0.017 | bis-μ-oxo | 94 |
| [{CuIII(iPr4dtne)}2(μ-O)2]2+ | 0.077,0.142 | bis-μ-oxo | 95 |
trigonal bypyramidal, τ = 1; square pyramidal, τ = 0.41
Electrophilic reactivity mechanisms such as hydrogen atom abstraction reactions are well-established for dicopper(II)-μ-η2:η2-(side-on)-peroxo and dicopper(III)-bis-μ-oxo species.3,17,67,96 In fact, the former is a better oxygen-atom transfer reagent,80 while the latter is a better hydrogen atom acceptor.97 Based on the chemical evidence presented here, it may be that an unobservable dicopper(III)-bis-μ-oxo [{OLCuIII}2(μ-O2-)2]2+ species that is structurally analogous to [{BzLCuIII}2(μ-O2-)2]2+ which forms during the decay of [{OLCuII}2(O22-)]2+; this could explain the equivalent toluene oxidation product yields starting either with OLCuI vs. BzLCuI. Furthermore, the geometry changes that result upon CuII/CuI redox changes based on X-ray crystallographic analysis of OLCuI and OLCuII, i.e. de-ligation of the anisole-ether-oxygen, support that a tridentate ligand framework is possible for the OL ligand that could potentially lead to [{OLCuIII}2(μ-O2-)2]2+. Supporting the notions described here is what we already mentioned, that the Cu2O2 species observed following oxygenation of OLCuI is [{OLCuII}2(O22-)]2+, but such dicopper(II)-μ-1,2-(end-on)-peroxo species typically are nucleophilic and not good oxidants.
Tolman and coworkers84 published an observation that is perhaps relevant to the present system involving chemistry with a tridentate tri-isopropyl substituted TACN triazacyclononane ligand with a pyridyl appendage (LPy) (Scheme 7). With their tetradentate ligand LPy (Scheme 7A), an end-on peroxide species with UV-visible and rR characteristics (Table 5) similar to those known for [{PyLCuII}2(O22-)]2+ were obtained. Thermal decay led to a ketone product, indicating a four-electron oxidation process. The chemistry was suggested to occur via a dicopper(II)-side-on-peroxide species transformed from the dicopper(II)-end-on-peroxide primary form.84 In a separate study, a modified LPy ligand possessing a sterically hindered 2-phenyl pyridyl donor formed a dicopper(III)-bis-μ-oxo species upon oxygenation and thermal decay instead led to a two-electron oxidative N-dealkylation reaction (Scheme 7B).85 The latter chemistry also occurs from the dicopper(III)-bis-μ-oxo species formed in the parent tridentate tri-isopropyl substituted TACN triazacyclononane ligand.17 Thus, different reactive species were probably involved in these two different reaction scenerios (Schemes 7A vs. 7B). The work from the Tolman labs may support the view that it is a dicopper(III)-bis-μ-oxo complex leads to the four-electron oxidation product (R1R2C-H to R1R2C=O, e.g., toluene to benzaldehyde) in the present system.
Scheme 7.

Summary/Conclusion
By employing a new anisole containing polypyridylamine ligand OL, a dicopper(II)-μ-1,2-end-on-peroxo species [{OLCuII}2(O22-)]2+ was obtained by reacting the copper(I) precursor OLCuI with molecular oxygen at -80 °C). The UV-vis and rR spectroscopic properties of [{OLCuII}2(O22-)]2+ closely mimic those of the analogous species [{PyLCuII}2(O22-)]2+, which possesses a pyridyl ligand PyL instead of an anisole-ether-oxygen donor as in [{OLCuII}2(O22-)]2+. Compared to [{PyLCuII}2(O22-)]2+, the νCu-o and νo-o shift to higher energy values for [{OLCuII}2(O22-)]2+ suggests that the Oanisole in that complex is a poorer donor than the corresponding Npyridyl atom in [{PyLCuII}2(O22-)]2+. This conclusion is further supported by the observed redox potential difference of ΔE½ = 0.11 V between PyLCuII and OLCuII with the former possessing a more negative value. Additionally, [{OLCuII}2(O22-)]2+ is very unstable in solvents possessing an oxygen-heteroatom as indicated by the lack of observable Cu2O2 species by UV-visible spectroscopy, suggesting competitive solvent binding. The poor electron-donating ability of OL as a result of the anisole-ether-oxygen moiety is also illustrated by the X-ray crystal structure of OLCuI and the carbon monoxide binding studies revealing that the Oanisole does not coordinate to the cuprous ion.
