

Conspectus and graphic
 Nitric oxide (NO), which is produced from L-arginine by the nitric oxide synthase (NOS) family of enzymes, is an important second-messenger molecule that regulates several physiological functions. In endothelial cells, it relaxes smooth muscle, which decreases blood pressure. Macrophage cells produce NO as an immune defense system to destroy pathogens and microorganisms. In neuronal cells, NO controls the release of neurotransmitters and is involved in synaptogenesis, synaptic plasticity, memory function, and neuroendocrine secretion.
Nitric oxide (NO), which is produced from L-arginine by the nitric oxide synthase (NOS) family of enzymes, is an important second-messenger molecule that regulates several physiological functions. In endothelial cells, it relaxes smooth muscle, which decreases blood pressure. Macrophage cells produce NO as an immune defense system to destroy pathogens and microorganisms. In neuronal cells, NO controls the release of neurotransmitters and is involved in synaptogenesis, synaptic plasticity, memory function, and neuroendocrine secretion.
NO is a free radical that is commonly thought to contribute to oxidative damage and molecule and tissue destruction, and thus it is somewhat surprising that it has so many significant beneficial physiological effects. However, the cell is generally protected from NO's toxic effects, except under certain pathological conditions in which excessive NO is produced. In that case, tissue damage and oxidative stress can result, leading to a wide variety of diseases, including rheumatoid arthritis, Alzheimer's disease, and Parkinson's disease, among others. In this Account, we describe research aimed at identifying small molecules that can selectively inhibit only the neuronal isozyme of NOS, nNOS. By targeting only nNOS, we attained the beneficial effects of lowering excess NO in the brain without the detrimental effects of inhibition of the two isozymes found elsewhere in the body (eNOS and iNOS).
Initially, in pursuit of this goal, we sought to identify differences in the second sphere of amino acids in the active site of the isozymes. From this study, the first class of dual nNOS-selective inhibitors was identified. The moieties important for selectivity in the best lead compound were determined by structure modification. Enhancement provided highly potent, nNOS-selective dipeptide amides and peptidomimetics, which were active in a rabbit model for fetal neurodegeneration. Crystal structures of these compounds bound to NOS isozymes showed a one-amino-acid difference between nNOS and eNOS in the second sphere of amino acids; this was the difference that we were searching for from the beginning of this project. With the aid of these crystal structures, we developed a new fragment-based de novo design method called “fragment hopping”, which allowed the design of a new class of nonpeptide nNOS-selective inhibitors. These compounds were modified to give low nanomolar, highly dual-selective nNOS inhibitors, which we recently showed are active in a rabbit model for the prevention of neurobehavioral symptoms of cerebral palsy. These compounds could also have general application in other neurodegenerative diseases for which excess NO is responsible.
Introduction
So, what do medicinal chemists do when there is an excess of a particular molecule that leads to a disease? They try to design compounds that lower the concentration of that molecule. But what if, for example, the goal were to lower nitric oxide (NO) production to prevent neurodegenerative diseases and all NO production were blocked? Then there may be the desired benefit to the target diseases, but there also would be detrimental effects resulting from the inability to produce the second messenger NO where it is needed in normal physiology. What is required in this case is an inhibitor of NO production only in neuronal tissue, not in other cells.
The enzyme that produces NO is nitric oxide synthase (NOS), and it occurs in three isozymic forms, two constitutive forms and one inducible form.i The one in endothelial cells (eNOS), which produces NO for regulation of the blood pressure, and the one in neuronal cells (nNOS), which produces NO for neurotransmission, are constitutive; the one in macrophage cells (iNOS) is induced by cytokines and pathogens to produce NO to combat infection and microorganisms. The three NOS isozymes are unusual in that all require five cofactors for catalysis. NADPH in the reductase domain transfers two electrons to FAD, then to FMN, which transfers one electron to a heme in the oxygenase domain (actually to the heme in the other subunit); tetrahydrobiopterin also is present in the oxygenase domain to help catalyze the conversion of L-arginine to L-citrulline and NO.
It has been demonstrated with transgenic mice that loss of each of the NOS isozymes would produce the effect expected of diminished NO in each of the respective cells.ii These experiments suggest that if selective inhibition of nNOS could be attained, there should be a protective effect on neurodegenerative diseases without the hypertensive effects of eNOS inhibition or potential immune system problems of iNOS inhibition.
Hypothesis for the Design of Selective Neuronal Nitric Oxide Synthase Inhibitors
Because of the potential benefit to the treatment of neurodegenerative disease, many pharmaceutical companies in the late 1980s and early 1990s initiated programs to identify nNOS-selective compounds.iii As there were no crystal structures available at that time, a common approach was to use the substrate, L-arginine, as the lead compound and make a large number of analogues in the hope that the appropriate structural change would produce a compound that preferentially bound to nNOS over eNOS and iNOS. However, for many years there was little or no success in finding highly dual-selective inhibitors. It seemed apparent to me that the reason for this lack of selectivity was that the active sites of all three of the isozymes were quite similar because the substrate and reaction for all three were identical. Therefore, any modifications that were made to L-arginine had similar effects on binding to the active sites of all three isozymes. What was needed was a compound that could be anchored into the active site and extend out to reach the second sphere of amino acid residues in search of a difference away from the heme-binding site. As there were no crystal structures reported for any of the NOS isozymes, it was not clear where differences, if any, might lie.
