

Abstract
A major bottleneck in the development of siRNA therapies is their delivery to the desired cell type or tissue, followed by effective passage across the cell membrane with subsequent silencing of the targeted mRNA. To address this problem, we describe the synthesis of core/shell hydrogel nanoparticles (nanogels) with surface-localized peptides that specifically target ovarian carcinoma cell lines possessing high expression levels of the Eph2A receptor. These nanogels are also demonstrated to be highly effective in the noncovalent encapsulation of siRNA and enable cell-specific delivery of the oligonucleotides in serum-containing medium. Cell toxicity and viability assays reveal that the nanogel construct is nontoxic under the conditions studied, as no toxicity or decrease in cell proliferation is observed following delivery. Importantly, a preliminary investigation of gene silencing illustrates that nanogel-mediated delivery of siRNA targeted to the EGF receptor results in knockdown of that receptor. Excellent protection of siRNA during endosomal uptake and endosomal escape of the nanogels is suggested by these results since siRNA activity in the cytosol is required for gene silencing.
INTRODUCTION
Significant effort has been invested in the design of colloidal drug carriers in order to improve drug localization and bioavailability (1-3). Ideally, an actively targeted particulate drug carrier will increase the therapeutic efficacy of a drug by delivery to the diseased site, while reducing drug-associated side effects. Attainment of this goal would greatly advance treatment of diseases (e.g., cancer) where the toxic effects of therapeutics administered systemically may outweigh their benefit. To date, many types of delivery vehicles have been explored for in vitro and in vivo drug delivery applications, including inorganic nanoparticles (4, 5), polyelectrolyte complexes (6), liposomes (7, 8), block copolymer micelles (9-11), and polymeric nanoparticles (12-15).
A particularly compelling phenomenon from the standpoint of cancer therapy is RNA interference (RNAi). RNAi is a relatively new approach to gene silencing, which has been demonstrated effective both in vitro and in vivo (16, 17). This technique employs small 21−25 nucleotide long double stranded small interfering RNAs (or siRNAs) to inhibit gene expression through degradation of a targeted mRNA (18). Whereas the potential for therapeutic oncology applications exist where siRNA would be used to specifically shut down genes necessary for tumor growth, the lack of efficient methods for in vivo siRNA delivery prevent widespread therapeutic use (16, 17). In addition to the confounding issues associated with systemic, intraveneous delivery of siRNA, its polyanionic nature and high molecular weight (∼13 kDa) prevent transport across the cell membrane (16, 17). Thus, effective siRNA carriers must enable efficient transport through the vasculature to the tumor and then must additionally enable intracellular delivery of the cargo. A common method currently used for siRNA delivery in vitro employs cationic lipid-based carriers (16, 17, 19) or polyelectrolytes (6). These charged moieties form polyplexes with the siRNA, forming aggregates that can be taken up into the cells, thereby delivering the siRNA to the cytosol. However, these carriers can have notable drawbacks with respect to toxicity and difficulties in specific cell targeting (16, 17, 20), thereby giving rise to a need for alternative delivery methods. A number of new approaches have been reported that overcome some of the shortcomings of lipid-based approaches. For example, Schiffelers et al. used an RGD (Arg-Gly-Asp peptide ligand)-PEG-PEI complex to target siRNA to tumor neovasculature (21). Song et al. presented the use of a protamine-antibody fusion protein using the Fab fragment of HIV-1 envelope antibody for siRNA delivery (22). Another targeting motif has been the use of liposomes in the form of an immunoliposome complex reported by Pirollo et al. (23). A number of other similar approaches have been taken (6, 20, 24-29), and these siRNA carriers have enabled certain degrees of success. However, issues of toxicity, leakiness, and payload capacity still persist, especially in the context of in vivo gene silencing (16, 17).
Building upon many of the lessons learned from these approaches, we and others have developed drug delivery methods that employ the synthetic hydrogel nanoparticle (nanogel) (13, 15, 30, 31). Nanogels possess a high degree of porosity, permitting a high payload capacity, and can also be selectively surface-functionalized to enable tumor-specific targeting. Thus, we have developed straightforward, scalable syntheses of surface-functionalized, ∼100-nm diameter, core/shell nanogels composed of poly(N-isopropylmethacrylamide) (pNIPMAm) (32, 33), an amphiphilic polymer that is strongly hydrated at physiological temperature and is likely therefore to resist protein adsorption relative to more hydrophobic carriers. This polymer has also garnered interest because of its dramatic thermoresponsivity; it undergoes an entropically driven coil-to-globule (swollen-to-collapsed) transition at ∼43 °C, which may have utility for thermally triggered delivery (32, 33). However, in the present demonstration, this thermoresponsivity is only used to enable the synthesis of monodispersed core/shell nanogels via precipitation polymerization, as we have discussed previously (32, 34). The core/shell pNIPMAm nanogel construct used to encapsulate and deliver siRNA to ovarian cancer cells is illustrated in Scheme 1. A previously described 12 amino acid peptide (YSAYPDSVPMMS or YSA) (35) was coupled to the surface of ∼100-nm diameter core/shell nanogels to permit cell-specific targeting and subsequent delivery of high concentrations of siRNA to the target cells. The YSA peptide mimics the ligand ephrin-A1, which binds to the erythropoietin-producing hepatocellular (Eph) A2 receptor. In addition to specific expression in neovasculature (36, 37), EphA2 is highly expressed by a number of tumor cells including those derived from ovarian (38, 39), prostate (40, 41), breast (42, 43), and colon (44, 45) cancers, making it an excellent target for tumor-specific delivery. Thus, we demonstrate herein that pNIPMAm nanogels have a high loading capacity for siRNA, and that these nanogels are delivered to the cytoplasm of ovarian cancer cells via ligand-receptor binding mediated endocytosis. Importantly, overt cytotoxicity was not observed to arise from the nanocarrier, suggesting that this approach could be a highly efficacious one. In addition, delivery of siRNA to cells in culture can be performed in the presence of serum, suggesting that nanogels may be of particular advantage for in vivo delivery.
Scheme 1.
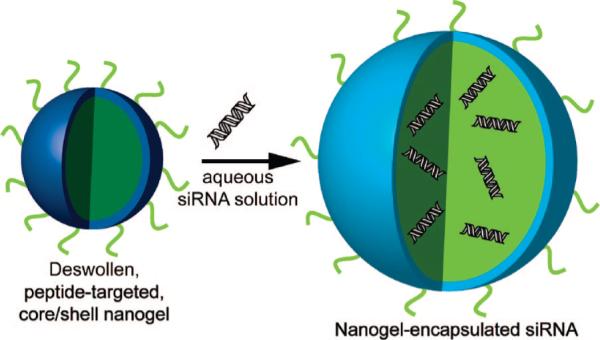
Noncovalent Encapsulation of siRNA in Peptide-Targeted Core/Shell Nanogels
MATERIALS AND METHODS
All materials were purchased from Sigma-Aldrich (St Louis, MO) and used as received unless otherwise noted.