The most striking characteristic of [{OLCuII}2(O22-)]2+ is the aliphatic alkylbenzene C-H bond oxidative ability. To our knowledge, this is the first example of such oxidation reactions starting with a dicopper(II)-μ-1,2-end-on-peroxo species. . Dioxygen 18O-labeling experiments demonstrate that the oxygen atom in the product species is derived from molecular oxygen. An apparent kH/kD = 7.5 ± 1 isotope effect suggests that α-H-atom abstraction from the alkylbenzene to yield a corresponding organic radical is likely to be the first as well as the rate-determining step of the reaction. Benzylic radical combination with O2 (free or derived from a copper complex) and well established ROO• and ROO•/metal (here CuI) chemistry can account for the distribution of observed organic products.
As discussed, unobservable Cu2O2 derived species may be the actual oxidant species. Chemical precedence suggests that substrate reactions of [{OLCuII}2(O22-)]2+ occur via its thermal transformation to a dicopper(III)-bis-μ-oxo isomer form [{OLCuIII}2(μ-O2-)2]2+. Such an equilibration is supported by conducting parallel toluene-oxidation reactions with [{BzLCuIII}2(μ-O2-)2]2+ giving identical product distributions and yields. The weakly coordinated axial Oanisole may deligate to form a tridentate η3-chelate, like exhibited by the cuprous species OLCuI, that would support a dicopper(III)-bis-μ-oxo coordination mode. A copper(II)-superoxo intermediate generated from the loss of loss of (OL)CuI from [{OLCuII}2(O22-)]2+ may alternatively be responsible for the observed H-atom abstraction reactivity. Further experimental or theoretical research is needed to test the validity of these hypotheses and to shed further light on the nature of the differing reactivity of CuIn/O2 derived species.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the National Institutes of Health (K.D.K., GM28962, and E.I.S., DK31450) for support of this research.
Footnotes
Supporting Information Available. X-ray structure determination summaries, figures and X-ray data files (CIF), GC and GC/MS product analyses from experiments with isotopically labeled materials. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Solomon EI, Sundaram UM, Machonkin TE. Chem. Rev. 1996;96:2563–2606. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- (2).Hatcher LQ, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- (3).Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- (4).Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- (5).Itoh S. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- (6).Rolff M, Tuczek F. Angew Chem Int Edit. 2008;47:2344–2347. doi: 10.1002/anie.200705533. [DOI] [PubMed] [Google Scholar]
- (7).Magnus KA, Tonthat H, Carpenter JE. Chem. Rev. 1994;94:727–735. [Google Scholar]
- (8).Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TD. Science. 2005;308:1890–2. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- (9).Klinman JP. Chem. Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- (10).Klinman JP. J. Biol. Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- (11).Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- (12).Chen P, Solomon EI. J. Am. Chem. Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- (13).Evans JP, Ahn K, Klinman JP. J. Biol. Chem. 2003;278:49691–49698. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]
- (14).Lieberman RL, Rosenzweig AC. Nature. 2005;434:177–82. doi: 10.1038/nature03311. [DOI] [PubMed] [Google Scholar]