Initial Design of Dual-Selective Neuronal Nitric Oxide Synthase Inhibitors
It was reported by a group at Glaxo Wellcome Laboratories in 1993iv that L-nitroarginine was a 250-fold selective inhibitor of nNOS over iNOS, but not selective over eNOS. Because of the lack of selectivity over eNOS, it produces extensive hypertension in animals.v However, L-nitroarginine was shown to be a competitive inhibitor, indicating that it was bound in the active site, as would be expected because of its structural similarity to the substrate. The nNOS/iNOS selectivity was impressive, so nitroarginine was selected as the anchor molecule to which additional amino acids could be added to extend into the second sphere of amino acids. It was already known that some dipeptides acted as substrates for NOS.vi Eventually, these compounds would have to cross the blood-brain barrier, so instead of making dipeptides, we started with a few analogues of nitroarginine- and phenylalanine-containing dipeptide esters and amides. Two of the analogues gave selective inhibition in the thousand-fold range.vii This provided the impetus for a larger library comprised of all of the possible nitroarginine-containing dipeptide amides containing the commonly encoded amino acids in addition to some non-commonly encoded and synthetic amino acids.viii Also, all four stereochemistries (L,L; L,D; D,L; D,D) and both regiochemistries (N- and C-terminal) of each dipeptide amide were prepared. About 185 analogues were synthesized and screened against the three isozymes of NOS in search of an analogue that inhibited nNOS without inhibiting iNOS or eNOS. At the end of the screen there were 10 analogues that were the first highly dual-selective inhibitors of nNOS over both iNOS and eNOS (Table 1). One common feature of most of the analogues was an amino group in the side chain of the amino acid attached to nitroarginine. The most potent analogue, L-nitroargininyl-L-2,4-diaminobutyramide (1, Figure 1), had a Ki of 130 nM towards nNOS with a selectivity over eNOS of 1538 and over iNOS of 192. It was interesting that the retro-inverso dipeptide amides, L-nitroargininyl-L-lysinamide (2) and D-lysyl-D-nitroargininamide (3) (Figure 2) had similar activities, although the selectivities varied. A possible binding rationalization for why 2 and 3 are both active is depicted in Figure 2. Electron-nuclear double resonance (ENDOR) spectroscopy is used to identify nuclei that interact weakly with an electron spin to give detailed information about atoms at a paramagnetic site. For example, it can be used to determine distances and orientations of atoms surrounding paramagnetic centers, such as the iron atom in heme. ENDOR spectroscopy supported the hypothesis that these two peptide amides were bound in nNOS in a 180° relationship.ix The retro-inverso analogue D-2,4-diaminobutyryl-D-nitroargininamide (4) was less potent and selective than 1.x 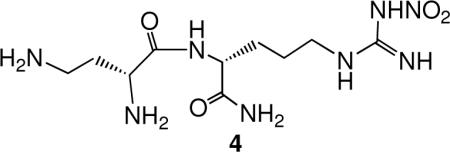
Table 1.
Dipepeptide Amides with Dual nNOS Selectivitya
| Compound |
Ki (μM) |
Selectivityb |
||||
|---|---|---|---|---|---|---|
 |
nNOS | iNOS | eNOS | nNOS/eNOS | nNOS/iNOS | |
| L-ArgNO2-L-ARgNO2-NH2 | 0.77 | 62 | 33 | 43 | 80 | |
| L-ArgNO2-L-Lys-NH2 | 0.45 | 100 | 140 | 310 | 230 | |
| L-ArgNO2-L-Orn-NH2 | 0.33 | 97 | 250 | 740 | 290 | |
| L-ArgNO2-L-Dbu-NH2 | 0.13 | 25 | 200 | 1500 | 200 | |
| L-ArgNO2-L-Dpr-NH2 | 1.1 | 61 | 260 | 240 | 55 | |
| L-ArgNO2-D-Asn-NH2 |  |
0.32 | 8.9 | 410 | 1300 | 28 |
| D-ArgNO2-L-Ser-NH2 | 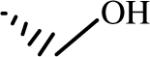 |
1.25 | 1200 | 220 | 170 | 940 |
| L-ArgNO2-D-Orn-NH2 | 2.0 | 100 | 1300 | 640 | 51 | |
| L-Lys-D-ArgNO2-NH2 | 1.7 | 4700 | 230 | 140 | 2800 | |
| D-Lys-D-ArgNO2-NH2 | 0.89 | 910 | 30 | 34 | 1000 | |
The enzymes used for the Ki determinations are bovine brain nNOS, recombinant murine iNOS, and recombinant bovine eNOS.
The ratio of Ki (eNOS or iNOS) to Ki (nNOS); all are nNOS selective.
Figure 1.

Structure of Lead Compound and Sites of Modification
Figure 2.

Hypothesis for the Activity of Retro-inverso NOS inhibitors
Structure-Activity Relationships (SAR) of the Lead Compound
The important question about these newly discovered dual-selective inhibitors, however, was why were they potent and dual selective? Consequently, a study was carried out to determine which moieties of lead molecule 1 were responsible for potency and selectivity.xi The biggest surprise was that the corresponding dipeptide analogue of 1 (instead of the dipeptide amide) exhibited poor potency (28 μM) and had little selectivity (3.5 fold over eNOS and 43 fold over iNOS). It was found that the free primary amino group was essential; acylation or alkylationxii strongly diminished both potency and selectivity. Alkylation of the peptide NH (5) or conversion to a peptoid (6) led to a sharp decrease in potency and selectivity (Figure 3).
Figure 3.

Modifications Made to Lead Compound 1
However, excision of the carboxamido group (7) only decreased potency and selectivity by less than a factor of four. We wondered if the importance of the nitroguanidino group derived from a low pKa guanidine or if the nitro group itself was beneficial. Conversion to the corresponding cyanoguanidine (8, pKa similar to that of nitroguanidine), however, destroyed both potency and selectivity.xiii
Conformationally-Rigid Analogues of the Lead Compound
While we were carrying out SAR studies to determine what moieties of the lead molecule was essential for potency and selectivity, we initiated structure modification studies. Because a side chain amino group was important, we prepared a family of conformationally-rigid analogues of 1 by replacing the diaminobutyramide group with 4-aminoprolinamides (9).xiv The syntheses of  these compounds required the development of new synthetic methodologies.xv These analogues have three stereogenic centers; therefore, we synthesized all eight stereoisomers. When the 4-aminoprolinamide was at the N-terminus (10), potencies were very weak. Also, we made the series of analogues in which the 4-aminoprolinamide was at the C-terminus and attached at the pyrrolidine nitrogen (11) before we determined that the peptide amide NH was essential for activity; since these compounds do not have a peptide amide NH, none was active. However, when the 4-aminoprolinamide was at the C-terminus and attached at the 4-amino nitrogen instead of the pyrrolidino nitrogen, one of the stereoisomers (12) was more potent (100 nM) than 1 with comparable selectivities. Substitution at the pyrrolidine nitrogen decreased the potency, but the 3-amino regioisomer of 12 (compound 13) was comparable in potency and selectivity to 12.xvi
these compounds required the development of new synthetic methodologies.xv These analogues have three stereogenic centers; therefore, we synthesized all eight stereoisomers. When the 4-aminoprolinamide was at the N-terminus (10), potencies were very weak. Also, we made the series of analogues in which the 4-aminoprolinamide was at the C-terminus and attached at the pyrrolidine nitrogen (11) before we determined that the peptide amide NH was essential for activity; since these compounds do not have a peptide amide NH, none was active. However, when the 4-aminoprolinamide was at the C-terminus and attached at the 4-amino nitrogen instead of the pyrrolidino nitrogen, one of the stereoisomers (12) was more potent (100 nM) than 1 with comparable selectivities. Substitution at the pyrrolidine nitrogen decreased the potency, but the 3-amino regioisomer of 12 (compound 13) was comparable in potency and selectivity to 12.xvi
Another conformationally-rigid class of peptidomimetic structures related to 12 that we prepared had pyrrolidino (14/15) and piperidino (16−18) moieties without the terminal amido group.12 The descarboxamido analogue (14) of 12 was slightly more potent and with enhanced selectivity over both eNOS and iNOS. Two of the piperidino analogues (16 and 18) were only slightly less potent than 12. 