Nanogel Core Synthesis
Nanogel core particles were synthesized by free-radical precipitation polymerization, as previously reported (32). The use of thermally phase separating polymers enables the use of precipitation polymerization for the synthesis of highly monodispersed nanogels (32). The molar composition was 98% N-isopropylmethacrylamide (NIPMAm), 2% N,N′-methylenebis(acrylamide) (BIS), with a total monomer concentration of 140 mM. The solution also contained a small amount (∼0.1 mM) of acrylamidofluorescein (AFA) to render the nanogels fluorescent for visualization via confocal microscopy (30, 32). In a typical synthesis, 100 mL of a filtered, aqueous solution of NIPMAm, BIS, and sodium dodecyl sulfate (SDS, 8 mM total concentration) was added to the reaction flask, which was then heated to 70 °C. The solution was purged with N2 gas and stirred vigorously until the temperature remained stable. The AFA was added, and after 10 min the reaction was initiated by the addition of a 1 mL solution of 800 mM ammonium persulfate (APS) to make the final concentration of APS in the reaction ∼8 mM. The solution turned turbid, indicating successful initiation. The reaction was allowed to continue for 4 h under an N2 blanket. After synthesis, the solution was filtered through Whatman filter paper to remove a small amount of coagulum.
Nanogel Shell Synthesis
The core nanogels described above were used as seeds for the addition of a hydrogel shell in a seeded precipitation polymerization scheme. The detailed procedure of the shell synthesis has been reported previously (32). Briefly, 10 mL of the core nanogel solution and 0.0577 g of SDS were first added to a three-neck round-bottom flask and heated under N2 gas to 70 °C. A 50 mM monomer solution with molar ratios of 97.5% NIPMAm, 2% BIS, and 0.5% aminopropyl methacrylate (APMA, Polysciences, Warrington, PA) was prepared in 39.5 mL of dH2O. The solution was added to the three-neck round-bottom flask, and the temperature was stabilized at 70 °C while continuously stirring. The reaction was initiated by a 0.5 mL aliquot of 0.05 M APS. The reaction proceeded for 4 h under N2 gas. Following the synthesis, the solution was filtered through Whatman filter paper, and the nanogels were purified by centrifugation followed by resuspension in dH2O.
Nanogel Characterization
Multiangle laser light scattering (MALLS) (Wyatt Technology Corporation, Santa Barbara, CA) detection following asymmetric field flow fractionation (AFFF) was used to determine the distribution of z-average radii (Rz) for all nanogels. For all separations, a cross-flow of 0.30 mL/min was used with a channel flow of 1.0 mL/min. The MALLS detector is equipped with a Peltier device to maintain a flow cell temperature of 25 °C and collects scattered light from 16 different fixed angles to determine the Rz of the nanogels. By measuring Rz as a function of elution time, we constructed a chromatogram that permits the determination of the weight fraction of nanogels as a function of radius, thereby providing a sample polydispersity. ASTRA 5.1.5.0 software was used to determine Rz values using the Debye fit method. The core/shell nanogels synthesized using the methods described above were determined to have Rz values of ∼54 nm with size polydispersities of <10%, as described previously (32); representative AFFF/MALLS data are shown in Supporting Information.
Characterization of the refractive increment (dn/dc) of nano-gels was performed to determine particle molecular weight by static light scattering. Differential refractive index analysis (dRI, OptiLab rEX, Wyatt Technologies, Inc.) was performed in batch mode. To ensure accurate data, the refractive index was calibrated prior to each measurement using sodium chloride concentrations ranging from 0.1 mg/mL to 15.0 mg/mL. All nanogel dilutions were prepared in dust-free vials, which were rinsed sequentially with deionized water, absolute ethanol, and HPLC-grade acetone. Nanogels were resuspended in distilled, deionized water over a concentration range from 2.5 × 10−6 g/mL to 3.75 × 10−4 g/mL. The use of MALLS in conjunction with the rEX differential refractometer permitted the measurement of the z-average molecular mass (Mz) from the determined dn/dc values and the angle-dependent light scattering data.
YSA Synthesis
The YSA peptide (YSAYPDSVPMMSC) was synthesized using standard Fmoc chemistry as described previously (46). Peptide synthesis was carried out by K. D. Clark, University of Georgia. Following synthesis, the peptide was cleaved from the resin and deprotected for 4 h in reagent K after air-drying. The peptide was purified using a series of 5 mL injections onto a preparatory HPLC column (10-m; particle size, 21.2 mm 25 cm, Jupiter C18; Phenomenex Inc., Torrance, CA) using HPLC-grade H2O and a linear gradient of acetonitrile (0−70 min, 10−80%) at 5 mL per min. Both the acetonitrile and H2O contained 0.05% trifluoroacetic acid. The desired peak was identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry, and the peaks from multiple runs were pooled, lyophilized, and stored at 4 °C in solid form. A scrambled form (SCR) of the YSA peptide (DYPSMAMYSPSVC) was also synthesized via this method for use as a control. On other occasions, the YSA and SCR peptides were purchased from GenScript Corp (Piscataway, NJ).
Peptide Conjugation
In this study we produced a maleimide-functionalized nanogel through the EDC coupling of ε-maleimidocaproic acid (EMCA) to the primary amines in the shell of the nanoparticle. As described in the nanogel shell synthesis, primary amines were introduced through the copolymerization of APMA (0.5% molar ratio). Given that APMA is efficiently incorporated at these low molar ratios, we can estimate the amine equivalents available for bioconjugation (∼2.2 × 10−6 amines per 88.3 mg of lyophilized particles). From this estimate, peptide coupling was performed by introducing YSA peptide in a 1:1 molar ratio with amine (YSA molecular weight = 1450.66 g/mol). The YSA peptide was then conjugated to the nanogels via maleimide coupling to the cysteine residue on the C-terminal end of the peptides.
First, 88.3 mg of nanogels (∼2.2 × 10−6 amine equivalents) was resuspended in 35.0 mL of pH 6.0 MES buffer and allowed to shake for 2 h. A second solution was prepared where 4.4 × 10−6 mol (0.68 mg) of 1-ethyl-3-methyl-(3-dimethylaminopropyl)carbodiimide (EDC, Pierce, Rockford, IL), 4.4 × 10−6 mol (0.96 mg) N-hydroxysulfosuccinimide (NHSS), and 2.2 × 10−6 mol (0.46 mg) of EMCA were dissolved in 3.0 mL of pH 6.0 MES buffer. This solution was reacted for 30 min at room temperature to activate the EMCA acid groups, which permits amide coupling to take place between the EMCA acid groups and the amines on the nanogel surface. This activated EMCA solution was then added to the nanogel solution and reacted for 2 h on a shaker table. The nanogels were centrifuged three times to remove any unreacted material, with resuspension in pH 6.0 MES buffer following each centrifugation. Finally, 3.2 mg of the appropriate peptide was added to the activated nanogels and reacted overnight. Peptide-functionalized nanogels were purified by centrifugation and resuspended in distilled, deionized water.
The number of bioconjugated YSA targeting peptides per particle was estimated by considering the number of primary amines available for conjugation and the number density of nanogels used during bioconjugation (as measured by static light scattering). Through differential refractometry, the nanogel refractive increment was determined to be 0.176 ± 0.002 mL/g. Measurement of z-average molecular weight through multi-angle static light scattering provided the z-average mass of nonconjugated particles, Mz = 2.19 × 107 g/mol (1° Debye fitting, 0.1% fit error). Thus, a total mass of 88.3 mg of lyophilized particles used during conjugation is equivalent to 2.43 × 1015 particles. Assuming a 50% peptide conjugation efficiency (47) and 2.2 × 10−6 amine equivalents available for bioconjugation, we conservatively estimate a peptide density of ∼225 YSA peptides/particle.