- (15).Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. J. Am. Chem. Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. [DOI] [PubMed] [Google Scholar]
- (16).Itoh S, Nakao H, Berreau LM, Kondo T, Komatsu M, Fukuzumi S. J. Am. Chem. Soc. 1998;120:2890–2899. [Google Scholar]
- (17).Mahapatra S, Halfen JA, Tolman WB. J. Am. Chem. Soc. 1996;118:11575–11586. [Google Scholar]
- (18).Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J. Am. Chem. Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- (19).Shearer J, Zhang CX, Hatcher LQ, Karlin KD. J. Am. Chem. Soc. 2003;125:12670–12671. doi: 10.1021/ja0359409. [DOI] [PubMed] [Google Scholar]
- (20).Itoh K, Hayashi H, Furutachi H, Matsumoto T, Nagatomo S, Tosha T, Terada S, Fujinami S, Suzuki M, Kitagawa T. J. Am. Chem. Soc. 2005;127:5212–5223. doi: 10.1021/ja047437h. [DOI] [PubMed] [Google Scholar]
- (21).Li L, Narducci Sarjeant AA, Vance MA, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2005;127:15360–15361. doi: 10.1021/ja054948a. [DOI] [PubMed] [Google Scholar]
- (22).Paul PP, Tyeklar Z, Jacobson RR, Karlin KD. J. Am. Chem. Soc. 1991;113:5322–5332. [Google Scholar]
- (23).Karlin KD, Zuberbühler AD. In: Bioinorganic Catalysis. Second Edition, Revised and Expanded Reedijk J, Bouwman E, editors. Marcel Dekker, Inc.; New York: 1999. pp. 469–534. [Google Scholar]
- (24).Karlin KD, Kaderli S, Zuberbühler AD. Acc. Chem. Res. 1997;30:139–147. [Google Scholar]
- (25).Karlin KD, Wei N, Jung B, Kaderli S, Niklaus P, Zuberbuhler AD. J. Am. Chem. Soc. 1993;115:9506–9514. [Google Scholar]
- (26).Baldwin MJ, Ross PK, Pate JE, Tyeklar Z, Karlin KD, Solomon EI. J. Am. Chem. Soc. 1991;113:8671–8679. [Google Scholar]
- (27).Jacobson RR, Tyeklar Z, Farooq A, Karlin KD, Liu S, Zubieta J. J. Am. Chem. Soc. 1988;110:3690–3692. [Google Scholar]
- (28).Kim C, Dong YH, Que L. J. Am. Chem. Soc. 1997;119:3635–3636. [Google Scholar]
- (29).Kaizer J, Klinker EJ, Oh NY, Rohde JU, Song WJ, Stubna A, Kim J, Munck E, Nam W, Que L. J. Am. Chem. Soc. 2004;126:472–473. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- (30).Maiti D, Woertink JS, Sarjeant AAN, Solomon EI, Karlin KD. Inorg Chem. 2008;47:3787–3800. doi: 10.1021/ic702437c. [DOI] [PubMed] [Google Scholar]
- (31).Wasser IM, Huang HW, Moenne-Loccoz P, Karlin KD. J. Am. Chem. Soc. 2005;127:3310–3320. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- (32).Liang HC, Kim E, Incarvito CD, Rheingold AL, Karlin KD. Inorg Chem. 2002;41:2209–2212. doi: 10.1021/ic010816g. [DOI] [PubMed] [Google Scholar]
- (33).Sanyal I, Ghosh P, Karlin KD. Inorg. Chem. 1995;34:3050–3056. [Google Scholar]
- (34).Masarwa A, Rachmilovich-Calis S, Meyerstein N, Meyerstein D. Coordin Chem Rev. 2005;249:1937–1943. [Google Scholar]
- (35).Li L, Karlin KD, Rokita SE. J. Am. Chem. Soc. 2005;127:520–521. doi: 10.1021/ja044209e. [DOI] [PubMed] [Google Scholar]
- (36).Tyeklar Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD. J. Am. Chem. Soc. 1993;115:2677–2689. [Google Scholar]
- (37).Lim BS, Holm RH. Inorg. Chem. 1998;37:4898–4908. doi: 10.1021/ic9801793. [DOI] [PubMed] [Google Scholar]
- (38).Osako T, Tachi Y, Taki M, Fukuzumi S, Itoh S. Inorg. Chem. 2001;40:6604–6609. doi: 10.1021/ic010625d. [DOI] [PubMed] [Google Scholar]
- (39).Osako T, Tachi Y, Doe M, Shiro M, Ohkubo K, Fukuzumi S, Itoh S. Chem. Eur. J. 2004;10:237–246. doi: 10.1002/chem.200305263. [DOI] [PubMed] [Google Scholar]
- (40).τ values (α-β/60°) calculated based on the X-ray crystallographic data reported in cited references.
