

Abstract
In this paper, we describe an optimized reconstitution of the thiamin thiazole synthase (ThiG) catalyzed reaction and demonstrate that the enzymatic product is an unanticipated dearomatized thiazole tautomer.
INTRODUCTION
Thiamin pyrophosphate (TPP) is an essential cofactor in all living systems1,2. Most prokaryotes and eukaryotes biosynthesize TPP, but humans cannot and require it (1.4mg/day) from dietary sources3. TPP consists of a thiazole ring attached to a pyrimidine ring. The biosynthesis of TPP involves separate enzymatic routes for producing each of these heterocycles. Furthermore, these enzymatic routes for production of the thiazole ring and the pyrimidine ring are different in prokaryotes and eukaryotes. The early steps in the biosynthesis of the thiamin thiazole in B. subtilis have been studied extensively and the mechanism outlined in Figure 1 now has substantial experimental support.4-11 In this mechanism, DXP 1 forms an imine with lysine 96 of the thiazole synthase. This imine then tautomerizes to aminoketone 3. Addition of ThiS-thiocarboxylate 6, formed separately by reactions catalyzed by ThiF and NifS, to the ketone of 3 gives 7, which undergoes an S/O acyl shift to 8 followed by loss of water to give 9. Elimination of ThiS gives 12. Addition of the thiol of 12 to the glycine imine, formed by ThiO-catalyzed oxidation of glycine, gives 13. Cyclization via a transimination gives 14, which could then aromatize by protonation/ deprotonation to give 15 or by decarboxylation to give 16. The late steps (12 to product) in the biosynthesis have not yet been experimentally characterized.
Figure 1.
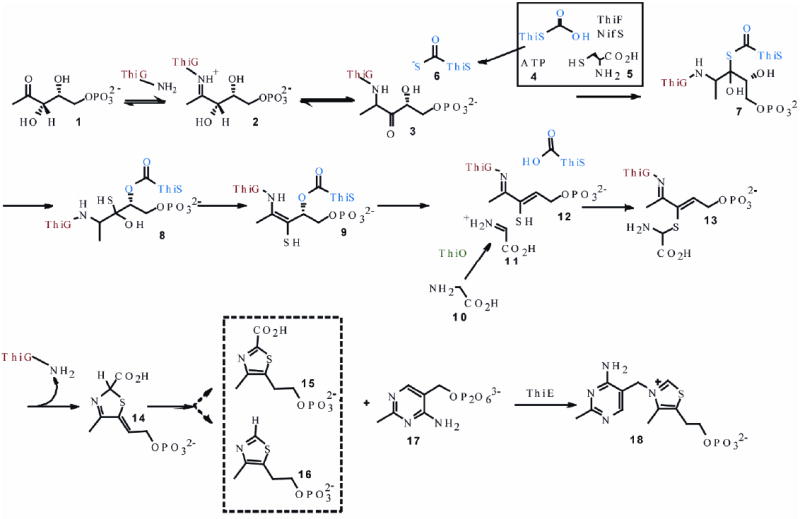
Currently proposed mechanism for the formation of the thiamin thiazole in B. subtilis.
It was not possible previously to directly characterize the final product of thiazole biosynthesis because our initial reconstitution yielded very low levels of thiazole and required a highly sensitive but indirect assay for product detection. This assay (Figure 2), involved alkylation of the thiazole product with pyrimidine 17, followed by the oxidation of the resulting thiamin phosphate 18 to the highly fluorescent thiochrome phosphate 19.
Figure 2.
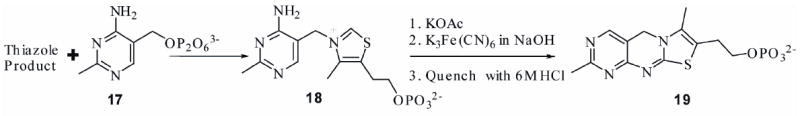
The thiochrome assay previously used for the detection of thiazole formation.