In Vitro siRNA Encapsulation and Release
Our group employs a “breathing-in” method for the encapsulation of various macromolecules within nanogels. In a typical method, lyophilized nanogels are resuspended in an aqueous solution containing the macromolecule to be loaded. Importantly, this is done using a loading solution volume that is almost completely imbibed by the swelling nanogels. In this fashion, the hydrogel network imbibes the payload with high efficiency and without relying on simple equilibrium partitioning to determine the maximum loading level. To determine the rate of siRNA release from nanogels loaded in this fashion, a mixture of oligonucleotide was prepared containing 0.250 mL of 20 μM siGLO red transfection indicator and 1.00 mL of 20 μM siGENOME Lamin A/C control siRNA (Dharmacon, Lafayette, CO). Particles were resuspended in this mixture at a concentration of 4 mg per 250 μL siRNA solution. This concentration of particles is near the solubility limit for the nanogels in PBS, ensuring a high degree of solvent and solute uptake into the hydrogel network. The particles were allowed to resuspend for 12 h at room temperature while shaking.
The encapsulation efficiency was determined via ultracentrifugation of the nanogel loading solution and measurement of supernatant siRNA concentration by UV-vis spectroscopy (Shimadzu UV-1601). The moles of siRNA in the loading solution (msiRNA,loading) and in the supernatant (msiRNA,supernatant) were determined via interpolation from a separately constructed standard curve of absorbance vs concentration (R2 > 0.99). The encapsulation efficiency (EE) of the system could then be calculated through analysis of the amount of siRNA in the loading solution and the remaining moles of siRNA in the supernatant after nanogel swelling was complete, as illustrated by eq 1 and in similar encapsulation experiments (48).
| (1) |
The release of solutes from nanogels was performed in 10% serum to simulate physiological conditions. Release experiments were performed by dispersing 200 μL of loaded nanogels in 2.20 mL of 0.01 M phosphate-buffered saline containing 10% fetal bovine serum (equilibrated at 37 °C) in 3.2 mL of polycarbonate centrifuge tubes (Beckman Coulter, Fullerton, CA). The nanogel suspension was allowed to incubate at 37 °C while being shaken. At specific time points, the tubes were centrifuged for 90 min at 687 000g (at 37 °C), and an aliquot of supernatant (0.75 mL) was removed for UV–vis analysis. This volume was replaced with fresh buffer. Upon centrifugation, the gel pellet had a homogenously distributed bright pink color, indicating significant retention of siRNA throughout the experiment. The cumulative siRNA released was calculated by calculating the total moles detected in the supernatant as a function of time, as described in eq 2.
| (2) |
All release studies were performed in triplicate for statistical analysis, using identical nanogel loading and release conditions.
Zeta-Potential Determination
Excluding the 0.5 mol% APMA copolymerized into the shell of our nanogel particles, the nanogels are composed of largely nonionic monomers. To confirm their suspected electroneutrality, which should be critical for reducing nonspecific cell and protein interactions, we measured the zeta-potential of both YSA-conjugated and nonconjugated core/shell nanogels (Zeta-Sizer Nano, Malvern, U.K.). All nanoparticles used in this investigation demonstrated zeta-potential values < +0.300 mV, suggesting that they are only weakly charged and should therefore not interact strongly with serum proteins or cell surfaces via Coulombic forces.
Cell Culture
Hey cells were provided by Gordon B. Mills, Department of Systems Biology, the University of Texas, M. D. Anderson Cancer Center. Hey cells were cultured in RPMI 1640 (Mediatech, Manassas, VA) supplemented with 10% v/v heat-inactivated fetal calf serum (Invitrogen), 2 mM l-glutamine (Mediatech), 10 mM HEPES buffer (Mediatech), penicillin (100 U/mL), and streptomycin (100 μg/mL). The BG-1 cell line was provided by Julie M. Hall and Kenneth S. Korach, Receptor Biology Section, Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental Health Sciences, NIH, Division of Intramural Research, Environmental Disease and Medicine Program, Research Triangle Park, NC. BG-1 cells were propagated in DMEM:F12/50:50 (Mediatech) supplemented with 10% v/v heat-inactivated fetal calf serum, penicillin, and streptomycin.
siRNA Encapsulation for Cell Studies
Using the “breathing-in” method for encapsulation (as described above), dried nanogels were reswollen in the presence of the siRNA, thereby imbibing the solute within the hydrogel network. In a typical procedure for in vitro cell delivery, a 20 μM solution (250 μL) of a fluorescent siRNA transfection indicator, siGLO (Dhar-macon), or EGFR siRNA (Dharmacon, Lafayette, CO) was prepared in phosphate-buffered saline (PBS). Lyophilized nano-gels were dissolved in the siRNA solution at a concentration of 4 mg in 250 μL and allowed to shake overnight at room temperature. Importantly, this nanogel concentration results in nearly all of the solvent being taken up by the nanogels. This volume-filling approach ensures a maximal uptake of siRNA within the nanogels. After being shaken, the nanogels were centrifuged to remove any free siRNA and resuspended in PBS. A standard curve for increasing concentrations of siRNA was made by measuring the absorbance at 260 nm using a Shimadzu UV 1601 spectrophotometer. After siRNA was encapsulated in the nanogels, they were centrifuged, and the absorbance of the supernatant was measured to determine the amount of incorporated siRNA.
Cell Transfection Using Nanogels
Hey or BG-1 cells were plated onto an eight-well chamber slide (5 × 103 cells/well) and the cells allowed to adhere overnight at 37 °C in a 5% CO2 atmosphere. After the wells were washed with PBS and the media were replaced, siGLO-loaded/YSA-conjugated nanogels, unloaded YSA-conjugated nanogels, pNIPMAm nanogels, or siGLO only were added to wells. Cells were incubated in each case for 4 h. In experiments where preincubation of ephrin-A1 was used to initiate internalization and degradation of EphA2, ephrin-A1 was added to the media at a final concentration of the ligand of 2 μg/mL. After incubation, the cells were washed with PBS, and the medium replaced. For fixation prior to confocal imaging, the cells were incubated with 2% (v/v) paraformaldehyde for 30 min.
Immunoblotting
Hey cells were plated into six-well plates (5 × 105/well) and allowed to adhere overnight at 37 °C, 5% CO2. The cells were lysed with 100 μL of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA (Fisher), 2 mM EGTA (Fisher), 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM β-gycerol phosphate, 1 mM phenylmethanesulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1% Triton X-100, and 5% glycerol), and the cell lysates were sonicated four times for 5 s each. The lysates were cleared by centrifugation at 11 000g rcf for 15 min at 4 °C. Cell lysates were prepared for analysis by the addition of an equal volume of Laemmli 2X sample buffer. The samples were heated to 95 °C for 5 min to denature the proteins. The proteins were separated on a 10% SDS-PAGE gel and transferred onto nitrocellulose. The blots were blocked with either 5% nonfat dry milk (NFDM) or 5% bovine serum albumin (BSA) in 10 mM Tris-buffered saline, pH 7.5, plus 1% Tween 20 (TBST, BioRad), for 1 h at room temperature. The blots were probed with anti-EGFR antibody (Cell Signaling, Danvers, MA; cat. no. 4405) or with a β-actin antibody (Millipore, Billerica, MA; Mab1501) diluted in 5% NFDM or 5% BSA overnight, with shaking at 4 °C. For EphA2 detection, the blots were probed with an anti-EphA2 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA; sc-294). The blots were washed three times with TBST and probed with goat antirabbit IgG (Santa Cruz, sc-2004) or with goat antimouse IgG (Santa Cruz, sc-2005) linked to horseradish peroxidase (HRP). Bands were visualized on film (Pierce) using the ECL reagent, SuperSignal West Pico (Pierce).