The side chain amino group could provide electrostatic stabilization or hydrogen bonding interactions with an nNOS residue. To test which of those interactions is more relevant, a series of hydroxyl-containing analogues of 7 was synthesized (19).xvii The most potent of the three compounds was one-eighth as potent as 7 with about one-third the eNOS selectivity, suggesting that an electrostatic interaction is more important than hydrogen bonding. 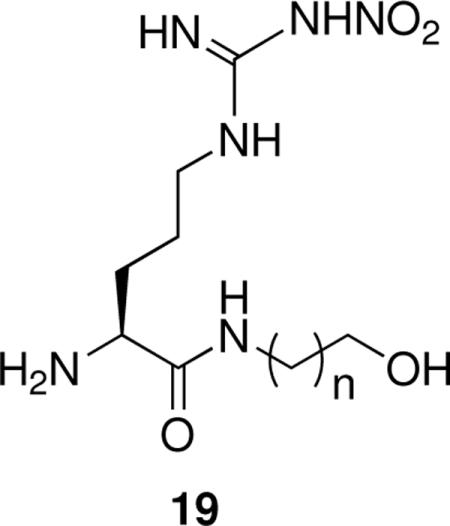
Reduced Peptide Bond Peptidomimetics
Peptides are notoriously susceptible to metabolic proteolysis; to avoid this potential problem, the peptide carbonyl was modified. The simplest modification was a reduction to the corresponding amine (20).xviii These reduced amide bond peptidomimetics were comparable in potency (120 nM), but the selectivities for nNOS over eNOS and iNOS were enhanced relative to 1. However, these compounds were still highly hydrophilic and, because of the number of amino groups, were multi-charged. Both of these properties could prohibit the compounds from entering the brain. To lower the charge, a related series of compounds with a hydroxyl in place of the terminal amino group (21) was prepared and found to have very poor potency and little selectivity, again supporting the importance of an electrostatic interaction in the second amino acid or amine.17 
To enhance potential bioavailability, the lipophilicity of the compounds had to be increased and the charge decreased. Those aims led to the design of a library of aromatic reduced peptide bond peptidomimetics (22 and 23).xix In addition to the added lipophilicity provided by the aromatic moieties, they also served to reduce the pKa values of many of the amino groups in the hope of reaching both goals simultaneously. One of these analogues (24) was the most potent nNOS-selective inhibitor we had prepared to that point (Ki 50 nM) with outstanding eNOS (2121-fold) selectivity.
Another common approach for replacing a peptide bond with a more bioavailable moiety is the use of a hydroxyethylene isostere.xx A series of reverse hydroxyethylene isosteres of 7 was synthesized (25, n = 1,2); the most potent (50% more potent than 7, n = 1) and selective (same selectivity over eNOS and half the iNOS selectivity) was 26.xxi As in the case of the reduced amide series, the hydroxyethylene isosteres with a terminal hydroxyl group instead of amino group had very poor potency and little or no selectivity. 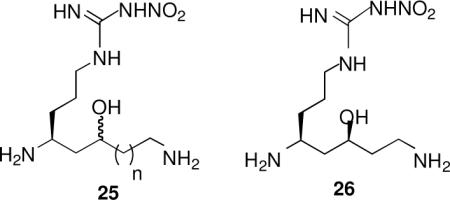
X-Ray Crystallography Supports the Initial Hypothesis
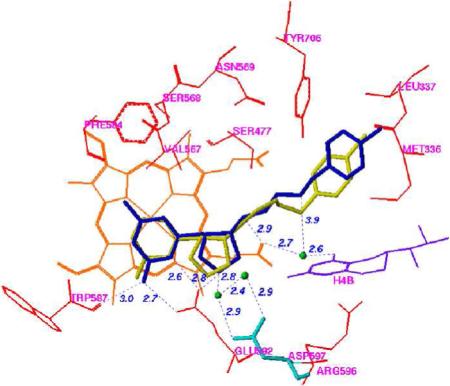
The first crystal structures of iNOSxxii and eNOSxxiii,xxiv were reported in the late 1990's. Both isozyme structures, particularly in the active site, were strikingly similar, as expected. The structure that was needed to elucidate the inhibitor selectivity we observed was that of nNOS, but that structure did not become available until 2002.xxv A fruitful collaboration between my group and the Poulos group ensued. Crystal structures of 1, 12, and 20 (n = 1) bound to nNOS and eNOS provided the answer to the question of selectivity.xxvi The structures shown in Figure 4, in which 1 is bound to nNOS and eNOS (the structures with 12 and 20 (n = 1) bound are similar), rationalize the SAR findings. In Figure 4A, it is apparent that the primary α-amino group of the L-nitroargininyl could not be modified, either by acylation or alkylation, because in nNOS it is engaged in an electrostatic interaction with Asp597 and Glu592. The peptide amide NH is important because of a hydrogen bond to a water molecule, and the C-terminal carboxamide undergoes hydrogen bonding with Gln478, Arg481, and Ser477. The other key interaction is the side chain amino group, which interacts via a water molecule to one of the heme propionate groups, presumably an electrostatic interaction because, as noted above, replacement of the amino group with a hydroxyl group leads to major losses in potency and selectivity. The nitroguanidino group of all three inhibitors binds to nNOS the same as the guanidino group of arginine analogues (including L-nitroarginine), amidines, and thioureas,xxvii namely, in a bifurcated hydrogen bonding interaction with the conserved Glu592. The nitro group strengthens this interaction with an additional hydrogen bond. From Figure 4A and 4B it can be seen that, unlike the curled conformation that 1 adopts when bound to nNOS, 1 adopts an extended conformation when bound to eNOS. Whereas the curled conformation in nNOS places the α-amino group in position for a direct interaction with Glu592, the extended conformation in eNOS places the α-amino group too far from Glu363 for direct hydrogen bonding; furthermore, an additional water molecule inserts between Glu363 and the α-amino group of 1. It appears that this conformational difference and the isozyme selectivity favoring nNOS binding over eNOS binding result from a single-residue difference between these two isozymes in the second sphere of amino acids, namely, Asp597 in nNOS is Asn368 in eNOS. Therefore, the protonated primary α-amino group of 1 in eNOS cannot participate in the electrostatic interaction that it does in nNOS because of the lack of the anionic aspartate residue at that position. This difference between nNOS and eNOS was the initial goal of this project, i.e., to see if there is a difference among the isozymes in the second sphere of amino acid residues in (or near) the active site. This accounts for why the earlier attempts by other groups to identify compounds that show isozyme selectivity had failed. They were making substrate analogues that bound directly in the active site over the heme-binding pocket, and the Asp597/Asn368 difference is at the periphery of the active site.