Since thiazole 16 was, at that time, the only identified substrate for the well-characterized thiamin phosphate synthase, it seemed reasonable to assume that thiochrome formation was a reliable way to measure the formation of this thiazole.7 However, the possibility remained that 14 or 15 could also be substrates for thiamin phosphate synthase, thus leaving unresolved the true identity of the reaction product of the bacterial thiazole synthase. Here we describe an improved reconstitution procedure which enables us to directly characterize the product of the bacterial thiazole synthase as the thiazole tautomer 14. The unexpected stability of 14 permits its characterization by 1-D and 2-D NMR studies and clarifies the later steps of the thiazole biosynthetic pathway in B. subtilis.
RESULTS and DISCUSSION
The product of thiazole biosynthesis is not thiazole phosphate
The previously reported reconstitution procedure was optimized and scaled up to produce larger quantities of the thiazole product. His-tagged proteins ThiF, NifS, ThiO and ThiSG were overexpressed in E. coli BL21(DE3). ThiS-COOH (in complex with ThiG), NifS and ThiF were incubated with L-cysteine 5 in the presence of dithiothreitol and ATP to form ThiS-COSH 6. This was then added to DXP 1 and glycine 10 in the presence of ThiO and ThiG to produce the product of the thiazole synthase-catalyzed reaction. The resulting reaction mixture was heat denatured, filtered and analyzed by reverse phase HPLC (Figure 3).
Figure 3.
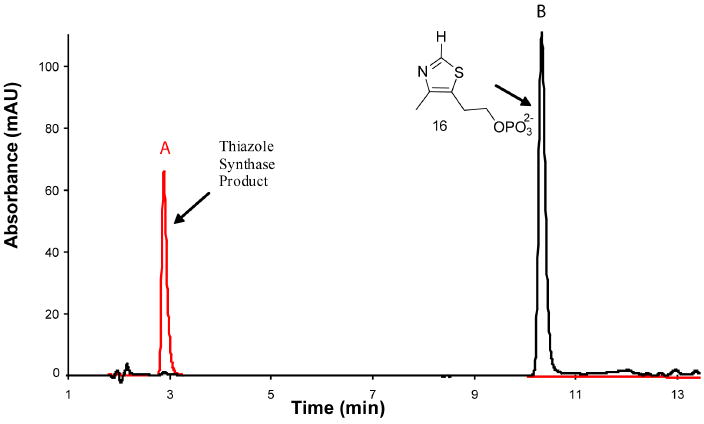
HPLC analysis of the product of the bacterial thiazole synthase reaction mixture. A - The enzymatic reaction mixture, B - Thiazole phosphate 16, the previously assumed reaction product.
The product of the reconstitution (peak A) was readily detected and did not comigrate with an authentic sample of thiazole phosphate 16 (peak B). The peak A compound had a UV absorption maximum at 300 nm and when treated with the pyrimidine 17 in the presence of thiamin phosphate synthase followed by thiochrome derivitization (Figure 2) produced a fluorescent product, which comigrated with an authentic sample of thichrome phosphate 19 (Figure 4 and Supplementary Figure 3). This experiment clearly demonstrates that the product of the thiazole reconstitution is not the anticipated thiazole phosphate 16.
Figure 4.
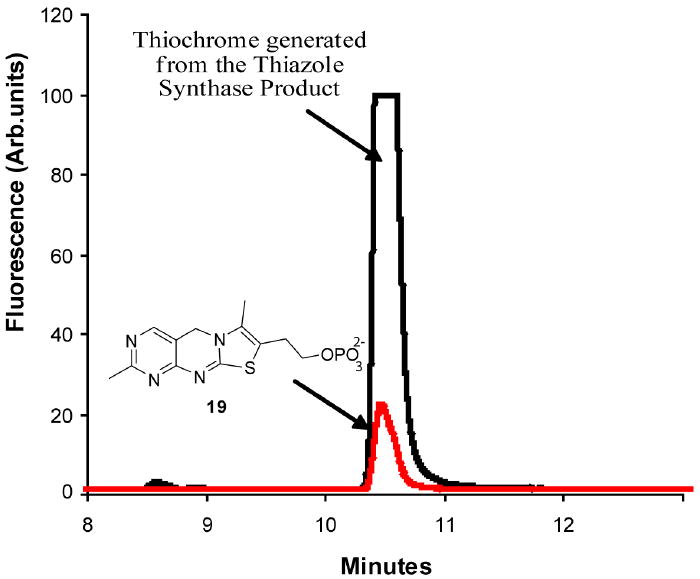
HPLC analysis showing the conversion of the peak A compound to thiochrome phosphate (red trace).