Confocal Microscopy
A Zeiss LSM510 confocal microscope was used to take cell images. Cells were incubated with nanogels for 4 h. After 4 h, the cells were washed and then fixed on the slide. An Ar+ laser was used to excite the AFA-labeled nanogels, whereas a HeNe laser was used to excite the fluorescently labeled siGLO. LSM510 software was used to view the images.
Flow Cytometry
Hey cells were plated at 2.5 × 105 cells/well in a 12-well, cell culture plate. Cells were allowed to adhere overnight in an incubator at 37 °C in a 5% CO2 atmosphere. Cells were washed, and fresh medium was added containing YSA-pNIPMAm or SCR-pNIPMAm nanogels at a concentration of 0.8 mg/mL and incubated for 4 h. Following incubation, the cells were washed with PBS and removed from the plate by trypsin–EDTA treatment. The cells were washed with PBS and fixed with 2% (v/v) paraformaldehyde. Cells were analyzed using a LSR Flow Cytometer (BD Biosciences). Data analysis was carried out using FlowJo software.
Toxicity Studies
Trypan Blue Exclusion Assay
Hey cells were plated onto an eight-well chamber slide (1 × 104 cells/well) and allowed to adhere overnight at 37 °C and 5% CO2. The medium was removed, the wells were washed with PBS, and the medium was replaced. PNIPMAm nanogels, YSA-conjugated nanogels, and SCR-conjugated nanogels were added to cells and incubated for 72 h. Untreated cells were used as controls. After 72 h, the cells washed with PBS, and a 1:1 solution of trypan blue was added to each well. After 1 min, the trypan blue was removed, the cells were washed with PBS, fixed with 2% (v/v) paraformaldehyde, and air-dried. Each well was then viewed via bright field microscopy to determine the number of stained (dead) versus unstained cells. Five fields were viewed for each treatment. Representative images are shown in Supporting Information.
Tox 8 Assay
Hey cells were plated onto 96-well plates (1 × 104 cells/well) and allowed to adhere overnight at 37 °C and 5% CO2. The media was removed, and the cells were washed with PBS followed by replacement of the medium. Cells incubated with EGFR siRNA-loaded YSA-labeled nanogels, unloaded YSA-labeled pNIPMAm nanogels, unlabeled pNIPMAm, or YSA peptide alone were tested using this assay. The cells were incubated under all conditions for 4 h. The cells were then washed with PBS, the medium was replaced, and the cells were incubated for an additional 72 h in medium. The Tox 8 reagent (Sigma) was added to the cells according to the manufacturer's instructions. The fluorescence at 590 nm was read after 1 h and the extent of cellular viability/proliferation determined.
RESULTS AND DISCUSSION
The nanogels described in this contribution were developed around two main design criteria, as depicted in Scheme 1. For this application, both peptide-based targeting of ovarian cancer and efficient encapsulation and delivery of RNA inhibitors (RNAi's) are required. The core/shell nanogels synthesized using the methods described above were determined to have Rz values of ∼54 nm with size polydispersities of <10%, as described previously (32). Representative AFFF/MALLS chromatograms for both the core and core/shell nanogels are shown in Supporting Information. To determine the time scale for retention of siRNA within the pNIPMAm nanogels, we investigated siRNA leakage using simulated physiological conditions. As described above, nanogels were loaded using a model mixture of siRNA, containing both the siGLO red transfection indicator and the siGENOME Lamin control. The nanogel was observed to encapsulate the siRNA with high efficiency (93 ± 1%), which is equivalent to a loading level of 1.6 wt% or 16 μg siRNA/mg of nanogels. As shown in Figure 1, only ∼33% of the siRNA is observed to leak from the nanogels within the first 12 h (67% retained). Indeed, this approximate level of retention persists out to 35 h, suggesting very efficient entrapment of the siRNA within the nanogel network. Retention of this magnitude is promising for intravenous oligonucleotide delivery given previously determined time scales (∼6 h) for extravasation via the enhanced permeability and retention effect (49).
Figure 1.
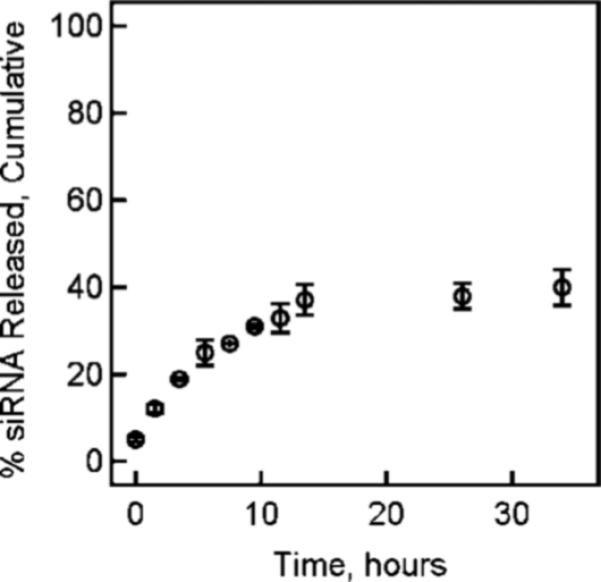
siRNA release profile from nanogels at 37 °C in PBS containing 10% fetal bovine serum. The error bars represent ± 1 standard deviation about the mean value (n = 3).
To establish the efficacy of targeting in vitro, we determined the uptake of nanogels by two ovarian cancer cell lines, Hey and BG-1. We previously demonstrated high expression of EphA2 by Hey cells and low expression of the receptor by the BG-1 cell line (see Supporting Information) (50). Because of these differences in EphA2 expression, we expected to see higher levels of nanogel uptake via receptor-mediated endocytosis with Hey cells as compared to BG-1 cells. Furthermore, we expected that the degree of siRNA delivery to those cells would be dependent on the cell type and the presence of the peptide ligand. To load siRNA into the nanocarrier, lyophilized nanogels were loaded with siGLO (a fluorescently labeled siRNA delivery tracker) by reswelling them in a concentrated solution of the siRNA, as described above. To obtain a relative concentration of the siGLO taken up by the nanogels, absorbance measurements were compared to a standard curve of siGLO in solution (R2 > 0.99). We determined in a series of three trials that 80−95% (by mass) of the siGLO was incorporated into the nanogels by this method, in agreement with the loading levels calculated in the release kinetics experiment described above.