Figure 4.


Stereoview of Crystal Structures of 1 Bound to (A) nNOS and (B) eNOS
To demonstrate the importance of this single amino acid difference, two mutants were made, one in which Asp597 of wild-type nNOS was mutated to an asparagine residue, which would make the nNOS have binding properties more similar to those of wild-type eNOS, and one in which Asn368 of wild-type eNOS was mutated to an aspartate, making eNOS more wild-type nNOS-like. These changes had no effect on the protein structures, but had a large effect on inhibitor binding. The crystal structure of the D597N nNOS mutant with 1 bound (Figure 5A) shows that the inhibitor switches to the extended conformation and a water molecule inserts between Glu592 and 1, exactly as if it were wild-type eNOS. Likewise, the N368D eNOS mutant binds 1 in a curled conformation (Figure 5B), exactly as in wild-type nNOS. Consistent with these conformational changes observed in the crystallographic analysis, it was found that the Ki for the three inhibitors increased (decreased potency) by 210−227 fold just from the single amino acid mutation! Conversely, the Ki values for the three inhibitors with the N368D mutant of eNOS decreased (increased potency) by a factor of 11−22 relative to wild-type eNOS. A second, less significant, difference is Val106 in eNOS, which corresponds to Met336 in nNOS, so a double eNOS mutant, N368D/V106M, was expressed, and the Ki value for 12 in the double mutant was shown to decrease (increased potency) by a factor of 100 fold.xxviii Clearly, the major binding difference between nNOS and eNOS resides in the electrostatic difference between Asp597 and Asn363, respectively.
Figure 5.


Stereoview of Crystal Structure of 1 Bound to (A) D597N nNOS Mutant and (B) N368D eNOS Mutant
As noted above, D-lysyl-D-nitroargininamide (3) appears to be a retro-inverso dipeptide amide of L-nitroargininyl-L-lysinamide (2) based on kinetic and ENDOR spectroscopic studies.9 Crystal structures of 3 bound to the heme domain of nNOS and eNOS show that the nitroguanidino group of the C-terminal D-nitroargininamide occupies the same binding site as the nitroguanidino group of the N-terminal L-nitroargininyl (2), as was predicted by the ENDOR spectroscopic measurements in support of a retro-inverso binding model.xxix The early model predicting that 2 and 3 bind at the same site (Figure 2) was not too far off from reality, except that two different carboxylates are involved in the binding of the two compounds.
Crystal structures of nNOS complexed with 12 and 20 (n = 1) (Figure 6A and 6B, respectively) reveal a conserved structural water molecule that is hydrogen bonded between the two heme propionate groups and the inhibitors. Based on computer modeling and docking experiments, we hypothesized that by attachment of a hydrogen bond donor group, such as a hydroxyl or amino group, to the nitrogen atoms on the inhibitors that interact with that water molecule, the inhibitor molecules may be able to displace the structural water molecule and interact directly with the heme cofactor. This should enhance the potencies of both of these inhibitors. Consequently, compounds 27 and 28 were synthesized and tested for inhibition of the three isozymes.xxx Surprisingly, there is little, if any, difference between the potency and selectivities for 20 (n = 1) compared with 27 (R = OH; when R = NH2, it was least potent) and  for 12 compared with 28. X-ray crystal structures of 27 (R = OH) and 28 bound to nNOS (Figure 7A and 7B, respectively) clearly showed that the N-hydroxyl groups had, indeed, displaced the structural water molecule, resulting in a direct interaction with the heme propionate, as was predicted by computer modeling. One possible explanation for the lack of increased potency is that the binding energy gain in the direct interaction with the cofactor was erased by the binding energy loss from disruption of the stable structural water molecule. Another explanation comes from inspection of the crystal structures with 27 (R = OH) and 28 bound, which show that the distance between the nitroguanidino group and Glu592 and that between the primary amino group and another structural water molecule are longer by 0.2 Å and 0.3 Å, respectively. This smaller H-bond binding energy compensates for the binding energy gained by direct interaction with the heme. These observations could rationalize why the hydroxyethylene isostere analogues (25) also did not exhibit greater improvement of potency than 7.21
for 12 compared with 28. X-ray crystal structures of 27 (R = OH) and 28 bound to nNOS (Figure 7A and 7B, respectively) clearly showed that the N-hydroxyl groups had, indeed, displaced the structural water molecule, resulting in a direct interaction with the heme propionate, as was predicted by computer modeling. One possible explanation for the lack of increased potency is that the binding energy gain in the direct interaction with the cofactor was erased by the binding energy loss from disruption of the stable structural water molecule. Another explanation comes from inspection of the crystal structures with 27 (R = OH) and 28 bound, which show that the distance between the nitroguanidino group and Glu592 and that between the primary amino group and another structural water molecule are longer by 0.2 Å and 0.3 Å, respectively. This smaller H-bond binding energy compensates for the binding energy gained by direct interaction with the heme. These observations could rationalize why the hydroxyethylene isostere analogues (25) also did not exhibit greater improvement of potency than 7.21
Figure 6.

Crystal Structure of nNOS Complexed with (A) 12 and (B) 20 (n = 1)
Figure 7.


Stereoview of Crystal Structure of (A) 27 (R = OH) and (B) 28 Bound to nNOS. Shows that the N-hydroxyl groups displace the structural water molecule
Computer Modeling to Design New Selective nNOS Inhibitors
With crystal structures of nNOS-selective inhibitors bound to NOS isozymes in hand, it was possible to do computer modeling to determine which regions in the active sites of the isozymes were important for isozyme selectivity.xxxi The active sites were characterized by examination of molecular interaction fields (MIFs) obtained by 10 different GRIDxxxii probes, and the MIFs were evaluated by the consensus principal component analysis (CPCA) method.xxxiii Twenty-five inhibitors were docked into this model, and regions identified by this method as being important for selectivity agreed with the SAR results. The two most important physicochemical parameters seem to be electrostatic and hydrophobic interactions.