- (41).Addison AW, Rao TN, Reedijk J, Vanrijn J, Verschoor GC. J. Chem. Soc. Dalton. 1984:1349–1356. [Google Scholar]
- (42).Kitajima N, Fujisawa K, Fujimoto C, Morooka Y, Hashimoto S, Kitagawa T, Toriumi K, Tatsumi K, Nakamura A. J. Am. Chem. Soc. 1992;114:1277–1291. [Google Scholar]
- (43).Jonas RT, Stack TDP. Inorg. Chem. 1998;37:6615–6629. doi: 10.1021/ic9713487. [DOI] [PubMed] [Google Scholar]
- (44).Pasquali M, Marini G, Floriani C, Gaetanimanfredotti A, Guastini C. Inorg. Chem. 1980;19:2525–2531. [Google Scholar]
- (45).Osako T, Terada S, Tosha T, Nagatomo S, Furutachi H, Fujinami S, Kitagawa T, Suzuki M, Itoh S. Dalton Trans. 2005:3514–21. doi: 10.1039/b500202h. [DOI] [PubMed] [Google Scholar]
- (46).Zhang CX, Kaderli S, Costas M, Kim E, Neuhold YM, Karlin KD, Zuberbuhler AD. Inorg Chem. 2003;42:1807–1824. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- (47).Laitar DS, Mathison CJN, Davis WM, Sadighi JP. Inorg. Chem. 2003;42:7354–7356. doi: 10.1021/ic035056j. [DOI] [PubMed] [Google Scholar]
- (48).Klaui W, Berghahn M, Frank W, Reiss GJ, Schonherr T, Rheinwald G, Lang H. Eur. J. Inorg. Chem. 2003:2059–2070. [Google Scholar]
- (49).Imai S, Fujisawa K, Kobayashi T, Shirasawa N, Fujii H, Yoshimura T, Kitajima N, Moro-oka Y. Inorg. Chem. 1998;37:3066–3070. [Google Scholar]
- (50).Kretzer RM, Ghiladi RA, Lebeau EL, Liang HC, Karlin KD. Inorg. Chem. 2003;42:3016–3025. doi: 10.1021/ic020521i. [DOI] [PubMed] [Google Scholar]
- (51).Fry HC, Lucas HR, Sarjeant AAN, Karlin KD, Meyer GJ. Inorg Chem. 2008;47:241–256. doi: 10.1021/ic701903h. [DOI] [PubMed] [Google Scholar]
- (52).Wei N, Murthy NN, Karlin KD. Inorg. Chem. 1994;33:6093–6100. [Google Scholar]
- (53).Schatz M, Becker M, Thaler F, Hampel F, Schindler S, Jacobson RR, Tyeklar Z, Murthy NN, Ghosh P, Chen Q, Zubieta J, Karlin KD. Inorg. Chem. 2001;40:2312–2322. doi: 10.1021/ic000924n. [DOI] [PubMed] [Google Scholar]
- (54).Rorabacher DB. Chem. Rev. 2004;104:651–97. doi: 10.1021/cr020630e. [DOI] [PubMed] [Google Scholar]
- (55).Karlin KD, Hayes JC, Juen S, Hutchinson JP, Zubieta J. Inorg. Chem. 1982;21:4106–4108. [Google Scholar]
- (56).Komiyama K, Furutachi H, Nagatomo S, Hashimoto A, Hayashi H, Fujinami S, Suzuki M, Kitagawa T. Bull. Chem. Soc. Jpn. 2004;77:59–72. [Google Scholar]
- (57).Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew. Chem. Int. Ed. 2001;40:4570–4590. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- (58).Liang HC, Karlin KD, Dyson R, Kaderli S, Jung B, Zuberbuhler AD. Inorg. Chem. 2000;39:5884–5894. doi: 10.1021/ic0007916. [DOI] [PubMed] [Google Scholar]
- (59).Formation of [{BzLCuIII}2(μ-O2-)2]2+ is very sensitive to solvent dryness. In very dry toluene, the reaction can take up to 2 hours to reach completion. In other solvents, the dicopper(III)-bis-μ-oxo species forms quickly and reaches λmax in one to two minutes.