Following loading with siGLO, nanogels were incubated with either Hey (high EphA2 expression) or BG-1 (low EphA2 expression) cells in order to compare the levels of targeted uptake by ovarian cancer cells. Uptake of the nanogels into the cells was followed using a fluorescent tag (AFA) incorporated into the nanogel core as well as by the fluorescence of the siGLO. In previous studies, we determined that high levels of nanogel uptake by cells occurred after 4 h (data not shown). As a result, cells were incubated for 4 h with siGLO-loaded/YSA-conjugated nanogels to monitor specific targeting to EphA2. Unloaded YSA-conjugated nanogels, nontargeted pNIPMAm nanogels, and siGLO only were used as controls, with identical 4-h incubation times. In all experiments described in this manuscript, we maintained a constant nanogel/cell ratio of 1 mg nanogels/5 × 105 cells. For siRNA-loaded nanogels, this corresponds to 16.6 μg siRNA/5 × 105 cells. After incubation, the cells were washed and the slides fixed for confocal microscopy imaging. Figure 2a shows that Hey cells targeted with YSA-conjugated nanogels have high levels of nanogel uptake as indicated by the presence of green fluorescence. At this time point, siGLO was retained at high levels within the internalized nanogels as indicated by the cell-localized red fluorescence. Merging of the two fluorescence channels showed strong overlap, further indicating delivery of the siGLO by the nanogels into the Hey cells. Hey cells incubated with YSA-targeted but unloaded nanogels showed strong green fluorescence, indicating cell uptake. A small amount of nontargeted uptake was observed for nanogels lacking the YSA peptide. Note that it was extremely difficult to find evidence of nonspecific uptake, and the fluorescence shown in the figure represents the appearance of the rare uptake event observed and does not represent the overall fluorescence from the entire population of cells. When Hey cells were incubated with siGLO alone, no cell-localized red fluorescence was detected; this is expected since RNA does not easily permeate the cell membrane in the absence of a carrier vehicle (16, 17).
Figure 2.

Confocal microscopy images of (a) Hey cells and (b) BG-1 cells following exposure to siGLO-loaded/YSA-conjugated pNIPAMAm nanogels, YSA-nanogels alone, unlabeled nanogels, and siGLO alone. The AFA and siGLO fluorescence channels are shown individually, along with a merge of the two channels. Scale bar = 40 μm.
Targeting experiments were also performed using low EphA2 expressing BG-1 cells (Figure 2b). Decreased levels of green fluorescence were observed in BG-1 cells when compared to the fluorescence observed in the Hey cell cultures. The lower amount of nanogel uptake by the BG-1 cells was most likely because of the reduced EphA2 receptor expression; we have demonstrated a ∼2.5-fold difference in EphA2 expression levels between these two cell lines (Supporting Information). Control studies using nontargeted pNIPMAm nanogels or siGLO only showed no fluorescence in either the green or red fluorescent channels. These results indicate that the YSA peptide imparts targeting properties to the nanogels in the case of both the high (Hey) and low (BG-1) EphA2 expressing cells, and that the amount of nanogel uptake was dependent upon the level of EphA2 receptor expression. These results also indicate that nonspecific or nontargeted uptake of nanogels by cultured cells is low, and that the siRNA is unable to penetrate the cell membrane in the absence of a carrier vehicle. Together, these initial results illustrate the promise of the targeted nanogel construct for targeted delivery of oligonucleotide cargo.
To further establish the mechanism of nanogel targeting and uptake, we took advantage of the known receptor internalization properties of the EphA2 receptor. Specifically, it has been shown that binding of ephrin-A1, a ligand for EphA2, to EphA2 receptor causes internalization and degradation of the receptor–ligand complex (51). Figure 3 shows the results of studies wherein this receptor recycling process was used to establish the nanogel target by preincubating Hey cells with ephrin-A1 before YSA-targeted nanogel incubation. We hypothesized that if uptake of nanogels is EphA2 receptor-mediated, YSA-targeted uptake after cell exposure to ephrin-A1 should be reduced, as the EphA2 receptor will be internalized and less available for binding to the nanogels. Hey cells were incubated overnight in an eight-well chamber slide. Two μg/mL of ephrin-A1 was added, and the cells were incubated for 1 h at 37 °C. After ephrin-A1 incubation, siGLO-loaded/YSA-conjugated nanogels were added to both ephrin-A1 and control (PBS) treated wells. The cells were incubated for 4 h, washed, and processed for imaging. Figure 3 shows the nanogel uptake in Hey cells preincubated with ephrin-A1. Whereas these cells (top three panels) show some uptake of nanogels and encapsulated siGLO, the amount of uptake is greatly diminished compared with untreated cells (lower three panels). These results suggest that YSA-conjugated uptake by Hey cells is conducted to a large extent through EphA2; however, a small amount of uptake may occur through nonspecific mechanisms or via binding of YSA to other Eph receptors (35). This is not surprising, given the fact that ephrin and various small molecule ephrin mimics display binding affinities for multiple receptors of the Eph family (52, 53). Flow cytometry was also used to establish the EphA2-associated binding of the peptide-targeted nanogels. In this case, a scrambled (SCR) peptide sequence (DYPSMAMYSPSVC) possessing the same amino acid composition of the YSA peptide was tethered to the nanogels. The resultant nanogels should therefore possess the same physicochemical surface properties as the YSA-labeled nanogels but should not specifically bind to the EphA2 receptor. Figure 4 shows the results of these studies, where cells incubated with YSA-labeled nanogels display ∼10-fold greater fluorescence relative to those incubated with SCR-labeled nanogels. Furthermore, the fluorescence signal associated with cells incubated with SCR-labeled nanogels is only slightly greater than the cell autofluorescence background signal.
Figure 3.
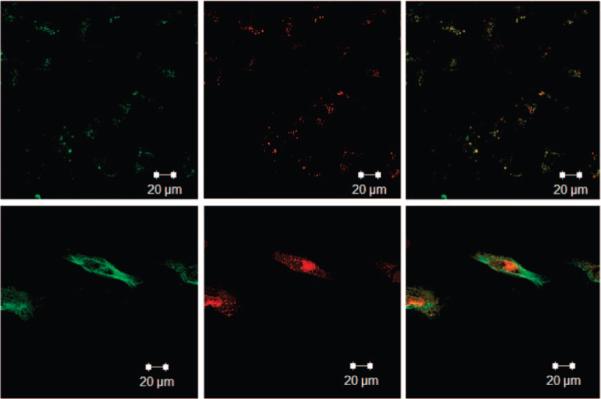
Confocal microscopy images of Hey cells (top) following exposure to siGLO-loaded/YSA-conjugated nanogels after 1 h ephrin-A1 incubation, and (bottom) following exposure to siGLO-loaded/YSA-conjugated nanogels alone. The fluorescence channels shown are as in Figure 2. Scale bar = 20 μm.
Figure 4.

Flow cytometry data comparing (a) cell autofluorescence (red) vs cells incubated with YSA-pNIPMAm nanogels (green) and (b) cell autofluorescence (red) vs cells incubated with SCR-pNIPMAm nanogels (green). The differential uptake between the YSA- and SCR-labeled nanogels indicates the Eph2A receptor-specific binding and uptake pathway.
The effect of nanogels on tumor cell toxicity and proliferation was examined using two cell viability assays. For the trypan blue exclusion assay, Hey cells were incubated with pNIPMAm nanogels, YSA-conjugated nanogels, or SCR-conjugated nano-gels for 72 h. The cells were then washed with PBS, and trypan blue was added to the cells. Five fields were observed via microscopy for each treatment group. Blue cells, indicating dead cells, were not observed in any of the fields examined for any of the treatment groups. Representative images are shown in the Supporting Information. To more precisely establish any negative effects associated with nanogel-based delivery, we used the Tox 8 viability proliferation assay. Hey cells were incubated in 96-well plates overnight, and nanogels were delivered and removed via the usual method. In the gene silencing data shown below, we chose siRNA targeting epidermal growth factor receptor (EGFR); knockdown of this receptor is nonlethal but has clinical relevance in the treatment of drug resistant ovarian carcinomas (54). This siRNA was therefore used in these toxicity studies as well. Again, we maintained a ratio of 1 mg nanogels/5 × 105 cells for all samples. For the EGFR siRNA-loaded nanogels, this corresponds to 16.6 μg siRNA/5 × 105 cells. Wells were washed with PBS, and 100 μL of cell culture medium was added to the wells. After 72 h, Tox 8 was added to the cells according to the manufacturer's instructions, and the cell viability was determined spectrophotometrically. This analysis (Figure 5) revealed no significant difference for any treatment when compared with control (untreated) cells, although exposure to nontargeted nanogels and siRNA-containing nanogels showed slight decreases in viability; the origin of this effect is currently under investigation. These results indicate that treatment of Hey cells with targeted nanogels does not greatly inhibit cell proliferation, indicating limited toxicity of the nanogels under these conditions.