The initial computer modeling studies were the impetus for a new, more comprehensive modeling approach directed at de novo inhibitor design of new classes of compounds having molecular diversity and NOS isozyme selectivity;xxxiv we termed this approach fragment hopping. The core of this approach is the derivation of the minimal pharmacophoric element for each pharmacophore. Sites for both ligand binding and isozyme selectivity are considered in deriving the minimal pharmacophoric elements. Five general-purpose libraries were established: a basic fragment library, a bioisostere library, rules for metabolic stability, a toxicophore library, and a side chain library. These libraries can be employed to generate focused fragment libraries to match the minimal pharmacophoric elements for each pharmacophore and to link the fragment to the desired molecule. Starting from the nitroarginine-containing inhibitors described above, this modeling method was applied to the design of new selective nNOS inhibitors, and the minimal pharmacophoric elements were derived (Figure 8A). Based on this model, a small nonpeptide molecule (29) was designed having nanomolar potency (390 nM) with nNOS/eNOS and 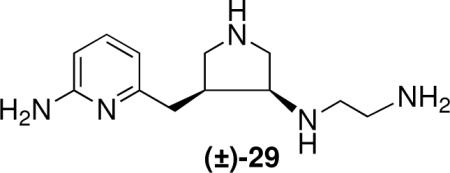 nNOS/iNOS selectivities of 1100 and 150, respectively. Unlike the nitroarginine-containing dipeptide amides, these compounds are racemates, suggesting that a single enantiomer should have greater potency and selectivity. A superimposition of the predicted bioactive conformation derived by this approach and the crystal structure of (the R,R-isomer of) 29 (Figure 8B) demonstrates why this compound exhibits excellent potency and nNOS selectivity. A comparison of the crystal structures of 29 and 20 (n = 1) (Figure 9), shows that the aminopyridine of 29 and the nitroguanidino group of 20 (n = 1) undergo electrostatic interactions with Glu592, and the pyrrolidino nitrogen of 29 and the α-amino group of 20 (n = 1) serve the same function of forming an electrostatic interaction with Asp597.
nNOS/iNOS selectivities of 1100 and 150, respectively. Unlike the nitroarginine-containing dipeptide amides, these compounds are racemates, suggesting that a single enantiomer should have greater potency and selectivity. A superimposition of the predicted bioactive conformation derived by this approach and the crystal structure of (the R,R-isomer of) 29 (Figure 8B) demonstrates why this compound exhibits excellent potency and nNOS selectivity. A comparison of the crystal structures of 29 and 20 (n = 1) (Figure 9), shows that the aminopyridine of 29 and the nitroguanidino group of 20 (n = 1) undergo electrostatic interactions with Glu592, and the pyrrolidino nitrogen of 29 and the α-amino group of 20 (n = 1) serve the same function of forming an electrostatic interaction with Asp597.
Figure 8.
(A) Minimal Pharmacophoric Elements for nNOS-Selective Binding (B) Superimposition of the Predicted Bioactive Conformation of 29 Derived by Fragment Hopping and the Crystal Structure of 29
Figure 9.

Stereoview of Overlay of the Crystal Structures of 29 and 20 (n = 1)
This method was refined further to take advantage of two steric and/or hydrophobic binding pockets, one lying just above the aminopyridine ring (F584/V587) and one forming a pocket beyond the terminal aminoethyl group (W306/M336/L337).xxxv Computer modeling suggested that attachment of a methyl group at the 4-position of the pyridine would be best accommodated in the F584/V587 pocket, and a series of arylalkyl groups could bind into the W306/M336/L337 pocket. A variety of structures were docked and tested, and two that looked promising had para-chlorobenzyl (30) and meta-fluorophenethyl (31) substituents attached to the terminal aminoethyl group.xxxvi They were much more potent and selective than the parent compound (29) having Ki values of 85 nM and 14 nM, respectively, with nNOS/eNOS selectivities of 1000-fold and 2000-fold, respectively, and nNOS/iNOS selectivities of 100-fold and 290-fold, respectively.
Chiral syntheses of the two cis- and two trans-isomers were developed; the (3’R, 4’R)-isomer (32) had a Ki of 5 nM and nNOS/eNOS and nNOS/iNOS selectivities of 3800 and 730, respectively.xxxvii Its enantiomer had one-tenth the potency with one-eighth and one-tenth the selectivities, respectively. One of the trans-isomers (33) was almost as potent and selective as 32. 
Recently, a crystallography/computer-based approach for the design of selective iNOS inhibitors called anchored plasticity was reported.xxxviii The general approach is that part of the inhibitor binds directly over the heme, where it is anchored in place by the invariant Glu residue. Bulky rigid substituents are built onto the inhibitor, which then extend out of the active site, where subtle differences in the NOS isoforms lead to isoform selectivity. This approach is fundamentally the same as the one we took in our earlier studies using nitroarginine as the anchor for the design of nNOS-selective inhibitors, which became the basis for fragment hopping.34
Although anchored plasticity and fragment hopping share common features, there are distinct differences. Unlike anchored plasticity, fragment hopping does not require prior identification of isozyme-selective inhibitors. The minimal pharmacophoric elements for ligand isozyme selectivity can be derived directly by comparison of the isozyme structures with the use of the GRID/CPCA approach. Another difference is that in the anchored plasticity model, conformational changes, which involve residues that both directly contact the bulky substituent of the inhibitor as well as second and third tier amino acids, are required to reveal new binding pockets and to control isoform selectivity; this is not the case with fragment hopping.
There also are examples of NOS inhibitors that would not have been identified by either of these approaches. The synthetic compound 1400W is a known iNOS-selective inhibitor (Ki = 0.14 μM for iNOS, 75 μM for eNOS, and 2 μM for nNOS).xxxix The crystal structures of the three NOS isozymes in complex with 1400W (iNOS: PDB code 1QW5,xl nNOS: PDB code 1QWC,40 and eNOS: PDB code 1FOIxli) exhibit little or no differences in the active site, even 8 Å away from the ligand. Therefore, residue movement is not the reason for isozyme selectivity. In this case, selectivity for iNOS probably derives from enzyme-ligand dynamics and 1400W serving as a unique irreversible inhibitor for iNOS without inactivator modificationxlii owing to the high turnover of iNOS compared to the other isoforms.
Animal Studies with Selective nNOS Inhibitors
The aromatic reduced peptidomimetic analogue 24 seemed to have sufficient potency and selectivity that we wanted to determine if it also had in vivo activity. In collaboration with Dr. Sidhartha Tan and Dr. Matthew Derrick, pediatricians at Evanston Hospital, who had developed a rabbit model for fetal neurodegeneration,xliii 24 was administered intrauterine to pregnant rabbits in which the oxygen supply to the fetuses was clamped off for 30 minutes to create a hypoxic-ischemic environment. Under these conditions, when saline was administered as a control, the fetuses all died prior to birth; removal of their brains revealed massive neurodegeneration that occurred under those conditions. However, when 24 was administered prior to hypoxia, there was a concentration-dependent protection of the fetal brain from neurodegeneration.xliv When fetal brain cells were cultured in the presence of 24, cell survival was concentration dependent, consistent with protection of the cells from apoptosis and death by 24.