Isolation and characterization of the Peak A compound
The most direct way to identify the Peak A compound was to compare its chromatographic behavior with that of authentic samples of thiazoles 14 and 15, the two most likely alternative products of the bacterial thiazole synthase-catalyzed reaction. Access to these compounds was greatly facilitated by our recent demonstration that species 24 and 25 copurify with the Saccharomyces cerevisiae thiazole synthase, an enzyme that catalyzes very different chemistry.12,13 Release of these metabolites from the S. cerevisiae thiazole synthase by heat denaturation, followed by purification by reverse-phase HPLC and treatment of these metabolites with nucleotide pyrophosphatase, generated authentic samples of the required reference compounds (Figure 5).
Figure 5.
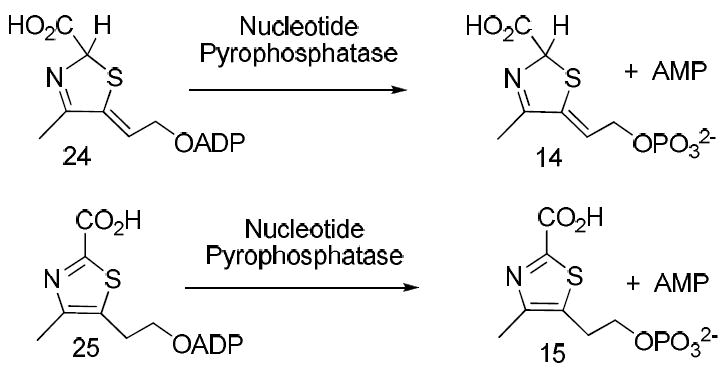
Procedures for the production of reference compounds 14 and 15
HPLC analysis, by strong anion-exchange, clearly demonstrated that the Peak A compound comigrated with 14 (Figure 6a). To further confirm this identity, 14, 15 and the Peak A compound were dephosphorylated by treatment with alkaline phosphatase and the resulting alcohols were reanalyzed by reverse phase HPLC. Again, the dephosphorylated Peak A compound comigrated with 22, the dephosphorylated product of 14 (Figure 6b).
Figure 6.
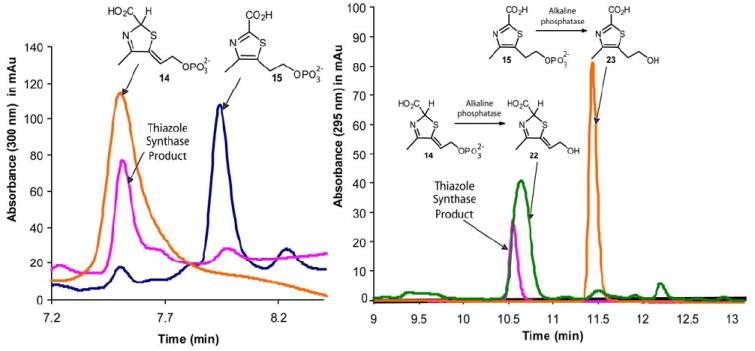
(a) - HPLC analysis of the thiazole synthase product (pink) and reference compounds 14 (orange) and 15 (blue). (b) - HPLC analysis of the dephosphorylated product of the thiazole synthase (pink) catalyzed reaction and reference compounds 22 (green) and 23 (orange).
The thiazole tautomer 14 (Peak A compound) is difficult to isolate in quantities suitable for NMR analysis because it decomposes extensively during the later stages of lyophilization presumably due to pH changes that occur during the lyophilization process. No cryoprotectants 18 could be used during lyophilization as added components would interfere with the NMR signals. The dilute samples of 14 used for the HPLC analysis however did not show this decomposition. Hence, alcohol 22 which is relatively stable during lyophilization, could be isolated in sufficient quantities and was used for spectroscopic analyses. 1D 1H-and 2D-1H-dqf-COSY spectra were collected, which are fully consistent with structure 22, Figure 7.