- (60).Solomon and coworkers have reported based on rR studies that a dicopper(II)-end-on-peroxo species reveals that the stronger the electron-donating ability of the chelate ligand, the lower the observed O-O and Cu-O stretching frequencies.61 An increase in the N-donor strength or electron density through ligand donation to the copper center leads to a compensating decrease in peroxide π* (σ) donation to the copper CuII ion (d 2z orbital it TBP geometry), thus weakening the Cu-O bond. As a result, more electron density is localized in the peroxide π* orbitals, weakening the O-O bond. Thus, the higher O-O and Cu-O frequencies observed in the rR spectrum of [{OLCuII}2(O22-)]2+ suggests that the anisole-ether-oxygen atom of OL is a weaker donor than the “corresponding” pyridyl nitrogen within [{PyLCuII}2(O2-)]2+. This conclusion is supported by the electrochemical studies carried out (vide supra). The weaker ligand donation by tetradentate ligand OL increases peroxide π* (σ) donation to the copper CuII ion (dz 2 orbital), which strengthens the Cu-O bond and consequently strengthens the O-O bond.
- (61).Henson MJ, Vance MA, Zhang CX, Liang HC, Karlin KD, Solomon EI. J. Am. Chem. Soc. 2003;125:5186–5192. doi: 10.1021/ja0276366. [DOI] [PubMed] [Google Scholar]
- (62).Kunishita A, Osako T, Tachi Y, Teraoka J, Itoh S. B Chem Soc Jpn. 2006;79:1729–1741. [Google Scholar]
- (63).Lucchese B. Johns Hopkins University; 2003. [Google Scholar]
- (64).Osako T, Karlin KD, Itoh S. Inorg. Chem. 2005;44:410–415. doi: 10.1021/ic048902e. [DOI] [PubMed] [Google Scholar]
- (65).Jacobson RR, Tyeklar Z, Karlin KD. Inorg. Chim. Acta. 1991;181:111–118. [Google Scholar]
- (66).Lucchese B, Humphreys KJ, Lee DH, Incarvito CD, Sommer RD, Rheingold AL, Karlin KD. Inorg. Chem. 2004;43:5987–5998. doi: 10.1021/ic0497477. [DOI] [PubMed] [Google Scholar]
- (67).Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. J. Am. Chem. Soc. 2002;124:9332–9333. doi: 10.1021/ja026905p. [DOI] [PubMed] [Google Scholar]
- (68).See Supporting Information.
- (69).Hilal SH, Bornander LL, Carreira LA. Qsar Comb Sci. 2005;24:631–638. [Google Scholar]
- (70).Guthrie JP. J. Am. Chem. Soc. 2000;122:5529–5538. [Google Scholar]
- (71).Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- (72).Bryant JR, Mayer JM. J. Am. Chem. Soc. 2003;125:10351–10361. doi: 10.1021/ja035276w. [DOI] [PubMed] [Google Scholar]
- (73).Resonance Raman spectroscopic attempts to observe [{OLCuIII}2(μ-O2-)2]2+ in solutions of end on peroxo complex [{OLCuII}2(O22-)]2+ were not successful. We also note that bubbling solutions of [{OLCuII}2(O22-)]2+ with CO, led to the appearance of a UV-Vis band at 410 nm (Figure 7), that characteristic of a superoxo-copper(II) complex with tetradentate ligand. However, rR spectroscopic interrogation68 did not provide proof for the presence of a species [OCuII-O2-]+.