After these initial neurodegeneration protection studies were completed, Tan and coworkers developed a modification of their rabbit model so that some of the fetuses came to term.xlv Inspection of the kits that survived showed symptoms and behaviors reminiscent of cerebral palsy, a family of neurodegenerative conditions; an important cause in humans is the blockage of oxygen to the fetus during pregnancy.xlvi There is no known cure and no treatment to protect the fetus from hypoxic brain injury leading to cerebral palsy,xlvii despite a reduction in the mortality of high-risk infants.xlviii Compounds 30 and 31 (racemates) were tested for their ability to prevent the symptoms of cerebral palsy in the rabbit model. Intrauterine administration of these compounds at a concentration of 100 × Ki 30 minutes prior to and immediately after uterine ischemia was carried out, and the effect on NOS and NO formation, on cardiovascular indicators, and on neurobehavioral effects were monitored. After birth the fetal brains were excised and frozen and half assayed for NOS and half for NO; both had decreased relative to the saline control animals. The blood pressure and heart rate of the rabbit dams were monitored during the experiment, and no change was observed relative to the saline control, suggesting little or no effect on eNOS (as expected from the large nNOS/eNOS selectivity). The most striking difference in the kits from saline-treated versus 30- or 31-treated rabbit dams was that almost half of the kits from saline-treated dams died prior to birth, but no deaths were observed from 30- and 31-treated animals. Of the kits from saline-treated dams that came to term, severe neurobehavioral abnormalities occurred in 67% of them compared to only 14% in those from dams treated with 30 or 31. Furthermore, the 30- and 31-treated animals exhibited a remarkably larger number (83 and 69%, respectively) of normal kits (in two litters, all 19 kits were normal); only 9% of the kits from saline-treated dams were born normal. None of the compounds caused any detectable systemic toxicity in the rabbit dams.
Summary
This research started with a basic science question: Are there any differences in the second sphere of amino acids in the active site of the isozymes of NOS that could be identified for nNOS-selective inhibitor design? From this study, the first class of dual nNOS-selective inhibitors was identified. The moieties of the best lead compound that were important for selectivity were determined by structure modification, then the potency and selectivity were enhanced to provide highly potent and nNOS-selective dipeptide amides and peptidomimetics, which were active in a rabbit model for fetal neurodegeneration. Crystal structures of these compounds bound to NOS isozymes showed that there was a one amino acid difference between nNOS and eNOS in the second sphere of amino acids; this was the difference that we were searching for from the beginning. With the aid of these crystal structures, a new fragment-based de novo design method was developed, called fragment hopping, which allowed the design of a new class of nonpeptide nNOS-selective inhibitors. These compounds have been modified to give low nanomolar, highly dual selective nNOS inhibitors, which were active in a rabbit model for the prevention of neurobehavioral symptoms of cerebral palsy. These compounds could have general application in neurodegenerative diseases because excessive NO leads to many of these diseases. However, these compounds are still too polar for good blood-brain barrier penetration, so future efforts are directed at increasing the bioavailability of these compounds, a common problem in drug design, which we are trying to resolve from basic principles important to bioavailability.
Acknowledgments
The author is most grateful to the past and current postdocs and students in the group who made this research possible. They include the following:
Postdocs: Robert P. Dixon, Henry (Qingwei) Zhang, Younghee Lee, José Antonio Gómez-Vidal, Haitao Ji, Bessie Mbadugha, Jianguo Fang, Fengtian Xue, and Jinwen Huang;
Graduate Students: Walter Fast, Hui Huang, Jung-Mi Hah, Timothy Roach, Erik Erdal, Elizabeth Litzinger, Jiwon Seo, Yaoqiu Zhu, Graham Lawton, and Kristin Jansen;
Undergraduate Students: Marc Levsky, Mary Beth Huff, Michael Forrester, Benjamin Stanton, Marc Sala, Meera Rao, George Michael, Jeffrey Martell, and Stephanie Choing.
We also are grateful to the National Institutes of Health (GM49725) for financial support of the research. The National Institutes of Health also supported collaborators in this research (Sidhartha Tan [NS43285 and NS051402], Thomas L. Poulos [GM57353], and Dr. Bettie Sue Masters [GM52419], and the Robert A. Welch Foundation also supported Dr. Bettie Sue Masters [AQ1192]).
Biography
Biographical Sketch
Richard B. Silverman was born in Philadelphia, PA in 1946, received a B.A. degree from Central High School of Philadelphia, then obtained a B.S. degree in chemistry at The Pennsylvania State University. His graduate studies at Harvard in organic chemistry were interrupted in 1968 by the military draft for two years, after which he returned to Harvard to complete his Ph.D. with David Dolphin in 1974. Following two years of NIH-supported postdoctoral work in mechanistic enzymology at Brandeis University with the late Robert Abeles, he began his independent career at Northwestern University in 1976. He is currently the John Evans Professor of Chemistry at Northwestern University. His research interests are in the design, synthesis, and study of enzyme inhibitors and receptor antagonists that are important for the treatment of various diseases. He is the inventor of Lyrica™, a blockbuster drug marketed by Pfizer for the treatment of neuropathic pain, fibromyalgia, and epilepsy.