Figure 7.
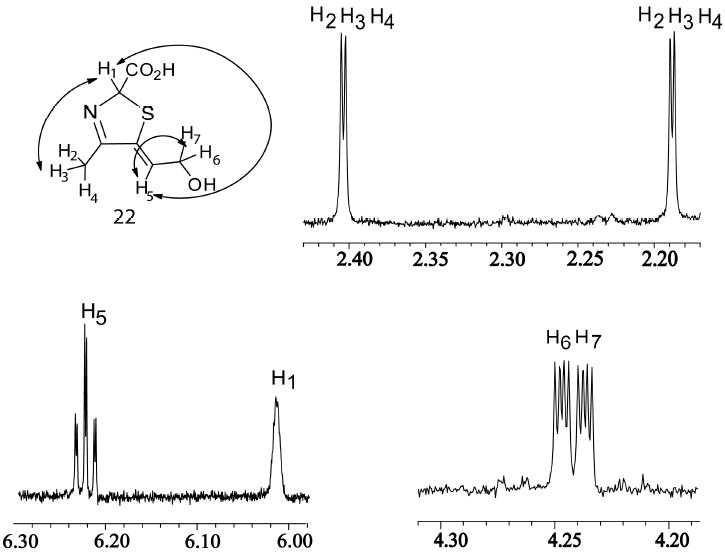
1H-NMR analysis of the dephosphorylated Peak A compound 22. The DXP sample used was labeled with 13C on the methyl group (unrelated reasons) hence the additional splitting of the H2/H3/H4 protons. Additional proton coupling, from the 2D dqf-COSY experiment, are indicated on the structure. (Supplementary Figure 4, 5 and 6).
Optimization of the complex reaction catalyzed by the bacterial thiazole synthase allowed for the unequivocal identification of the reaction product as the thiazole tautomer 14 rather than the thiazole 16. This identification underscores the problem of identifying trace metabolites by enzyme-catalyzed derivatization, even when the derivatizing enzyme has been very well-studied. The stability of compounds 14 and 22 is surprising, as thiazole tautomers should readily aromatize. However, 22 is stable in the experimental time scale of purification and buffer-exchange by HPLC over 10 hours and lyophilization over 48 hours. This stability suggests that an as yet unidentified enzyme may be involved in the catalysis of this aromatization reaction.
Experimental Methods
Source of Chemicals
All chemicals and snake venom nucleotide pyrophosphatase were purchased from Sigma-Aldrich Corporation (USA) unless otherwise mentioned. Calf intestinal phosphatase was obtained from New England Biolabs. LB medium was obtained from EMD Biosciences. Kanamycin, ampicillin and IPTG were purchased from LabScientific Inc. NTA resin was the NTA superflow by Qiagen. The microcon membrane filters were from Millipore. Analytical HPLC (Agilent 1100 instrument) was carried out using a Phenomenex Gemini C18 110A (150×4.6 mm, 5 μm ID) reverse phase column and a Phenosphere Strong Anion-Exchange (SAX) 80A (250×4.6 mm, 5 μm ID) column. HPLC purifications were carried out using a semi-prep Supelco LC-18-T (250×10 mm, 5 μm ID) column. HPLC grade solvents were obtained from Fisher Scientific. Previously synthesized stock of [1-13C]-DXP 14 was used as the substrate of the thiazole reconstitution reactions.