Figure 5.
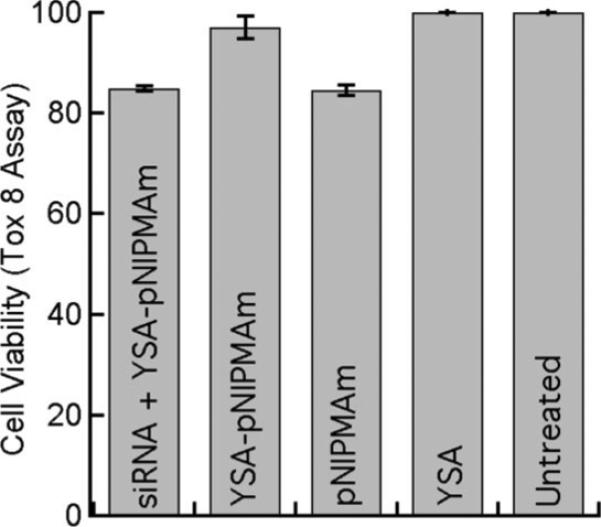
Cell viability as determined with a Tox 8 assay for untreated Hey cells and Hey cells followinga4h incubation with EGFR siRNA-loaded YSA-labeled nanogels, YSA-labeled pNIPMAm nanogels, unlabeled pNIPMAm, or YSA peptide alone. Error bars represent ±1 standard deviation about the average value (n = 3).
These promising preliminary studies clearly illustrate the efficacy of peptide-targeted delivery of siRNA cargos via nanogel carriers. The lack of toxicity observed is of particular interest, given the high toxicity observed for some cationic lipid-based siRNA targeting methods, which limits the maximum doses that can be delivered, and also compromises the potential for in vivo delivery. In the present studies, a ratio of 16 μg siRNA or 1 mg nanogels/5 × 105 cells was used throughout without significant toxicity being observed. These concentrations are somewhat higher than those suggested for common commercial regents such as RNAiFect (Qiagen) or DharmaFECT (Dharmacon), suggesting that the nanogel approach is capable of delivering siRNA amounts at or above those achievable by optimized commercial reagents. Another complicating factor in current methodologies is the frequent need for cellular delivery under serum free conditions; serum lipids and proteins compromise the stability of many liposomal formulations, making their efficacy significantly lower. In the studies described herein, we have illustrated that delivery of siGLO is excellent in serum-containing medium, further establishing the promise of this construct. As a final preliminary test of the efficacy of the approach, we performed a limited investigation of siRNA-based silencing. Clearly, any delivery approach must deliver functional siRNA to the cell interior in order for it to be truly useful. If the nanogel carrier were unable to protect the cargo against degradation in the endosomal or lysosomal compartments, or if the nanogels were unable to escape from endosomes in order to deliver the siRNA to the cytosol, the amount of RNAi would be very low. Thus, we have undertaken a preliminary study of gene silencing to illustrate a minimal requirement for siRNA delivery: the functional silencing of a target mRNA.
As described above, we chose siRNA targeting EGFR; knockdown of this receptor is nonlethal but has clinical relevance in the treatment of drug resistant ovarian carcinomas (54). To determine if we could effectively knockdown EGFR in vitro, EGFR siRNA was encapsulated at a concentration of 16.6 μg of EGFR siRNA/mg of nanogels, using the loading technique described above. Nanogels were then added to Hey cells (1 mg of nanogels or 16.6 μg siRNA/5 × 105 cells) and incubated at 37 °C for 4 h. The unincorporated nanogels were then removed by washing the cells, and the medium was replaced. Controls included cells incubated with unloaded YSA-targeted nanogels, pNIPMAm nanogels, and untreated cells. All cells were harvested at 48 h and assayed for EGFR expression by immunoblotting, as described in the Experimental Section. Figure 6 shows the results of this experiment; a significant reduction in EGFR expression is observed under these conditions relative to all controls (p < 0.01 relative to untreated sample by paired t test, n = 3). A small, statistically insignificant decrease in EGFR expression was noted in the unloaded, YSA-targeted nanogel control (p > 0.1). If this observation is indeed a real one, it may be due to cross talk between the EGFR and the EphA2 receptors, as described by Larsen and colleagues (55). In addition, a small decrease in EGFR expression was observed when cells were incubated with pNIPMAm nanogels alone, although the difference is not statistically significant (p > 0.3) in light of the large observed variability in expression.
Figure 6.
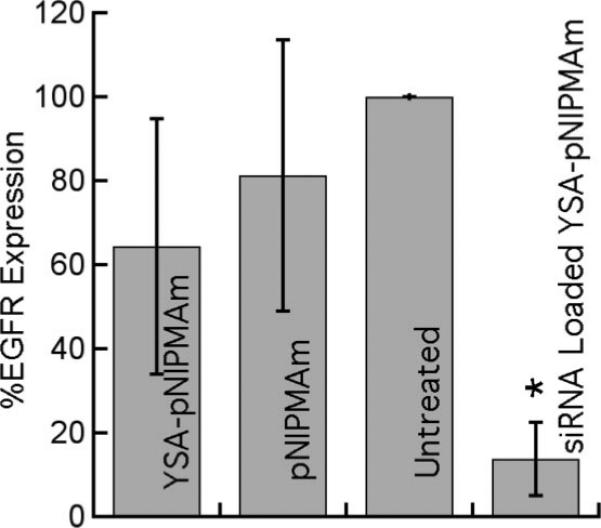
EGFR expression, as determined by immunoblot, in Hey cells following a 4 h incubation with either unloaded YSA-nanogels, unloaded nontargeted nanogels, or siRNA loaded YSA-nanogels. Untreated cells were set at 100% expression. All cells were harvested 48 h after removal of the nanogels. Error bars represent ±1 standard deviation about the average value (n = 3, *p < 0.01 relative to untreated sample).
These preliminary results illustrate that the targeted nanogels are capable of functional delivery of siRNA to ovarian carcinomas without overt toxic effects, and that the subsequently internalized siRNA is available for gene silencing. Whereas this preliminary demonstration is relatively focused in scope, it clearly shows the promise of the construct. We are currently exploring the generality of this approach in a detailed study of silencing timecourse, persistence, cell type dependence, dose response, and applicability to other known silencing targets.