References
- i.a Li H, Poulos TL. Structure-function studies on nitric oxide synthases. J. Inorg. Biochem. 2005;99(1):293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]; b Fleming I. Biology of nitric oxide synthases. Handbook of Physiology: Microcirculation. (2nd Edition) 2008:56–80. [Google Scholar]
- ii.a Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]; b Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]; c Ferriero DM, Holtzman DM, Black SM, Sheldon RA. Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol. Dis. 1996;3:64–71. doi: 10.1006/nbdi.1996.0006. [DOI] [PubMed] [Google Scholar]
- iii.Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Selective neuronal nitric oxide synthase inhibitors. Curr. Topics Med. Chem. 2005;5:603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]
- iv.Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective inhibition of constitutive nitric oxide synthase by L-NG-nitroarginine. Biochemistry. 1993;32:8512–8517. doi: 10.1021/bi00084a017. [DOI] [PubMed] [Google Scholar]
- v.Yamamoto K, Shimamura K, Sekiguchi F, Sunano S. Effects of NG-nitro-L-arginine on the blood pressure of spontaneously hypertensive rats with different degrees of hypertension. Clin. Exp. Hypertens. 2001;23(7):533–544. doi: 10.1081/ceh-100106824. [DOI] [PubMed] [Google Scholar]
- vi.a Iyengar R, Stuehr DJ, Marletta MA. Macrophage synthesis of nitrite, nitrate, and N-nitrosamines: precursors and role of the respiratory burst. Proc. Natl. Acad. Sci. USA. 1987;84:6369–6373. doi: 10.1073/pnas.84.18.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hecker M, Walsh DT, Vane JR. On the substrate specificity of nitric oxide synthase. FEBS Lett. 1991;294:221–4. doi: 10.1016/0014-5793(91)81434-a. [DOI] [PubMed] [Google Scholar]
- vii.Silverman RB, Huang H, Marletta MA, Martasek P. Selective inhibition of neuronal nitric oxide synthase by Nω-nitroarginine- and phenylalanine-containing dipeptides and dipeptide esters. J. Med. Chem. 1997;40:2813–2817. doi: 10.1021/jm970200u. [DOI] [PubMed] [Google Scholar]
- viii.Huang H, Martasek P, Roman LJ, Masters BSS, Silverman RB. Nω-nitroarginine-containing dipeptide amides. Potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- ix.Tierney DL, Huang H, Martásek P, Roman LJ, Silverman RB, Hoffman BM. ENDOR spectroscopic evidence for the geometry of binding of retro-inverso-Nω-nitroarginine-containing dipeptide amides to neuronal nitric oxide synthase. J. Am. Chem. Soc. 2000;122:7869–7875. [Google Scholar]
- x.Huang H, Martásek P, Roman LJ, Silverman RB. Syntheses and evaluation of dipeptide amides containing Nω-nitroarginine and D-2,4-diaminobutyric acid as inhibitors of neuronal nitric oxide synthase. J. Enzyme Inhib. 2001;16:233–239. doi: 10.1080/14756360109162371. [DOI] [PubMed] [Google Scholar]
- xi.Huang H, Martásek P, Roman LJ, Silverman RB. Synthesis and evaluation of peptidomimetics as selective inhibitors and active site probes of nitric oxide synthases. J. Med. Chem. 2000;43:2938–2945. doi: 10.1021/jm000127z. [DOI] [PubMed] [Google Scholar]
- xii.Seo J, Martásek P, Roman LJ, Silverman RB. Selective L-nitroargininylaminopyrrolidine and L-nitroargininylaminopiperidine neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. 2007;15:1928–1938. doi: 10.1016/j.bmc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xiii.Roach T, Silverman RB. unpublished results.
- xiv.Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. Potent and selective conformationally-restricted neuronal nitric oxide synthase inhibitors. J. Med. Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- xv.a Gómez-Vidal JA, Silverman RB. Short, highly efficient syntheses of 3-azido- and 4-azidoproline derivatives and their precursors. Organic Lett. 2001;3:2481–2484. doi: 10.1021/ol0161054. [DOI] [PubMed] [Google Scholar]; b Gómez-Vidal JA, Forrester MT, Silverman RB. Mild and selective sodium azide-mediated cleavage of p-nitrobenzoic esters. Organic Lett. 2001;3:2477–2479. doi: 10.1021/ol016104b. [DOI] [PubMed] [Google Scholar]
- xvi.Ji H, Gómez-Vidal JA, Martásek P, J. Roman LJ, Silverman RB. Conformationally-restricted dipeptide amides as potent and selective neuronal nitric oxide synthase inhibitors. J. Med. Chem. 2006;49:6254–6263. doi: 10.1021/jm0604124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvii.Mbadugha BNA, Seo J, Ji H, Martásek P, Roman LJ, Shea TM, Silverman RB. Hydroxyl-terminated peptidomimetic inhibitors of neuronal nitric oxide synthase. Bioorg. Med. Chem. 2006;14:3681–3690. doi: 10.1016/j.bmc.2006.01.044. [DOI] [PubMed] [Google Scholar]
- xviii.Hah J-M, Roman LJ, Martásek P, Silverman RB. Reduced amide bond isosteric peptidomimetics. (4S)-N-(4-Amino-5-[aminoalkyl]aminopentyl)-N'-nitroguanidines, potent and highly selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- xix.Hah J-M, Martásek P, Roman LJ, Silverman RB. Aromatic reduced amide bond peptidomimetics as selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2003;46:1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]
- xx.Wiley RA, Rich DH. Peptidomimetics derived from natural products. Med. Res. Rev. 1993;13:327. doi: 10.1002/med.2610130305. [DOI] [PubMed] [Google Scholar]
- xxi.Erdal EP, Martásek P, Linda J. Roman LJ, Silverman RB. Hydroxyethylene isosteres of selective neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. 2007;15:6096–6108. doi: 10.1016/j.bmc.2007.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxii.a Crane BR, Arvai AS, Gachhhui R, Wu C, Ghosh DK, Getzoff ED, Steuhr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278(5337):425–431. doi: 10.1126/science.278.5337.425. Washington D.C. [DOI] [PubMed] [Google Scholar]; b Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279(5359):2121–2126. doi: 10.1126/science.279.5359.2121. Washington D.C. [DOI] [PubMed] [Google Scholar]
- xxiii.Raman CS, Li H, Martasek P, Kral V, Masters BSS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase : a paradigm for pterin function involving a novel metal center. Cell. 1998;95(7):939–950. doi: 10.1016/s0092-8674(00)81718-3. Cambridge, Massachusetts. [DOI] [PubMed] [Google Scholar]
- xxiv.Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat. Struct. Biol. 1999;6(3):233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- xxv.a Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-Alkyl-N'-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41(47):13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]; b Bretscher LE, Li H, Poulos TL, Griffith OW. Structural Characterization and Kinetics of Nitric-oxide Synthase Inhibition by Novel N5-(Iminoalkyl)- and N5-(Iminoalkenyl)-ornithines. J. Biol. Chem. 2003;278(47):46789–46797. doi: 10.1074/jbc.M306787200. [DOI] [PubMed] [Google Scholar]