Overexpression and purification of enzymes
ThiSG, ThiF, NifS, ThiO and ThiE: E. coli BL21(DE3) containing the ThiSG overexpression plasmid (ThiG is co-purified with ThiS for stability) in pET16b was grown in LB medium containing ampicillin (40 μg/mL) with shaking at 37 °C until the OD600 reached 0.6. At this point, protein overexpression was induced with isopropyl-β-D-thiogalactopyranoside (IPTG) (final concentration = 2 mM) and cell growth was continued at 15 °C for 16 h. The cells were harvested by centrifugation and the resulting cell pellets were stored at -80 °C. To purify the protein, the cell pellets from 1L of culture were resuspended in 25 mL lysis buffer (10 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8) and lysed by sonication (Heat systems Ultrasonics model W-385 sonicator, 2 s cycle, 50% duty). The resulting cell lysate was clarified by centrifugation and the ThiSG protein was purified on Ni-NTA resin following the manufacturer’s instructions. After elution, the protein was desalted using a 10-DG column (BioRad) pre-equilibrated with 50 mM Tris-HCl buffer, pH 7.8. The remaining proteins ThiF (pET22), NifS (pET16), ThiO (pET22) ThiE (pQE32 and pREP4) were overexpressed and purified in a similar manner.15,16 NifS, ThiO and ThiE were stored in aliquots at -80 °C in 20% glycerol. ThiSG and ThiF were purified immediately before use.
Reconstitution of the thiazole synthase catalyzed reaction on an analytical scale
All solutions were made with 50 mM tris buffer, pH 8. Final concentrations of the reactants are given in parentheses. Cysteine (0.35 mM), DTT (0.70 mM), ATP (0.60 mM) and MgCl2 (3.5 mM) were incubated with purified ThiSG (1.25 μM), ThiF (1.24 μM) and 70 μL NifS (1.38 μM) for 1.5 hours. Total volume of this solution was 425 μL. Glycine (6.50 mM), DXP (0.33 mM), MgCl2 (3.5 mM) and ThiO (6.8 μM) were then added to this reaction mixture and the final volume of the reconstitution mixture now was 610μL. This mixture was incubated for an additional 2 hours. The reaction mixture was then analyzed for product formation using the thiochrome assay (see below). In this reconstitution, 16% of the DXP was converted to product. This is a 3-fold improvement over our previously reported reconstitution, and corresponds to about 12 turnovers by the thiazole synthase.
Thiochrome Assay
The thiochrome assay involves conversion of the thiazole product of the reconstitution to thiamin phosphate (18) and further to thiochrome phosphate. The product of the thiazole reconstitution is reacted with 4-amino-5-hydroxymethyl-2-methylpyrimidinyl pyrophosphate (17) (0.5 mM) in the presence of thiamin phosphate synthase (ThiE) (1.00 μM). The reaction is allowed to stand at room temperature for 2 hours and then quenched with an equal volume of 10% TCA. Potassium acetate (50 μL of 4M) is added to 100 μL of the quenched reaction followed by oxidative cyclization to thiochrome phosphate (10) using 50 μL of a saturated solution of K3Fe(CN)6 in 7M NaOH. The oxidation reaction is neutralized after 1 minute with 6M HCl and analyzed by reverse phase HPLC with fluorescence detection (excitation at 365 nm, emission at 450 nm). The following linear gradient, at a flow rate of 1 mL/min, was used. Solvent A is water, solvent B is 100 mM K2HPO4, pH 6.6, solvent C is methanol. 0 min: 100% B; 2 min: 10% A, 90%B; 10 min: 25% A, 15% B, 60% C; 12 min: 25% A, 15% B, 60%; 15 min: 100% B; 17 min: 100% B. A time-course for the thiazole reconstitution is shown in supplementary Figure 1.