CONCLUSIONS
Peptide-labeled nanogels with a high loading capacity for siRNA have been developed and can be effectively targeted to ovarian carcinomas by receptor–peptide binding. The encapsulated siRNA is transported into the cell interior, where it is available for gene silencing, as illustrated in this case by EGFR knockdown. Since the locus of siRNA-mediated gene silencing is the cytosol, the results are suggestive of the surprising conclusion that endosomal uptake of the nanogels is followed by endosomal escape, resulting in efficient transport/release of the siRNA to the cytosol. Whereas we do not currently know the exact mechanism by which endosomal escape occurs. It is plausible that the nanogels respond to endosomal changes in osmotic pressure and ionic strength by undergoing a volume change. This phenomenon, called osmotic swelling/deswelling (56) is fundamental to the phase behavior of gel networks and may serendipitously be responsible for the excellent delivery properties described above. In addition to the gene-silencing efficacy, the nanocarriers are demonstrated to be nontoxic under the conditions investigated and are effective even when delivered in serum-containing medium. As a result of these studies, we are currently investigating the fundamental mechanisms of nanogel endosomal release. Additionally, the gene silencing results are being validated in a broader study of RNAi with plans to extend their use to in vivo delivery and silencing in animal models.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge support from Emory–Georgia Tech Nanotechnology Center for Personalized and Predictive Oncology (5-40256-G11). L.A.L. acknowledges financial support from the DHHS (1 R21 EB006499-01). The ovarian cancer studies (E.B.D. and J.F.M.) were supported by the Georgia Cancer Coalition, The Debora Nash Harris Endowment Fund, and The Ovarian Cancer Institute. Thanks to Roman Mezencev for his help with the flow cytometry studies.
Footnotes
Supporting Information Available: Scanned immunoblot illustrating the expression levels of Eph2A receptor in Hey and BG-1 cells and representative AFFF/MALLS chromatograms showing the size and monodispersity of the core and core/shell nanogels, and representative trypan blue assay microscopy results. This material is available free of charge via the Internet at http://pubs.acs.org/BC.
LITERATURE CITED
- 1.Saad M, Garbuzenko OB, Ber E, Chandna P, Khandare JJ, Pozharov VP, Minko T. Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging? J. Controlled Release. 2008;130:107–114. doi: 10.1016/j.jconrel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003;21:1184–1191. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 3.Moses MA, Brem H, Langer R. Advancing the field of drug delivery: Taking aim at cancer. Cancer Cell. 2003;4:337–341. doi: 10.1016/s1535-6108(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 4.Derfus AM, Chen AA, Min DH, Ruoslahti E, Bhatia SN. Targeted quantum dot conjugates for siRNA delivery. Bioconjugate Chem. 2007;18:1391–6. doi: 10.1021/bc060367e. [DOI] [PubMed] [Google Scholar]
- 5.Lee SH, Bae KH, Kim SH, Lee KR, Park TG. Amine-functionalized gold nanoparticles as non-cytotoxic and efficient intracellular siRNA delivery carriers. Int. J. Pharm. 2008;364:94–101. doi: 10.1016/j.ijpharm.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 6.Kim S, Jeong JH, Lee SH, Kim S, Park TG. LHRH Receptor-Mediated Delivery of siRNA Using Polyelectrolyte Complex Micelles Self-Assembled from siRNA–PEG–LHRH Conjugate and PEI. Bioconjugate Chem. 2008;19:2156–2162. doi: 10.1021/bc800249n. [DOI] [PubMed] [Google Scholar]
- 7.Eavarone DA, Yu X, Bellamkonda RV. Targeted drug delivery to C6 glioma by transferrin-coupled liposomes. J. Biomed. Mater. 2000;51:10–14. doi: 10.1002/(sici)1097-4636(200007)51:1<10::aid-jbm2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 8.Lee RJ, Low PS. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J. Biol. Chem. 1994;269:3198–204. [PubMed] [Google Scholar]
- 9.van Nostrum CF. Polymeric micelles to deliver photosensitizers for photodynamic therapy. Adv. Drug Delivery Rev. 2004;56:9–16. doi: 10.1016/j.addr.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Zhang K, Fang H, Wang Z, Taylor JS, Wooley KL. Cationic shell-crosslinked knedel-like nanoparticles for highly efficient gene and oligonucleotide transfection of mammalian cells. Biomaterials. 2008;30:968–977. doi: 10.1016/j.biomaterials.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 11.Sun TM, Du JZ, Yan LF, Mao HQ, Wang J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29:4348–55. doi: 10.1016/j.biomaterials.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 12.Mao HQ, Leong KW. Design of polyphosphoester-DNA nanoparticles for non-viral gene delivery. Adv. Genet. 2005;53:275–306. [PubMed] [Google Scholar]
- 13.Vinogradov SV. Colloidal microgels in drug delivery applications. Curr. Pharm. Des. 2006;12:4703–4712. doi: 10.2174/138161206779026254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng J, Teply Benjamin A, Sherifi I, Sung J, Luther G, Gu Frank X, Levy-Nissenbaum E, Radovic-Moreno Aleksandar F, Langer R, Farokhzad Omid C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–76. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das M, Mardyani S, Chan WCW, Kumacheva E. Biofunctionalized pH-responsive microgels for cancer cell targeting: Rational design. Adv. Mater. (Weinheim, Ger.) 2006;18:80–83. [Google Scholar]
- 16.Akhtar S, Benter IF. Nonviral Delivery of synthetic siRNAs in vivo. J. Clin. Invest. 2007;117:3623–3632. doi: 10.1172/JCI33494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sepp-Lorenzino L, Ruddy M. Challenges and Opportunities for Local and Systemic Delivery of siRNA and Antisense Oligonucleotides. Clin. Pharmacol. Ther. 2008;84:628–632. doi: 10.1038/clpt.2008.174. [DOI] [PubMed] [Google Scholar]
- 18.Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110:563–74. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 19.Spagnou S, Miller AD, Keller M. Lipidic carriers of siRNA: Differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry. 2004;43:13348–13356. doi: 10.1021/bi048950a. [DOI] [PubMed] [Google Scholar]
- 20.Jeong JH, Mok H, Oh YK, Park TG. siRNA Conjugate Delivery Systems. Bioconjugate Chem. 2009;20:5–14. doi: 10.1021/bc800278e. [DOI] [PubMed] [Google Scholar]
- 21.Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang QQ, Storm G, Molema G, Lu PY, Scaria PV, Woodle MC. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004;32:e149. doi: 10.1093/nar/gnh140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song E, Zhu P, Lee S-K, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005;23:709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 23.Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G, Hogrefe RI, Palchik G, Chang EH. Materializing the Potential of Small Interfering RNA via a Tumor-Targeting Nanodelivery System. Cancer Res. 2007;67:2938–2943. doi: 10.1158/0008-5472.CAN-06-4535. [DOI] [PubMed] [Google Scholar]