- xxvi.Flinspach M, Li H, Jamal J, Yang W, Huang H, Hah J-M, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Structural basis for dipeptide amide isoform-selective inhibition of neuronal nitric oxide synthase. Nature (Struct. Mol. Biol.) 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- xxvii.a Li H, Raman CS, Martasek P, Kral V, Masters BSS, Poulos TL. Mapping the active site polarity in structures of endothelial nitric oxide synthase heme domain complexed with isothioureas. J. Inorg. Biochem. 2000;81:133–139. doi: 10.1016/s0162-0134(00)00099-4. [DOI] [PubMed] [Google Scholar]; b Li H, Raman CS, Martasek P, Masters BSS, Poulos TL. Crystallographic studies on endothelial nitric oxide synthase complexed with nitric oxide and mechanism-based inhibitors. Biochemistry. 2001;40:5399–5406. doi: 10.1021/bi002658v. [DOI] [PubMed] [Google Scholar]
- xxviii.Li H, Flinspach ML, Igarashi J, Jamal J, Yang W, Gómez-Vidal JA, Litzinger EA, Huang H, Erdal EP, Silverman RB, Poulos TP. Exploring the binding conformations of bulkier dipeptide amide inhibitors in constitutive nitric oxide synthases. Biochemistry. 2005;44:15222–15229. doi: 10.1021/bi0513610. [DOI] [PubMed] [Google Scholar]
- xxix.Flinspach M, Li H, Jamal J, Yang W, Huang H, Silverman RB, Poulos TL. Structures of the neuronal and endothelial NOS heme domain with D-nitroarginine-containing dipeptide inhibitors bound. Biochemistry. 2004;43:5181–5187. doi: 10.1021/bi0361867. [DOI] [PubMed] [Google Scholar]
- xxx.Seo J, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Structure-based design and synthesis of Nω-nitro-L-arginine-containing peptidomimetics as selective inhibitors of neuronal nitric oxide synthase. Displacement of the heme structural water. J. Med. Chem. 2007;50:2089–2099. doi: 10.1021/jm061305c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxi.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. Computer modeling of selective regions in the active site of nitric oxide synthases: implication for the design of isoform-selective inhibitors. J. Med. Chem. 2003;46:5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- xxxii.GRID version 20. Molecular Discovery, Ltd.; 4 Chandos Street, London, U.K.: 2002. [Google Scholar]
- xxxiii.a GOLPE 4.5. Multivariate Informetric Analysis Srl.; Viale del Castagni 16, Perugia, Italy: 1999. [Google Scholar]; b Westerhuis JA, Kourti T, Macgregor JF. Analysis of multiblock and hierarchical PCA and PLS models. J. Chemomet. 1998;12:301–321. [Google Scholar]
- xxxiv.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J. Am. Chem. Soc. 2008;130(12):3900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxv.Ji H, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. Discovery of highly potent and selective inhibitors of neuronal nitric oxide synthase by fragment hopping. Submitted. [DOI] [PMC free article] [PubMed]
- xxxvi.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martásek P, Roman LJ, Vásquez-Vivar J, Poulos TL, Silverman RB. Selective neuronal nitric oxide synthase inhibitors for prevention of cerebral palsy. Ann. Neurol. doi: 10.1002/ana.21555. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxvii.Ji H, Silverman RB. unpublished results.
- xxxviii.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nature Chem. Biol. 2008;4(11):700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxix.Collins JL, Shearer BG, Oplinger JA, Lee S, Garvey EP, Salter M, Duffy C, Burnette TC, Furfine ES. N-Phenylamidines as selective inhibitors of human neuronal nitric oxide synthase: Structure-activity studies and demonstration of in vivo activity. J. Med. Chem. 1998;41(15):2858–2871. doi: 10.1021/jm980072p. [DOI] [PubMed] [Google Scholar]
- xl.Fedorov R, Hartmann E, Ghosh DK, Schlichting I. Structural basis for the specificity of the nitric-oxide synthase inhibitors W1400 and Nω-propyl-L-Arg for the inducible and neuronal isoforms. J. Biol. Chem. 2003;278(46):45818–45825. doi: 10.1074/jbc.M306030200. [DOI] [PubMed] [Google Scholar]
- xli.Li H, Raman CS, Martásek P, Masters BS, Poulos TL. Crystallographic studies on endothelial nitric oxide synthase complexed with nitric oxide and mechanism-based inhibitors. Biochemistry. 2001;40(18):5399–5406. doi: 10.1021/bi002658v. [DOI] [PubMed] [Google Scholar]
- xlii.a Zhu Y, Nikolic D, Van Breemen RB, Silverman RB. Mechanism of inactivation of inducible nitric oxide synthase by amidines. Irreversible enzyme inactivation without inactivator modification. J. Am. Chem. Soc. 2005;127(3):858–868. doi: 10.1021/ja0445645. [DOI] [PubMed] [Google Scholar]; b Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272(8):4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- xliii.a Derrick M, He J, Brady E, Tan S. The in vitro fate of rabbit fetal brain cells after acute in vivo hypoxia. J. Neurosci. 2001;21(7):RC138/1–RC138/5. doi: 10.1523/JNEUROSCI.21-07-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tan S, Zhou F, Nielsen VG, Wang Z, Gladson CL, Parks DA. Increased injury following intermittent fetal hypoxia-reoxygenation is associated with increased free radical production in fetal rabbit brain. J. Neuropath. Exp. Neur. 1999;58(9):972–981. doi: 10.1097/00005072-199909000-00007. [DOI] [PubMed] [Google Scholar]
- xliv.Tan S, Derrick M, Ji H, Silverman RB. unpublished results.
- xlv.a Derrick M, Luo NL, Bregman JC, Jilling T, Ji X, Fisher K, Gladson CL, Beardsley DJ, Murdoch G, Back SA, Tan S. J. Neurosci. 2004;24:24–34. doi: 10.1523/JNEUROSCI.2816-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tan S, Drobyshevsky A, Jilling T, Ji X, Ullman LM, Englof I, Derrick M. J. Child Neurol. 2005;20:972–979. doi: 10.1177/08830738050200120801. [DOI] [PubMed] [Google Scholar]; c Derrick M, Drobyshevsky A, Ji X, Tan S. A model of cerebral palsy from fetal hypoxia-ischemia. Stroke. 2007;38(2 Suppl):731–735. doi: 10.1161/01.STR.0000251445.94697.64. [DOI] [PubMed] [Google Scholar]
- xlvi.Robinson S, Petelenz K, Li Q, Cohen ML, DeChant A, Tabrizi N, Bucek M, Lust D, Miller RH. Developmental changes induced by graded prenatal systemic hypoxicischemic insults in rats. Neurobiol. Dis. 2005;18(3):568–581. doi: 10.1016/j.nbd.2004.10.024. [DOI] [PubMed] [Google Scholar]
- xlvii.Winter S, Autry A, Boyle C, Yeargin-Allsopp M. Trends in the prevalence of cerebral palsy in a population-based study. Pediatrics. 2002;110:1220–1225. doi: 10.1542/peds.110.6.1220. [DOI] [PubMed] [Google Scholar]
- xlviii.Tyson JE, Gilstrap LC. Hope for perinatal prevention of cerebral palsy. JAMA. 2003;290:2730–2732. doi: 10.1001/jama.290.20.2730. [DOI] [PubMed] [Google Scholar]