Reconstitution of the thiazole synthase-catalyzed reaction on a preparative scale
All solutions were made with 50 mM tris buffer, pH 8. Cysteine (0.35 mM), DTT (0.70 mM), ATP (0.60 mM) and MgCl2 (3.5 mM) were incubated with purified ThiSG (1.25 μM), ThiF (1.24 μM) and NifS (1.38 μM) for 1.5 hours. Total volume of this solution was 1.3 mL. Glycine (6.50 mM), DXP (0.33 mM), MgCl2 (3.5 mM) and ThiO (6.8 μM) were added to this reaction mixture and the reconstitution solution now had a final volume of 1.8mL. This mixture was incubated for an additional 2 hours. The reaction mixture was then analyzed for product formation by HPLC analysis, with UV detection. The following linear gradient, at a flow rate of 3 mL/min, was used: Solvent A is water, solvent B is 100 mM KPi, pH 6.6, solvent C is methanol. 0 min: 100% B; 5 min: 10% A, 90% B; 12 min: 25% A, 15% B, 60% C; 18 min: 25% A, 15% B, 60% C; 22 min: 100% B; 25 min: 100%B. A product, eluting at 2.8 min was observed. This product did not comigrate with thiazole phosphate (16) (Figure 3). The compound eluting at 2.8 min was collected and buffer exchanged into a low concentration of volatile ammonium acetate buffer by HPLC. The following linear gradient was used at a flow rate of 3 mL/min: Solvent A is water, solvent B is 25 mM NH4OAc, pH 6.6, solvent C is methanol. 0 min: 100% B; 2 min: 10% A, 90%B; 6 min: 15% A, 20% B, 65% C; 8 min: 15% A, 20% B, 65%; 11 min: 100% B; 14 min: 100%B. The collected fractions were then pooled and lyophilized to successfully obtain the product of the bacterial thiazole synthase.
1D-1H NMR and 2D-dqf-COSY NMR analyses
To prepare the thiazole tautomer alcohol 22 for 1D-1H NMR and 2D-dqf-COSY NMR studies, the product 14 that eluted out from the HPLC purification at 2.8min was collected and treated with 1 unit of calf intestinal phosphatase for 20 min at room temperature to form 22. This was then buffer exchanged into a low concentration of volatile ammonium acetate buffer by HPLC. The following linear gradient was used at a flow rate of 3 mL/min: Solvent A is water, solvent B is 25 mM NH4OAc, pH 6.6, solvent C is methanol. 0 min: 100% B; 2 min: 10% A, 90%B; 6 min: 15% A, 20% B, 65% C; 8 min: 15% A, 20% B, 65%; 11 min: 100% B; 14 min: 100%B. The collected fractions were then pooled and lyophilized to obtain the thiazole tautomer alcohol 22, which was then used for 1D-1H NMR and 2D-dqf-COSY NMR studies. A Shigemi NMR tube (susceptibility-matched for D2O) was used for all the experiments, which were carried out on a Varian INOVA 600 MHz instrument equipped with a 5 mm triple gradient inverse-detection HCN probe.
Preparation of HPLC standards of 14, 22, 15 and 23
Compound 14 was obtained by the following procedure - overexpressed S. cerevisiae THI4p (thiazole synthase) protein was denatured as follows: THI4p from 4 L of culture (~200 mg, 10 mL) was divided into twenty 500 μL aliquots and heat denatured (100 °C, 2minutes). The precipitated protein was removed by centrifugation and the supernatants were combined and filtered through a 10 kDa MW cut off microcon filter. Adenylated 14 was purified by HPLC using the following linear gradient at a flow rate of 3 mL/min: solvent A is water, solvent B is 100 mM KPi, pH 6.6, solvent C is methanol. 0 min: 100% B; 3 min: 10% A, 90%B; 17 min: 34% A, 60% B, 6% C; 21 min: 35% A, 25% B, 40% C; 23 min: 100%B and the collected fractions were pooled. A second HPLC purification, using a low concentration of volatile ammonium acetate buffer, was performed on the pooled fractions using the following linear gradient at a flow rate of 3 mL/min: Solvent A is water, solvent B is 25 mM NH4OAc, pH 6.6, solvent C is methanol. 0 min: 100% B; 2 min: 10% A, 90%B; 6 min: 15% A, 20% B, 65% C; 8 min: 15% A, 20% B, 65%; 11 min: 100% B; 14 min: 100%B. The collected fractions were then lyophilized to yield micromolar quantities of adenylated 14. This was then treated with 1 unit nucleotide pyrophosphatase at pH 7.2 to yield 14 (Figure 5) and further with 1 unit calf intestinal phosphatase in phosphate buffer, pH 7.8 for 20 min. to yield 22 (Supplementary Figure 2). Adenylated compound 15 is also found bound to S. cerevisiae THI4p and 15 was obtained by the same purification procedure as described above for the preparation of compound 14. Compound 23 was synthesized by carboxylation of thiazole alcohol 26 using the reported literature procedure17 (Supplementary Figure 2).