- 24.Mok H, Park TG. Self-crosslinked and reducible fusogenic peptides for intracellular delivery of siRNA. Biopolymers. 2008;89:881–8. doi: 10.1002/bip.21032. [DOI] [PubMed] [Google Scholar]
- 25.Juliano RL. Peptide-oligonucleotide conjugates for the delivery of antisense and siRNA. Curr. Opin. Mol. Ther. 2005;7:132–136. [PubMed] [Google Scholar]
- 26.Kang HM, DeLong R, Fisher MH, Juliano RL. Tat-conjugated PAMAM dendrimers as delivery agents for antisense and siRNA oligonucleotides. Pharm. Res. 2005;22:2099–2106. doi: 10.1007/s11095-005-8330-5. [DOI] [PubMed] [Google Scholar]
- 27.Simeoni F, Morris MC, Heitz F, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31:2717–2724. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zatsepin TS, Turner JJ, Oretskaya TS, Gait MJ. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr. Pharm. Des. 2005;11:3639–3654. doi: 10.2174/138161205774580769. [DOI] [PubMed] [Google Scholar]
- 29.Zhang CL, Tang N, Liu XJ, Liang W, Xu W, Torchilin VP. siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J. Controlled Release. 2006;112:229–239. doi: 10.1016/j.jconrel.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nayak S, Lee H, Chmielewski J, Lyon LA. Folate-Mediated Cell Targeting and Cytotoxicity Using Thermoresponsive Microgels. J. Am. Chem. Soc. 2004;126:10258–10259. doi: 10.1021/ja0474143. [DOI] [PubMed] [Google Scholar]
- 31.Shin Y, Chang JH, Liu J, Williford R, Shin YK, Exarhos GJ. Hybrid nanogels for sustainable positive thermosensitive drug release. J. Controlled Release. 2001;73:1–6. doi: 10.1016/s0168-3659(01)00247-4. [DOI] [PubMed] [Google Scholar]
- 32.Blackburn WH, Lyon LA. Size-controlled synthesis of monodisperse core/shell nanogels. Colloid Polym. Sci. 2008;286:563–569. doi: 10.1007/s00396-007-1805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berndt I, Popescu C, Wortmann F-J, Richtering W. Mechanics versus thermodynamics: swelling in multiple-temperature-sensitive core-shell microgels. Angew. Chem., Int. Ed. 2006;45:1081–1085. doi: 10.1002/anie.200502893. [DOI] [PubMed] [Google Scholar]
- 34.Jones CD, Lyon LA. Synthesis and Characterization of Multiresponsive Core-Shell Microgels. Macromolecules. 2000;33:8301–8306. [Google Scholar]
- 35.Koolpe M, Dail M, Pasquale EB. An ephrin mimetic peptide that selectively targets the EphA2 receptor. J. Biol. Chem. 2002;277:46974–9. doi: 10.1074/jbc.M208495200. [DOI] [PubMed] [Google Scholar]
- 36.Lin YG, Han LY, Kamat AA, Merritt WM, Landen CN, Deavers MT, Fletcher MS, Urbauer DL, Kinch MS, Sood AK. EphA2 overexpression is associated with angiogenesis in ovarian cancer. Cancer. 2007;109:332–40. doi: 10.1002/cncr.22415. [DOI] [PubMed] [Google Scholar]
- 37.Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 2005;19:1884–6. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]
- 38.Han L, Dong Z, Qiao Y, Kristensen GB, Holm R, Nesland JM, Suo Z. The clinical significance of EphA2 and Ephrin A-1 in epithelial ovarian carcinomas. Gynecol. Oncol. 2005;99:278–86. doi: 10.1016/j.ygyno.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 39.Thaker PH, Deavers M, Celestino J, Thornton A, Fletcher MS, Landen CN, Kinch MS, Kiener PA, Sood AK. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin. Cancer Res. 2004;10:5145–50. doi: 10.1158/1078-0432.CCR-03-0589. [DOI] [PubMed] [Google Scholar]
- 40.Walker-Daniels J, Coffman K, Azimi M, Rhim JS, Bostwick DG, Snyder P, Kerns BJ, Waters DJ, Kinch MS. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate. 1999;41:275–80. doi: 10.1002/(sici)1097-0045(19991201)41:4<275::aid-pros8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 41.Zeng G, Hu Z, Kinch MS, Pan CX, Flockhart DA, Kao C, Gardner TA, Zhang S, Li L, Baldridge LA, Koch MO, Ulbright TM, Eble JN, Cheng L. High-level expression of EphA2 receptor tyrosine kinase in prostatic intraepithelial neoplasia. Am. J. Pathol. 2003;163:2271–6. doi: 10.1016/S0002-9440(10)63584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–52. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- 43.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumori-genesis of mammary epithelial cells. Cancer Res. 2001;61:2301–6. [PubMed] [Google Scholar]
- 44.Kataoka H, Igarashi H, Kanamori M, Ihara M, Wang JD, Wang YJ, Li ZY, Shimamura T, Kobayashi T, Maruyama K, Nakamura T, Arai H, Kajimura M, Hanai H, Tanaka M, Sugimura H. Correlation of EPHA2 overexpression with high microvessel count in human primary colorectal cancer. Cancer Sci. 2004;95:136–41. doi: 10.1111/j.1349-7006.2004.tb03194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito T, Masuda N, Miyazaki T, Kanoh K, Suzuki H, Shimura T, Asao T, Kuwano H. Expression of EphA2 and E-cadherin in colorectal cancer: correlation with cancer metastasis. Oncol. Rep. 2004;11:605–11. [PubMed] [Google Scholar]
- 46.Clark KD, Volkman BF, Thoetkiattikul H, King D, Hayakawa Y, Strand MR. Alanine-scanning mutagenesis of plasmatocyte spreading peptide identifies critical residues for biological activity. J. Biol. Chem. 2001;276:18491–6. doi: 10.1074/jbc.M100579200. [DOI] [PubMed] [Google Scholar]
- 47.Hermanson GT. Bioconjugate Techniques. 1st ed. Vol. 1. Academic Press; San Diego: 1996. [Google Scholar]
- 48.Betancourt T, Shah K, Brannon-Peppas L. Rhodamine-loaded poly(lactic-co-glycolic acid) nanoparticles for investigation of in vitro interactions with breast cancer cells. J. Mater. Sci. Mater. Med. 2009;20:387–395. doi: 10.1007/s10856-008-3594-z. [DOI] [PubMed] [Google Scholar]
- 49.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 50.Scarberry KE, Dickerson EB, McDonald JF, Zhang ZJ. Magnetic nanoparticle-peptide conjugates for in vitro and in vivo targeting and extraction of cancer cells. J. Am. Chem. Soc. 2008;130:10258–10262. doi: 10.1021/ja801969b. [DOI] [PubMed] [Google Scholar]
- 51.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2000;2:62–9. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 52.Noberini R, Koolpe M, Peddibhotla S, Dahl R, Su Y, Cosford ND, Roth GP, Pasquale EB. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J. Biol. Chem. 2008;283:29461–72. doi: 10.1074/jbc.M804103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Surawska H, Ma PC, Salgia R. The role of ephrins and Eph receptors in cancer. Cytokine Growth Factor Rev. 2004;15:419–33. doi: 10.1016/j.cytogfr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 54.Thaker PH, Yazici S, Nilsson MB, Yokoi K, Tsan RZ, He J, Kim SJ, Fidler IJ, Sood AK. Antivascular therapy for orthotopic human ovarian carcinoma through blockade of the vascular endothelial growth factor and epidermal growth factor receptors. Clin. Cancer Res. 2005;11:4923–33. doi: 10.1158/1078-0432.CCR-04-2060. [DOI] [PubMed] [Google Scholar]
- 55.Larsen AB, Pedersen MW, Stockhausen MT, Grandal MV, van Deurs B, Poulsen HS. Activation of the EGFR gene target EphA2 inhibits epidermal growth factor-induced cancer cell motility. Mol. Cancer Res. 2007;5:283–93. doi: 10.1158/1541-7786.MCR-06-0321. [DOI] [PubMed] [Google Scholar]
- 56.Saunders BR, Vincent B. Osmotic deswelling of microgel particles in the presence of free polymer. Prog. Colloid Polym. Sci. 1997;105:11–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.