- (74).Goldstein S, Meyerstein D. Acc. Chem. Res. 1999;32:547–550. [Google Scholar]
- (75).(a) VonSonntag C, Schuchmann H-P. In: Peroxyl radicals. Alfassi ZB, editor. Wiley; Chichester; New York: 1997. pp. 173–234. [Google Scholar]; (b) Russell GA. J. Am. Chem. Soc. 1957;79:3871–3877. [Google Scholar]
- (76).Masarwa A, Rachmilovich-Calis S, Meyerstein N, Meyerstein D. Coord. Chem. Rev. 2005;249:1937–1943. [Google Scholar]
- (77).Meyerstein D. Met. Ions Biol. Syst. 1999;36:41–77. [Google Scholar]
- (78).Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- (79).Maiti D, Lee DH, Gaoutchenova K, Wurtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew Chem Int Edit. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- (80).Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. J. Am. Chem. Soc. 998;120:7841–7847. [Google Scholar]
- (81).Corsi DM, Murthy NN, Young VG, Karlin KD. Inorg. Chem. 1999;38:848–858. doi: 10.1021/ic981091s. [DOI] [PubMed] [Google Scholar]
- (82).Komeda N, Nagao H, Kushi Y, Adachi G, Suzuki M, Uehara A, Tanaka K. Bull. Chem. Soc. Jpn. 1995;68:581–589. [Google Scholar]
- (83).Schatz M, Becker M, Walter O, Liehr G, Schindler S. Inorg. Chim. Acta. 2001;324:173–179. [Google Scholar]
- (84).Halfen JA, Young VG, Tolman WB. J. Am. Chem. Soc. 1996;118:10920–10921. [Google Scholar]
- (85).Berreau LM, Halfen JA, Young VG, Tolman WB. Inorg. Chim. Acta. 2000;297:115–128. [Google Scholar]
- (86).Schneider JL, Halfen JA, Young VG, Tolman WB. New. J. Chem. 1998;22:459–466. [Google Scholar]
- (87).Addison AW, Rao TN, Reedijk J, Vanrijn J, Verschoor GC. J Chem Soc Dalton. 1984:1349–1356. [Google Scholar]
- (88).Kitajima N, Fujisawa K, Morooka Y, Toriumi K. J. Am. Chem. Soc. 1989;111:8975–8976. [Google Scholar]
- (89).Kodera M, Katayama K, Tachi Y, Kano K, Hirota S, Fujinami S, Suzuki M. J. Am. Chem. Soc. 1999;121:11006–11007. [Google Scholar]
- (90).Hu ZB, George GN, Gorun SM. Inorg. Chem. 2001;40:4812–+. doi: 10.1021/ic015529r. [DOI] [PubMed] [Google Scholar]
- (91).Lam BMT, Halfen JA, Young VG, Hagadorn JR, Holland PL, Lledos A, Cucurull-Sanchez L, Novoa JJ, Alvarez S, Tolman WB. Inorg. Chem. 2000;39:4059–4072. doi: 10.1021/ic000248p. [DOI] [PubMed] [Google Scholar]
- (92).Mahapatra S, Halfen JA, Wilkinson EC, Pan GF, Wang XD, Young VG, Cramer CJ, Que L, Tolman WB. J. Am. Chem. Soc. 1996;118:11555–11574. [Google Scholar]
- (93).Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Que L, Zuberbuhler AD, Tolman WB. Science. 1996;271:1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- (94).Hayashi H, Fujinami S, Nagatomo S, Ogo S, Suzuki M, Uehara A, Watanabe Y, Kitagawa T. J. Am. Chem. Soc. 2000;122:2124–2125. [Google Scholar]
- (95).Mahapatra S, Young VG, Kaderli S, Zuberbuhler AD, Tolman WB. Angew. Chem. Int. Ed. 1997;36:130–133. [Google Scholar]
- (96).Itoh S, Taki M, Nakao H, Holland PL, Tolman WB, Que L, Fukuzumi S. Angew. Chem. Int. Ed. 2000;39:398–+. doi: 10.1002/(sici)1521-3773(20000117)39:2<398::aid-anie398>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- (97).Mahadevan V, Henson MJ, Solomon EI, Stack TDP. J. Am. Chem. Soc. 2000;122:10249–10250. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.