Supplementary Material
Acknowledgments
We acknowledge the help of Frank Schroeder with the NMR dqf-COSY experiment and David Hilmey for the synthesis of the thiazole carboxylate alcohol 22. This research was supported by a grant from the National Institutes of Health to TPB (DK440843).
Footnotes
Supporting Information Available. Figures for time course of thiazole production by thiochrome assay, preparation of HPLC standards for 22 and 23 and 2-D dqf-COSY data for 14. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Butterworth RF. Nutr Res Rev. 2003;16:277–283. doi: 10.1079/NRR200367. [DOI] [PubMed] [Google Scholar]
- 2.Jordan F. Nat Prod Rep. 2003;20:184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- 3.Soriano JM, Moltó JC, Mañes J. Nutr Res. 2000;20:1249–1258. [Google Scholar]
- 4.Begley TP, Downs DM, Ealick SE, McLafferty FW, Van Loon APGM, Taylor S, Campobasso N, Chiu H-J, Kinsland C, Reddick JJ. J Xi Arch Microbiol. 1999;171:293–300. doi: 10.1007/s002030050713. [DOI] [PubMed] [Google Scholar]
- 5.Spenser ID, White RL. Angew Chem Int Ed Engl. 1997;36:1032–1046. [Google Scholar]
- 6.Begley TP. Nat Prod Rep. 1996;13:177–185. doi: 10.1039/np9961300177. [DOI] [PubMed] [Google Scholar]
- 7.Park J-H, Dorrestein PC, Zhai H, Kinsland C, McLafferty FW, Begley TP. Biochemistry. 2003;42:12430–12438. doi: 10.1021/bi034902z. [DOI] [PubMed] [Google Scholar]
- 8.Settembre EC, Dorrestein PC, Park J-H, Augustine AM, Begley TP, Ealick SE. Biochemistry. 2003;42:2971–2981. doi: 10.1021/bi026916v. [DOI] [PubMed] [Google Scholar]
- 9.Dorrestein PC, Zhai H, McLafferty FW, Begley TP. Chem Biol. 2004;11:1373–1381. doi: 10.1016/j.chembiol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Settembre EC, Dorrestein PC, Zhai H, Chatterjee A, McLafferty FW, Begley TP, Ealick SE. Biochemistry. 2004;43:11647–11657. doi: 10.1021/bi0488911. [DOI] [PubMed] [Google Scholar]
- 11.Dorrestein PC, Zhai H, Taylor SV, McLafferty FW, Begley TP. J Am Chem Soc. 2004;126:3091–3096. doi: 10.1021/ja039616p. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. J Am Chem Soc. 2006;128:7158–7159. doi: 10.1021/ja061413o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatterjee A, Schroeder FC, Jurgenson CT, Ealick SE, Begley TP. J Am Chem Soc. 2008;130:11394–11398. doi: 10.1021/ja802140a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor SV, Vu LD, Begley TP, Schoerken U, Grolle S, Sprenger GA, Bringer-Meyer S, Sahm H. J Org Chem. 1998;63:2375–2377. [Google Scholar]
- 15.Park J-H, Dorrestein PC, Zhai H, Kinsland C, McLafferty FW, Begley TP. Biochemistry. 2003;42:12430–12438. doi: 10.1021/bi034902z. [DOI] [PubMed] [Google Scholar]
- 16.Settembre EC, Dorrestein PC, Park J-H, Augustine AM, Begley TP, Ealick SE. Biochemistry. 2003;42:2971–2981. doi: 10.1021/bi026916v. [DOI] [PubMed] [Google Scholar]
- 17.Haginoya N, Kobayashi S, Komoriya S, Yoshino T, Suzuki M, Shimada T, Watanabe K, Hirokawa Y, Furugori T, Nagahara T. J Med Chem. 2004;47:5167–5182. doi: 10.1021/jm049884d. [DOI] [PubMed] [Google Scholar]
- 18.Rexroad J, Wiethoff CM, Jones LS, Middaugh CR. Cell Preservation Technology. 2002;1:91–104. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.